

Abstract
Ataxin-1 is a neurodegenerative disorder protein whose glutamine-repeat expanded form causes spinocerebellar ataxia type 1 (SCA1) in humans and exerts cytotoxicity in Drosophila and mouse. We report here that the cytotoxicity caused by ataxin-1 is modulated by association with a related protein, Brother of ataxin-1 (Boat). Boat and ataxin-1 share a conserved AXH (ataxin-1 and HMG-box protein 1) domain, which is essential for both proteins' interactions with the transcriptional corepressor SMRT and its Drosophila homolog, SMRTER. The Boat–ataxin-1 interaction is mediated through multiple regions in both proteins, including a newly identified NBA (N-terminal region of Boat and ataxin-1) domain. We investigated the physiological relevance of the Boat–ataxin-1 interaction in Drosophila and discovered that a mutant ataxin-1-mediated eye defect is suppressed by ataxin-1's association with Boat. Correspondingly, in transgenic SCA1 mouse, Boat expression is greatly reduced in Purkinje cells, the primary targets of SCA1. Our study thus establishes that Boat is an in vivo binding partner of ataxin-1 whose altered expression in Purkinje cells may contribute to their degeneration in SCA1 animals.
Keywords: ataxin-1, Boat, SCA1, SMRT, SMRTER
Introduction
Spinocerebellar ataxia-1 (SCA1) is a dominant inherited neurodegenerative disease whose pathology is characterized by ataxia, progressive motor deterioration, and loss of Purkinje cells in the cerebellum and neurons in the brainstem (Zoghbi and Orr, 2000). SCA1 is associated with expanded glutamine-repeats in ataxin-1 (Orr et al, 1993; Banfi et al, 1994), a nuclear foci-forming protein whose exact functions are not yet fully understood. Although it is well established that glutamine-repeat expansion in ataxin-1 is central to the pathogenesis of SCA1, emerging evidence indicates that the neurotoxicity of ataxin-1 is modulated by its non-glutamine-repeat regions and by its interacting factors. For example, a recent study indicated that altering the phosphorylated state of mutant ataxin-1 by substituting S776 with A776 greatly diminishes its neurotoxicity in mouse Purkinje cells (Emamian et al, 2003). Correspondingly, increasing the expression of 14-3-3—whose association with ataxin-1 depends on the phosphorylation of S776—enhances ataxin-1 aggregate formation in human cultured cells and causes a more severe mutant ataxin-1-mediated eye phenotype in fly (Chen et al, 2003). By demonstrating that the cytotoxicity of mutant ataxin-1 can be influenced both by regions outside its glutamine-repeat and by its interacting factors—in both the mouse and fly systems—these recent studies highlight not only the importance of understanding the normal function of the protein, but also the power of using model systems to unravel the molecular mechanisms underlying the detrimental effects of mutant ataxin-1.
Our recent study suggested that ataxin-1 is involved in transcriptional regulation (Tsai et al, 2004). This proposed property of ataxin-1 became evident through our observations that ataxin-1 associates directly with the transcriptional corepressor SMRT (Silencing Mediator of Retinoid and Thyroid hormone receptors), forms complexes with HDAC3 (histone deacetylase 3), and represses gene transcription in human cultured cells. We also reported an interaction between ataxin-1 and SMRTER, the Drosophila cognate of SMRT (Tsai et al, 1999), and the localization of ataxin-1 to SMRTER-positive chromosomal loci (Tsai et al, 2004). These lines of evidence indicate that interacting with SMRT and its related factors is a feature of ataxin-1 conserved in evolution, although it has been unclear which specific regions in ataxin-1 are responsible for such interactions. Recent reports have revealed that ataxin-1 encodes an AXH (ataxin-1 and HMG-box protein 1 (HBP1)) domain; such a domain is found in many vertebrate and invertebrate proteins (de Chiara et al, 2003). The conservation of this domain therefore led us to speculate that it may be involved in ataxins-1's interaction with SMRT and SMRTER.
In this paper, we confirm an AXH domain-dependent SMRT interaction for ataxin-1. Following this initial finding, we subsequently discovered that another AXH domain protein, which we have named Brother of ataxin-1 (Boat), also interacts with SMRT and SMRTER in an AXH domain-dependent manner. Our characterization of Boat further reveals that Boat also interacts with ataxin-1, suggesting that the biological properties of ataxin-1, Boat, and SMRT/SMRTER are interconnected. To investigate the relationships among these proteins, we employed various systems, including yeast, cultured human cells, transformed Drosophila and mouse. Our data reveal that Boat is an in vivo binding partner of ataxin-1 whose association with mutant ataxin-1 modulates its properties.
Results
The AXH domain of ataxin-1 is involved in SMRT interaction
Our previous results indicated that ataxin-1 interacts with SMRT in yeast (Tsai et al, 2004). While we mapped the ataxin-1 interacting domain to the C-terminal 1755–2518 region of SMRT, the specific region(s) in ataxin-1 responsible for SMRT association has not yet been identified. To determine the precise region in ataxin-1 required for SMRT interaction, we used the yeast two-hybrid method (Fields and Song, 1989). We transformed yeast Y190 cells with a Gal4 DNA binding domain-SMRT fusion construct, GBT-SMRT(1755–2518), along with the serial Gal4 activation domain (GAD)-ataxin-1 fusion constructs shown in Figure 1A. The transformed cells were tested for positive (in blue color) or negative protein–protein interactions. Whereas positive reporter activities were detected when ataxin-1(4–703) and ataxin-1(227–703) constructs were used, no reporter activity was observed for the ataxin-1(4–553) construct. Since the AXH domain, which corresponds to the 575–688 region of ataxin-1, is deleted from ataxin-1(4–553), this result suggests that the AXH domain is involved in SMRT interaction. To test this supposition further, we generated and tested a construct expressing an ataxin-1 variant whose AXH domain is specifically deleted. Indeed, no interaction was observed between ataxin-1(227–703ΔAXH) and SMRT(1755–2518), further confirming the AXH domain's involvement in SMRT interaction.
Figure 1.
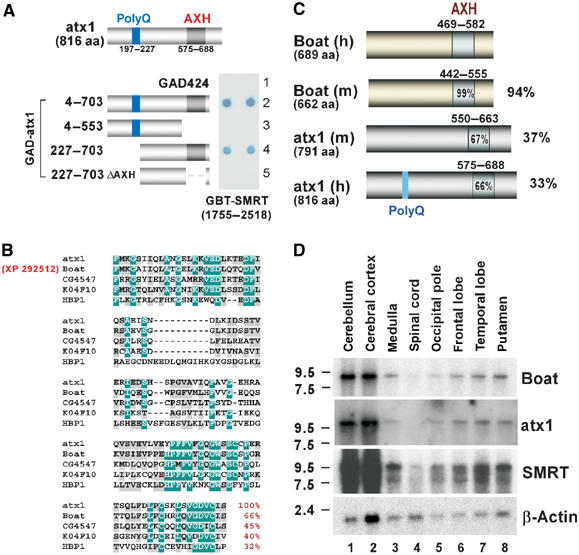
The identification of Boat, an AXH domain protein related to ataxin-1 (also referred to as atx1 here and in other figures). (A) Yeast two-hybrid assays mapping the regions in ataxin-1 responsible for SMRT interaction. The diagram shows the structure of ataxin-1 and the regions of ataxin-1 used to construct the GAD-ataxin-1 variants. ΔAXH represents deletion of the 575–688 region of ataxin-1. Positive β-galactosidase activity is based on blue color reaction from the dot blot assays. (B) Sequence alignment of the AXH domains of ataxin-1, XP 292512 (Boat), fly CG4547, worm K04F10.1, and human HBP1. The highly and moderately conserved residues are marked in green and gray, respectively. The percentage of similar residues shared by ataxin-1 and each of the listed proteins in their AXH domain is shown in red. (C) Schematic diagram showing the homologous regions shared by the human (h) and mouse (m) Boat and ataxin-1 proteins. The percentage represents the extent of sequence similarity shared between human Boat and human ataxin-1, mouse ataxin-1, or mouse Boat. (D) Northern blot analysis showing the Boat, ataxin-1, and Smrt transcripts detected in different human brain tissues. β-Actin is used as an internal control.
The identification of Boat
Since an AXH domain is found in several uncharacterized vertebrate and invertebrate proteins, including Drosophila CG4547, Caenorhabditis elegans K04F10.1 (de Chiara et al, 2003; Chen et al, 2004), and a novel human protein, XP 292512, present in the human genome database (Figure 1B), we investigated whether any of these proteins also interacts with SMRT. In this study, we concentrate on XP 292512 (689 amino acids) because this protein is closely related to ataxin-1 (Figure 2). The resemblance of this protein to ataxin-1 led us to name it Brother of ataxin-1 (Boat). A mouse protein (XM 146508) (662 amino acids), which is 94% similar to human Boat, was also identified in the mouse genome database (Figures 1C and 2). Unlike ataxin-1, neither human nor mouse Boat encodes any glutamine-repeat tract.
Figure 2.

Sequence alignment of human and mouse Boat and ataxin-1 and their functional domains. Identical and similar residues are shown in black and gray, respectively. The AXH domain, the polyglutamine-repeat tract, and the NBA domain are marked with a blue box, a red line, and a green box, respectively. The region deleted within the previously defined self-association domain of ataxin-1 is underlined with an orange line and is marked as ΔSAR (self-association region) in this study.
Dot blot analysis reveals that Boat transcripts are expressed in multiple human tissues and cell lines (Supplementary Result 1). In the human brain, Boat is detected as a single transcript (∼9 kb) with the highest expression levels observed in the cerebellum and the cerebral cortex (Figure 1D). Both ataxin-1 and Smrt display similar expression profiles in the human brain, implying that Boat, ataxin-1, and SMRT may cooperate in regulating the development or functioning of the brain.
Boat interacts with SMRT and its related factors
The sequence resemblance between Boat and ataxin-1 suggests that Boat may interact with SMRT. To test this possibility, we continued to use the yeast two-hybrid approach. Yeast Y190 cells were transformed with GBT-SMRT(1755–2518) and a GAD plasmid expressing full-length Boat. Strong reporter activity was detected (Figure 3A), demonstrating that Boat is another AXH domain protein that interacts with SMRT. We next tested whether Boat also interacts with other SMRT-related factors and, if so, whether the demonstrated interactions also involve the AXH domain.
Figure 3.
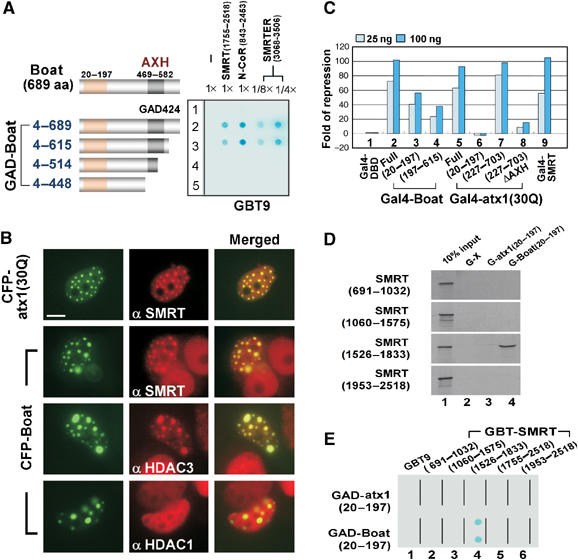
Boat and ataxin-1 share similar, but not identical, properties. (A) Yeast two-hybrid assays showing an AXH domain-dependent interaction between Boat and SMRT, N-CoR, or SMRTER, using the indicated plasmids. In the case of SMRTER, only 1/8 or 1/4 of the normal amount of yeast culture was used. The tested regions of SMRT, N-CoR, and SMRTER are indicated. The diagram shows the structure of Boat and the fragments used to generate the GAD-Boat constructs. The AXH domain is shown as a gray box. The 20–197 region (NBA) of Boat is shown as an orange box. (B) Immunostaining results showing the colocalization of SMRT or HDAC3, but not HDAC1, with CFP-Boat nuclear foci. MCF-7 cells transfected with plasmid expressing CFP-Boat or CFP-ataxin-1(30Q) were immunostained with the indicated antibodies (Texas red, red). CFP-ataxin-1(30Q) was used as a positive control. Scale bar: 10 μM. (C) Gal4 reporter assays showing the transcriptional properties of different Gal4-Boat and Gal4-ataxin-1 variants. HEK293 cells were co-transfected with a Gal4-responsive luciferase reporter (MH100 × 4), along with CMX-based Gal4-DBD ataxin-1 and Boat variants. Luciferase reporter activity was normalized by the concentration of total proteins in the cell extract. Both Gal4-SMRT and Gal4-ataxin-1(30Q) were used as positive controls. (D) Pull-down assays showing the specific interactions between Boat(20–197) and SMRT(1526–1833). In vitro pull-down assays were carried out using bacterially expressed GST fusion proteins and in vitro-translated 35S-methionine-labeled SMRT fragments. G: GST. (E) Yeast two-hybrid assays showing a specific interaction between Boat(20–197) and SMRT(1526–1833). The different combinations of plasmids used in the yeast assays are indicated.
A series of GAD-Boat constructs (Figure 3A) were tested against constructs expressing SMRT, nuclear receptor corepressor (N-CoR), and SMRTER. N-CoR is an SMRT-related factor (Horlein et al, 1995), whereas SMRTER is the homolog of SMRT and N-CoR in fly (Tsai et al, 1999). We tested the potential interaction between Boat and the C-terminal region of N-CoR and SMRTER because the corresponding region in SMRT is involved in ataxin-1 and Boat interactions. In all three cases, positive interactions were detected when we used a construct expressing either full-length Boat or Boat(4–615). In contrast, we detected negative interactions when constructs expressing Boat(4–514) and Boat(4–448) were used. Because Boat(4–448) lacks the entire AXH domain and Boat(4–514) lacks the C-terminal half of the AXH domain, these results reveal that the C-terminal half of the AXH domain is critical for SMRT, N-CoR, and SMRTER interaction.
Boat forms nuclear foci that recruit SMRT and HDAC3 in cultured human cells
In light of our observations that ataxin-1 forms nuclear foci that recruit endogenous SMRT and HDAC3, and that ataxin-1 exerts transcriptional repression in mammalian cells (Tsai et al, 2004), we predicted that Boat would recapitulate these observed properties of ataxin-1 in cultured human cells. Accordingly, we transfected MCF-7 cells with a plasmid expressing a cyan fluorescent protein (CFP)-tagged Boat fusion protein (CFP-Boat) and looked for any effects on the distribution of endogenous SMRT and HDAC3. CFP-Boat does form a nuclear focal pattern similar to that of CFP-ataxin-1(30Q) in transfected cells (Figure 3B). Indirect immunostaining experiments using either SMRT or HDAC3 antibody revealed that, upon formation of CFP-Boat nuclear foci, the nuclear patterns of endogenous SMRT and HDAC3 are affected. The newly formed punctate patterns of both proteins are virtually identical to the pattern displayed by CFP-Boat, thus suggesting their interactions. The interactions between Boat and SMRT and between Boat and HDAC3 are specific, because no alteration in the nuclear distribution of HDAC1 was observed in a parallel experiment.
Boat encodes two independent transcriptional repressive domains
Because SMRT and HDAC3 are involved in transcriptional repression (Chen and Evans, 1995; Ordentlich et al, 1999; Park et al, 1999; Privalsky, 2004; Tsai and Fondell, 2004), we looked for transcriptional repression associated with Boat, using the heterologous Gal4 system. In this assay, Boat is tethered to the target sites of Gal4 through fusion with the Gal4 DNA-binding domain (DBD); its transcriptional properties can therefore be measured by the Gal4 reporter assays. Like Gal4-ataxin-1(30Q) and Gal4-SMRT, which were used as positive controls, Gal4-Boat exerts potent transcriptional repressive effects (Figure 3C, lanes 2, 5, and 9). This result confirms our expectations. Transcriptional repression, although to a lesser degree, was also observed for two truncated Boat variants, Boat(20–197) and Boat(197–615) (lanes 3 and 4), suggesting that Boat encodes at least two transcriptional repression domains.
In parallel experiments, transcriptional repressive effects were also observed for ataxin-1(227–703) (lane 7). However, a substantial reduction in transcriptional repression was observed for ataxin-1(227–703ΔAXH) and for ataxin-1(20–197) (lanes 6 and 8). In the case of ataxin-1(227–703ΔAXH), its reduced transcriptional repression can be explained by the impaired interaction between this ataxin-1 variant and SMRT (Figure 1A). Our finding that ataxin-1(20–197) has no repressive activity surprised us, because the corresponding 20–197 region of Boat, despite resemblance in their sequences, does cause transcriptional repression. The difference in the transcriptional effects of Boat and ataxin-1 suggests that these two proteins are not functionally equivalent in their N-terminal regions.
The identification of a functional NBA domain
The transcriptional repression displayed by Boat 20–197 prompted us to investigate whether this region interacts with SMRT. We first employed an in vitro GST pull-down assay. We generated bacterial GST, GST-Boat(20–197), or GST-ataxin-1(20–197) fusion proteins and tested them against various in vitro-translated 35S-methionine-labeled SMRT proteins. As shown in Figure 3D, specific binding takes place only between GST-Boat(20–197) and SMRT(1526–1833). No other tested SMRT fragments show interaction with GST-Boat(20–197), nor does GST-ataxin-1(20–197) bind any of the tested SMRT fragments.
The specific interaction between Boat(20–197) and SMRT(1526–1833) was further confirmed in yeast using two-hybrid assays. The combination of GAD-Boat(20–197) and GBT-SMRT(1526–1833) gives rise to prominent reporter activation (Figure 3E). Because Boat(20–197) does not interact with SMRT(1755–2518) (lane 5), whereas full-length Boat does (Figure 3A, lane 2), these results suggest that Boat encodes at least two independent regions that enable SMRT interaction: its AXH domain is involved in SMRT(1755–2518) association, while its N-terminal 20–197 region is responsible for SMRT(1526–1833) interaction. Because the 20–197 region of Boat apparently encodes a functional domain, we named this N-terminal region of Boat and the corresponding region of ataxin-1 the NBA domain.
Boat interacts with itself and with ataxin-1
It has been shown that ataxin-1 interacts with itself (Burright et al, 1997). We therefore investigated whether Boat interacts with itself or with ataxin-1. Yeast cells were transformed with GBT-Boat or GBT-Boat(20–197), along with GAD constructs expressing ataxin-1(30Q), ataxin-1(82Q), and Boat. With the exception of GAD424, all other combinations of plasmids resulted in reporter activation (Figure 4A). These results reveal that (1) Boat is able to interact with itself, (2) Boat is able to interact with ataxin-1, (3) Boat self-association and Boat–ataxin-1 interaction are in part mediated through the NBA domain of Boat, and (4) the Boat–ataxin-1 interaction is not perturbed by glutamine-repeat expansion. Considering these findings and our earlier result that the 20–197 region of Boat also binds SMRT (Figure 3D and E), we conclude that the NBA domain of Boat is a versatile domain that may have pivotal roles in influencing the functions of Boat and ataxin-1.
Figure 4.
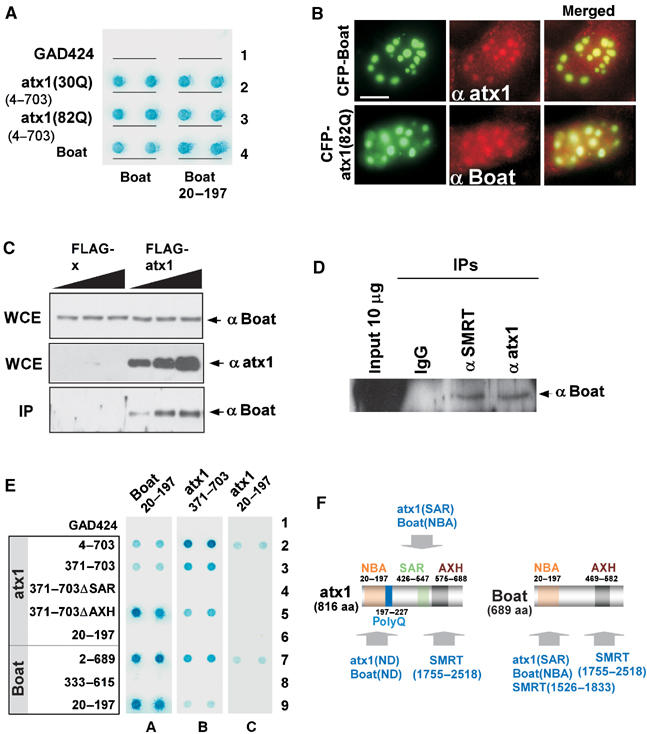
Boat interacts with itself and with ataxin-1 through multiple domains. (A) Yeast two-hybrid assays showing the interactions between Boat or Boat(20–197) and ataxin-1 variants, using the indicated plasmids. (B) Immunostaining images showing the colocalization of ataxin-1 and Boat in cultured human cells. MCF-7 cells were transfected with plasmids expressing CFP-Boat or CFP-ataxin-1(82Q), followed by immunostaining with an ataxin-1 or Boat antibody (Texas red, red). Scale bar: 10 μM. (C) Co-IP assay showing the presence of endogenous Boat in the precipitated FLAG-ataxin-1(82Q) protein complexes. HEK293 cells were transfected with various amounts of plasmids expressing either FLAG or FLAG-ataxin-1(82Q). The resulting cell extracts were immunoprecipitated with FLAG (M2) antibody-conjugated agarose beads. Western blot analysis for the precipitated protein complexes was carried out using Boat and ataxin-1 antibodies. WCE: whole cell extract; IP: immunoprecipitation. (D) Co-IP assay showing the interactions between Boat and ataxin-1 and between Boat and SMRT under normal physiological conditions. Nuclear extracts prepared from HeLa cells were subjected to immunoprecipitation using ataxin-1 or SMRT antibody-bound protein A/G agarose beads. Western blot analysis was carried out using a Boat antibody. (E) Yeast two-hybrid assays mapping the regions in Boat and in ataxin-1 responsible for their self- and mutual interactions. ΔSAR and ΔAXH represent the deletion of 426–547 and 575–688 from ataxin-1, respectively. (F) A summary of the results shown in (E). The locations of the mapped SAR, AXH, and NBA domains and their associated properties in Boat and ataxin-1 are indicated. ND: not yet fully defined.
We next examined whether the same Boat–ataxin-1 interactions can be recapitulated in human cultured cells. MCF-7 cells were first transfected with a plasmid expressing CFP-Boat or CFP-ataxin-1(82Q) and then immunostained with ataxin-1 antibody and Boat antibody, respectively. In both cases, we observed colocalization of the endogenous protein with the CFP fusion (Figure 4B). The mutual association between ataxin-1 and Boat in cultured cells was further investigated using a co-immunoprecipitation (Co-IP) approach. In the first experiment, cells were transfected with various amounts of plasmids expressing FLAG or FLAG-ataxin-1(82Q). The cell extracts derived from each set of transfected cells were subjected to immunoprecipitation, using FLAG antibody-conjugated beads. Indeed, a specific interaction between endogenous Boat and FLAG-ataxin-1(82Q)—but not between Boat and FLAG—was revealed by Western blot analysis (Figure 4C).
In the second experiment, we examined the interactions between Boat and ataxin-1 and between Boat and SMRT under normal physiological conditions. Since HeLa cells display a higher level of Boat, ataxin-1, and SMRT expression (Supplementary Result 1, spot B10, not shown for ataxin-1), nuclear extracts prepared from nontransfected HeLa cells were subjected to Co-IP experiments, using SMRT or ataxin-1 antibodies. Our Western blot analysis, using Boat antibody, reveals that Boat is specifically present in the protein complexes immunoprecipitated by ataxin-1 or by SMRT antibody, but not by the control IgG (Figure 4D). We therefore conclude that in vivo interactions between Boat and ataxin-1, and between Boat and SMRT, do take place in human cells under normal physiological conditions.
Boat and ataxin-1 interact with each other through multiple domains
We next mapped which specific domains of Boat and ataxin-1 are responsible for their self- and mutual interactions. We generated a series of truncated GAD constructs for ataxin-1 and Boat (Figure 4E) and tested them individually against GBT constructs for Boat(20–197), ataxin-1(20–197), or ataxin-1(371–703). Of the tested GAD-ataxin-1 constructs, ΔAXH and ΔSAR represent the deletions of the AXH domain and of the previously mapped self-association region (SAR), respectively. We included the GAD-ataxin-1ΔSAR and GBT-ataxin-1(371–703) constructs in our experiments because it has been shown that the removal of the SAR from ataxin-1 impairs its ability to self-associate (Klement et al, 1998) and that the 371–703 region of ataxin-1 harbors a self-association domain (Burright et al, 1997).
As shown in Figure 4E, our results reveal Boat's and ataxin-1's complex pattern of interactions. Notably, the NBA domain of Boat and the SAR domain of ataxin-1 are responsible for each protein's self-dimerization (spots A7–9 and B2–4) and for their mutual interaction (spots A2–4 and B7–9). In contrast, the AXH domain of ataxin-1 is dispensable for its self-association and for Boat interaction (spots A5 and B5). Importantly, we found that the NBA domain of ataxin-1 encodes an additional domain for self-association and for Boat interaction (spots C2 and C7). The specific interactions enabled by each of the analyzed domains in Boat and ataxin-1 are summarized in Figure 4F.
Boat and ataxin-1 interact with each other in Drosophila
Since we have established a direct protein–protein interaction between Boat and ataxin-1 in mammalian cells, we predict that their interaction can be visualized in Drosophila as well. Fly lines expressing either individual protein or different combinations of ataxin-1(82Q), FLAG-ataxin-1(0Q), FLAG-Boat, or HA-Boat(NBA) proteins were generated and tested in salivary glands. In this study, we used ataxin-1(0Q) as a control to Boat, because Boat lacks any glutamine-repeat tract. Indeed, our results shown in Supplementary Results 2 and 3 confirm that Boat is a specific in vivo binding partner of ataxin-1 and that their interaction is, in part, mediated through the NBA domain of Boat.
Boat modulates the eye phenotypes caused by ataxin-1 in Drosophila
The physical interaction between ataxin-1 and Boat prompted us to investigate the physiological relevance of their interaction. Drosophila eyes were used for such an investigation, because it has been shown that ataxin-1(82Q) causes defects in Drosophila eyes (Feany and Bender, 2000; Fernandez-Funez et al, 2000; Tsai et al, 2004). Specifically, we were interested in knowing whether the ataxin-1(82Q)-mediated eye phenotype is modulated by Boat. Accordingly, we crossed an eye-specific Gal4 line, GMR-Gal4, with fly lines expressing different combinations of ataxin-1 and Boat variants mentioned above. The resulting eye phenotypes were analyzed and compared using light microscopy, scanning electronic microscopy, and cross-sections that were immunostained with a neuronal specific 22C10 antibody.
We confirmed a wild-type eye structure for GFP (a control protein) and a severely deformed eye phenotype for ataxin-1(82Q) (Figure 5A and B). In the latter case, prominently glazed eye surface, loss of bristles and pigmentation, and disarrayed of ommatidia were observed. The ridge along the dorsal–ventral axis of the eye consistently displays the most severe defects (marked with an arrow). Severe degeneration of photoreceptors was also observed (Figure 5B′). Somewhat unexpectedly, expressing ataxin-1(0Q) is also detrimental to eye morphogenesis. This observation is consistent with our observation for ataxin-1(0Q) in salivary glands (Supplementary Result 2D). The defects include loss of bristles and pigmentation, dented ommatidium, and degeneration of photoreceptors (Figure 5C and C′). However, the eye's overall structure and the organization of ommatidia are still maintained. This observation indicates that ataxin-1 has intrinsic toxicity in fly independent of its glutamine-repeat. In contrast, Boat- or Boat(NBA)-expressing eyes are indistinguishable from those of wild-type flies (Figure 5D, D′, E, and E′), indicating that Boat is benign in fly tissues. The fact that Boat and Boat(NBA) do not cause malformation in the eye made it possible for us to investigate whether these two Boat variants can modulate the ataxin-1(82Q)-mediated eye phenotype.
Figure 5.
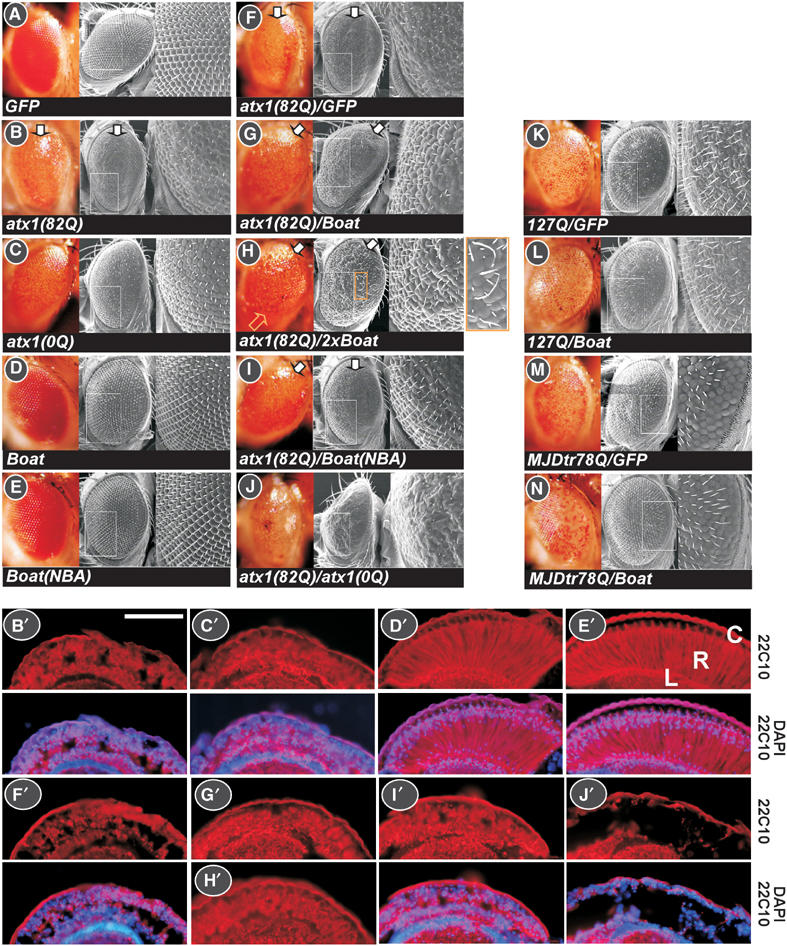
Boat modulates the eye phenotype caused by mutant ataxin-1 in Drosophila. (A–N) Light microscopy and scanning electron microscopy images, showing the surface of adult compound eyes. The genotype of each examined fly line is indicated. In these studies, an eye-specific Gal4 driver line, GMR-Gal4, was used. The UAS-GFP in (A, F, K, M), UAS-HA-127Q in (K, L), and UAS-MJDtr(78Q) in (M, N) were used as control fly lines. The enlarged image corresponds to the boxed area in each examined eye. The arrows in (B, F–I) indicate the ridge of the damaged compound eye along the dorsal–ventral axis. The yellow arrow and the enlarged image in (H) highlight the thickening and the elongated bristles. (B′–J′) Cross-sections of retinas corresponding to the eyes shown in (B–J). All frozen sections were immunostained with a 22C10 antibody to mark the photoreceptors and were counterstained with DAPI. R: retina; C: cornea; L: lamina. Scale bar: 1000 μM.
To facilitate our genetic experiments, we generated a recombined fly line, GMR-Gal4; UAS-ataxin-1(82Q), and tested it against fly lines expressing GFP, Boat, Boat(NBA), and ataxin-1(0Q). While no modulating effect was detected when the control GFP fly line was used (Figure 5F), moderate suppressing effects were obtained when fly lines expressing Boat or Boat(NBA) were used (Figure 5G, I, G′, and I′). A more significant suppressing effect was achieved when we used fly lines carrying two copies of the UAS-Boat transgene (Figure 5H and H′). We observed a restoration of bristle formation, pigmentation, and overall eye structure. These findings indicate that ataxin-1(82Q)-mediated cytotoxicity is partially neutralized by ataxin-1's association with Boat. Interestingly, a gain-of-function phenotype, showing thicker and longer bristles, was also observed (see enlarged image in Figure 5H). This phenomenon suggests that the Boat–ataxin-1 complex promotes bristle formation in eyes. In contrast to the observed effects of Boat, ataxin-1(0Q) aggravates the ataxin-1(82Q)-mediated eye phenotype, evidenced by collapsed eye structure and severely degenerated photoreceptors (Figure 5J and J′). These results reveal that the ataxin-1(82Q)–ataxin-1(0Q) complex and the ataxin-1(82Q)–Boat complex exert very different biological properties in fly.
To determine whether the observed suppressing effect of Boat is specific to ataxin-1(82Q), we additionally tested Boat against two other polyglutamine disease proteins with known toxicity. Whereas 127Q consists mainly of a long glutamine-repeat tract, MJDtr(78Q) represents a pathological form of spinocerebellar ataxia type 3 (SCA3/MJD) (Warrick et al, 1998; Kazemi-Esfarjani and Benzer, 2000). In both cases, their characteristic eye phenotypes are not affected by Boat (Figure 5K–N), demonstrating that the suppressing effect of Boat is specific to ataxin-1(82Q).
Boat and ataxin-1 display similar expression patterns in the mouse brain
Having established the relationship between ataxin-1 and Boat in human cells and Drosophila, we next investigated their relationship in the mouse brain. We first examined the expression patterns of Boat, ataxin-1, and SMRT in 8-week-old FVB/N (wild-type) mouse brains, using specific antibodies that we have developed for each of these proteins. To determine which brain tissues express these proteins, we employed two additional antibodies: Neuronal Nuclei (NeuN) antibody marks neuronal cells (but not Purkinje cells) in the CNS, while Calbindin antibody labels Purkinje cells.
Boat, ataxin-1, and SMRT are abundantly and similarly expressed in different regions of the mouse brain (Figures 6 and 7). In most of the examined regions, these three proteins are highly expressed in cells expressing NeuN, indicating that these three proteins are enriched in neuronal cells. In the cerebellum, Boat expression is elevated in Purkinje cells (Figure 6A). This expression profile of Boat resembles that of ataxin-1 (Clark et al, 1997) (also see Figure 6B). Their resemblance extends to the cellular level: whereas both Boat and ataxin-1 are expressed primarily as nuclear proteins in most brain cells, they are distributed beyond the nucleus into the cell body and dendrites in Purkinje cells (marked with arrows) and in inferior olive cells.
Figure 6.
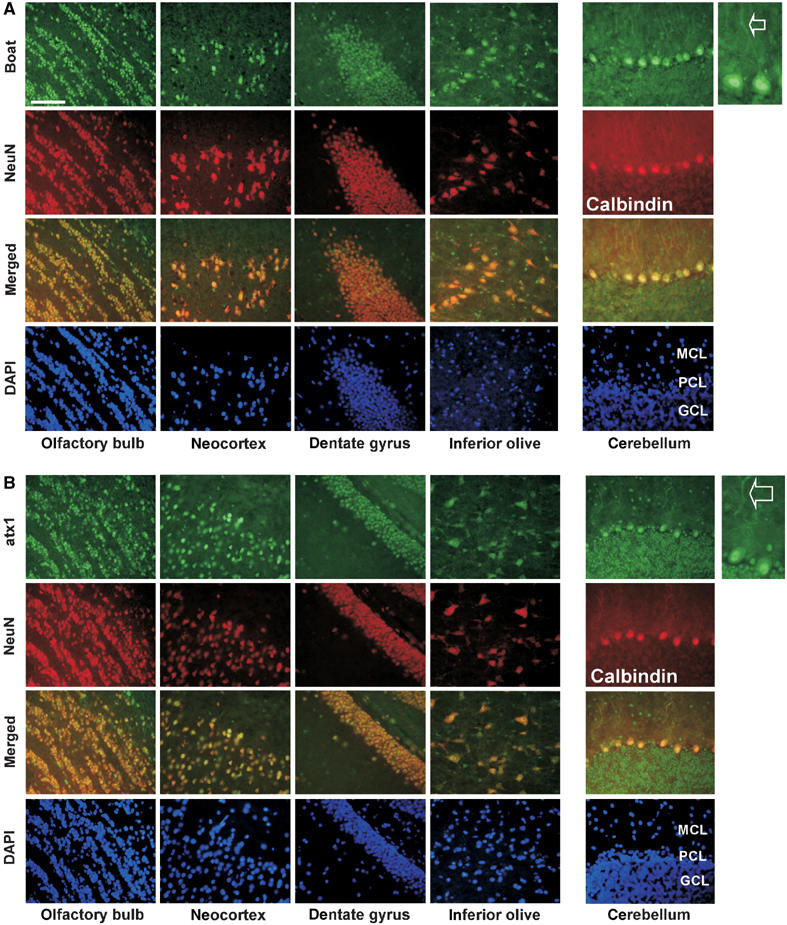
Boat and ataxin-1 share similar expression patterns in the mouse brain. Frozen brain sections prepared from 8-week-old wild-type (FVB/N) mice were co-immunostained with Boat(284–412) antibody (A) or ataxin-1(477–575) antibody (B) (FITC, green) and with NeuN or Calbindin antibody (Texas red, red). The regions of focus are olfactory bulb, neocortex, dentate gyrus, inferior olive, and cerebellum. The staining patterns of Boat and ataxin-1 in the dendritic regions of Purkinje cells in the cerebellum are indicated with arrows. The molecular cell layer, Purkinje cell layer, and granular cell layer are labeled MCL, PCL, and GCL, respectively. All tissues were counterstained with DAPI. Scale bar: 1000 μM.
Figure 7.
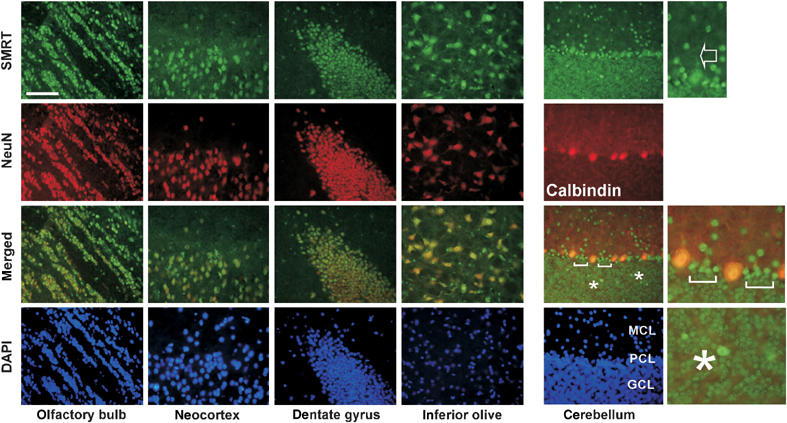
SMRT is enriched in neuronal cells in the mouse brain. Frozen brain sections prepared from 8-week-old wild-type (FVB/N) mice were co-immunostained with SMRT(972–1151) antibody (FITC, green) and NeuN or Calbindin antibody (Texas red, red). The staining of SMRT in the dendritic regions of Purkinje cells is indicated with an arrow. Brackets and asterisks mark elevated SMRT expression in the cells adjacent to Purkinje cells and within the granular cell layer, respectively. Scale bar: 1000 μM.
In comparison, SMRT displays a more divergent expression pattern in the cerebellum (Figure 7). Aside from a few cells within the granular layer and in between Purkinje cells that express much higher levels of SMRT (marked with brackets and asterisks), the level of SMRT in Purkinje cells is relatively lower than the levels of Boat and ataxin-1. Even so, SMRT can still be detected in the dendritic regions of Purkinje cells (marked with an arrow). The coexpression of Boat, ataxin-1, and SMRT in many brain cells, including Purkinje cells, indicates that these three proteins can function individually and collaboratively in the mouse brain.
Boat expression is reduced in the Purkinje cells of SCA1 mouse
Because degeneration of Purkinje cells is a hallmark of SCA1 patients and of SCA1 mouse (Zoghbi and Orr, 2000), we next examined whether Boat or SMRT expression is altered in the Purkinje cells of age-matched (8-week-old) SCA1 mice, which specifically express human mutant ataxin-1 in their Purkinje cells (Burright et al, 1995). In transgenic SCA1 mouse, the expression levels of both Boat and SMRT are reduced (Supplementary Results 4C and E). Because degeneration of Purkinje cells is already evident in 8-week-old SCA1 mouse (Skinner et al, 1997), we suspected that these observed alterations in Boat and SMRT expression might be secondary effects.
We therefore examined the expression of Boat and SMRT in the Purkinje cells of 3-week-old SCA1 mice. At this young age, no discernible degeneration of Purkinje cells and no alteration in Calbindin expression can be observed (Clark et al, 1997) (Figure 8A and B). Therefore, any change in the expression patterns of Boat and SMRT at this young age would more likely reflect a direct effect of SCA1. Indeed, the SMRT expression pattern is not altered in 3-week-old SCA1 mice. It is therefore possible that the observed reduction in SMRT expression in the Purkinje cells of older SCA1 mice is a secondary effect. However, this is not the case for Boat. Whereas Boat is abundantly expressed as a nuclear protein in the Purkinje cells of 3-week-old FVB/N mice (Figure 8A), it is barely detectable in the Purkinje cells of age-matched SCA1 mice (Figure 8B). As expected, such altered Boat expression only takes place in the Purkinje cells of SCA1 mice (Supplementary Results 4F and G), where mutant ataxin-1 is specifically expressed.
Figure 8.
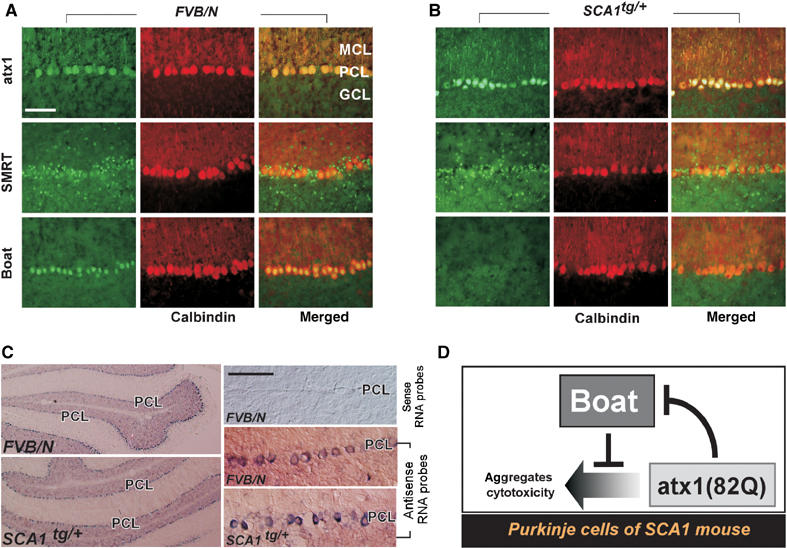
Boat expression is reduced in the Purkinje cells of SCA1 mice at the early stage. Frozen brain sections, prepared from 3-week-old wild-type (FVB/N) mice (A) or from age-matched SCA1tg/+ mice (B), were co-immunostained with Calbindin antibody along with ataxin-1, SMRT, or Boat antibody. (C) In situ hybridization for 3-week-old wild-type (FVB/N) and SCA1tg/+ mice, using antisense or sense RNA probes for Boat. The enlarged images show the regions adjacent to the Purkinje cell layer (PCL). (D) A model depicting the relationship between Boat and ataxin-1 in the Purkinje cells of SCA1 mice. Scale bars: 1000 μM.
To investigate whether the decreased Boat expression is regulated at the transcriptional or at the post-transcriptional level, in situ hybridization experiments for 3-week-old FVB/N and SCA1 mouse brains were carried out. In contrast to the response of Boat protein, no significant reduction of Boat transcripts was observed in the Purkinje cells of SCA1 mouse (Figure 8C). Therefore, the downregulation of Boat in the Purkinje cells of SCA1 mouse takes place at the post-transcriptional level. Since our results in fly indicate that Boat antagonizes ataxin-1(82Q)-mediated cytotoxicity, the disappearance of this safeguard protein in Purkinje cells may enable mutant ataxin-1 to bring about damaging effects in these primary target cells of SCA1.
Discussion
Ataxin-1 is a polyglutamine disease protein whose glutamine-repeat expanded form is involved in SCA1 (Orr et al, 1993; Banfi et al, 1994). Inspired by our recent finding that ataxin-1 associates with the transcriptional corepressors SMRT and SMRTER (Tsai et al, 2004), we investigated how these associations are established. We here report that the ataxin-1–SMRT interaction is mediated through a conserved AXH domain (Figure 1A). Extending from this initial finding, we probed the possibility whether any other AXH domain proteins also interact with SMRT in a similar manner. Indeed, we succeeded in identifying a novel human AXH domain protein, Boat, whose interaction with SMRT also depends on its AXH domain (Figure 3A). The Boat–SMRT interaction was further confirmed in human cultured cells (Figure 3B), in which Boat-enriched nuclear foci recruit endogenous SMRT. Boat also shares additional properties with ataxin-1, including interaction with other SMRT-related factors, such as N-CoR and SMRTER, forming complexes with HDAC3, and functioning as a potent transcriptional repressor (Figure 3A–C). The resemblances between ataxin-1 and Boat suggest that the two proteins are likely involved in overlapping transcriptional regulation pathways in vertebrate cells.
Significantly, we further discovered that Boat also interacts directly with ataxin-1. Their physical interaction was observed not only in yeast and human cultured cells (Figure 4), but also in Drosophila cells (Supplementary Result 2). In Drosophila, we were able to demonstrate the physiological relevance of the Boat–ataxin-1(82Q) interaction by showing that ataxin-1(82Q)-mediated cytotoxicity is suppressed when ataxin-1(82Q) interacts with Boat (Figure 5). In mouse, Boat and ataxin-1 share strikingly similar expression patterns in the brain, including in Purkinje cells (Figure 6). Most importantly, we found that Boat expression is dramatically decreased in the Purkinje cells of SCA1 mouse as early as 3 weeks old (Figure 8). Our research therefore establishes that Boat is an in vivo binding partner of ataxin-1, whose absence in Purkinje cells may contribute to their degeneration in SCA1 mouse.
The AXH domain
We discovered Boat because it shares an AXH domain with ataxin-1. This 114-amino-acid domain was first identified because it is common to ataxin-1, HBP1, and the Drosophila CG4547 and C. elegans K04F10.1 proteins (de Chiara et al, 2003). Although the protein structure of the AXH domain has been solved (Chen et al, 2004), very little is known about the exact functions of this conserved domain. In this study, we link the functions of the AXH domain to SMRT association and to transcriptional repression (Figures 1A, 3A, and C). The connection between the AXH domain and transcriptional repression correlates with an earlier report that a region encompassing the AXH domain of HBP1, an HMG-box chromatin remodeling transcription factor (Tevosian et al, 1997), is responsible for its transcriptional repressive effects. Based on this correlation, it is possible that a common feature of the AXH domain is transcriptional repression, and that additional AXH domain proteins may also associate with SMRT/SMRTER.
The AXH domain of ataxin-1 may also possess other functional properties. For example, structural analysis of the AXH domain indicates that it forms an oligomer-binding fold structure—a motif found in many oligonucleotide-binding proteins (Chen et al, 2004). Therefore, the ability of ataxin-1 to bind RNA (Yue et al, 2001) could be mediated through its AXH domain. In addition, the mapped regions in ataxin-1 responsible for LANP interaction (Matilla et al, 1997), USP7 ubiquitin–protease binding (Hong et al, 2002), and p80 coilin association (Hong et al, 2003) also encompass the AXH domain. These various studies suggest that the AXH domain is a molecular scaffold domain engaged in multiple protein–protein interactions and in RNA binding. The close resemblance between the AXH domains in ataxin-1 and Boat raises the possibility that some of the known features of ataxin-1 may be shared by Boat as well.
The NBA domain
Although we found substantial similarities between ataxin-1 and Boat (Figure 3), these two AXH domain proteins also encode distinct properties. Their respective N-terminal regions, which we called the NBA domain, may be in part responsible for their functional differences. Notably, we found that whereas the NBA domain of Boat is able to interact with the 1526–1833 region of SMRT and with itself, these two properties are absent from the NBA domain of ataxin-1 (Figures 3D, E, and 4E). Therefore, even though the NBA domains of ataxin-1 and Boat show sequence resemblance (Figure 2), their divergence is sufficient to enable Boat to adopt a different structural configuration for self-association and for SMRT interaction.
Our finding that ataxin-1 self-associates by means of its NBA domain is of particular significance (Figure 4E, panel C), since it has long been presumed that ataxin-1 self-association is dictated by a single self-association motif (Burright et al, 1997). Our results, on the contrary, reveal that ataxin-1 self-association depends on complex interactions among at least three domains in the protein. In addition to a previously mapped SAR domain (Burright et al, 1997; Klement et al, 1998), the NBA domain and a third domain targeted by the NBA domain are also involved. Based on these mapped domains, we propose that ataxin-1 self-association can be achieved not only through intermolecular interaction between separate ataxin-1 molecules, but also through intramolecular interaction between the NBA domain and the third region of the same ataxin-1 molecule.
Significantly, both the NBA and SAR domains of ataxin-1 are also targeted by Boat (Figure 4E): the SAR domain of ataxin-1 is a target of Boat's NBA domain, whereas the NBA domain of ataxin-1 is a target of full-length Boat. The coincidence between the domains for ataxin-1 self-association and ataxin-1–Boat interaction therefore provides a conceptual framework to explain why Boat and Boat(NBA) can suppress ataxin-1(82Q)-mediated phenotypes (cytotoxicity) in Drosophila eyes (Figure 5G–I): the formation of ataxin-1–Boat heterodimer takes place at the expense of self-association of mutant ataxin-1. Various studies in fly on different polyglutamine disease proteins have shown that the levels of protein expression correlate with their toxicity (Zoghbi and Botas, 2002). Therefore, a reduced level of self-associated mutant ataxin-1, as a result of its dimerization with Boat, may be a reason for its reduced toxicity.
The relevance of Boat to SCA1
Our finding that Boat, ataxin-1, and SMRT are all expressed in many NeuN-positive cells in the mouse brain (Figures 6 and 7) demonstrates that these three proteins can in fact encounter one another and modulate one another's functions in such neuronal cells. Because Boat and SMRT are both expressed in Purkinje cells—the primary targets of SCA1—it is likely that these two ataxin-1-associating factors are relevant to SCA1 pathogenesis as well. Most revealing is that the expression level of Boat in the Purkinje cells of 3-week-old SCA1 mouse is significantly reduced (Figure 8B). This reduction in Boat expression takes place prior to the appearance of nuclear inclusions and dendritic thinning of Purkinje cells (Clark et al, 1997; Skinner et al, 1997), therefore representing an early event in SCA1 pathogenesis. This initial loss of Boat protein could result in increased levels of self-associated mutant ataxin-1 protein in Purkinje cells, hence aggravating the mutant protein's toxicity. This in turn might set off a feed-forward mechanism for increasing damage, since increased ataxin-1 self-association would cause further reduction of Boat expression (see the model in Figure 8D).
Reduced Boat expression in SCA1 is regulated at the post-transcriptional level, since Boat transcripts are still abundantly present in the Purkinje cells of SCA1 mouse (Figure 8C). Intriguingly, this phenomenon appears to be unique to Purkinje cells, since Boat expression is unaffected by ataxin-1(82Q) in HEK293 cells (Figure 4C, top panel) and in Drosophila cells (Supplementary Result 2M). Therefore, future studies on how Boat is downregulated by ataxin-1(82Q) in the Purkinje cells of SCA1 mice may yield critical information on how tissue specificity is established for SCA1.
Conclusion
We here identify Boat as an AXH domain-dependent SMRT interacting factor and as an ataxin-1 interacting factor. Our functional analysis and comparison of the properties of Boat and ataxin-1 define important functions for the AXH and NBA domains of both proteins. The properties of Boat that we reveal in this study suggest that degenerative processes in the Purkinje cells of SCA1 animals may arise from a sequence of decreased Boat expression followed by an increased presence of toxic mutant ataxin-1 aggregates. Our finding that Boat can modulate the cytotoxicity of ataxin-1 uncovers an important piece of the mechanism involved in modulating the pathological effects of mutant ataxin-1, thus opening the path for new therapeutic strategies for treating SCA1.
Materials and methods
Constructs
Full-length Boat cDNA fragments were first isolated from a human cerebellum polyA RNA library (Clontech, 636122) by PCR reaction using two primers corresponding to the 5′ and 3′ ends of the predicted human Boat gene (XP 292512). The enzyme-digested PCR DNA fragments corresponding to full-length Boat, truncated Boat, full-length ataxin-1, or truncated ataxin-1 were then subcloned into various vectors to generate GBT9, GAD424, CFP, FLAG, HA, and GST fusion constructs. Detailed information for each of the described constructs will be available upon request.
Generation of Boat, ataxin-1, and SMRT antibodies
DNA fragments encoding Boat(284–412), ataxin-1(4–197), ataxin-1(477–575), or SMRT(972–1151) were subcloned into pGEX-4T1 (Pharmacia) to generate the GST-Boat, GST-ataxin-1, and GST-SMRT fusion constructs. The resulting fusion proteins were induced and purified from BL21 bacteria using standard protocols. The recombinant GST fusion proteins, including Boat(284–412), ataxin-1(477–575), and SMRT(972–1151), were used for generating guinea-pig polyclonal antibodies; GST-ataxin-1(4–197) fusion protein was used to generate rabbit polyclonal antibody. The sera were purified using the Antibody Spin Purification Kit (Pierce) after the third inoculation. Western blot analysis and immunostaining experiments show that the Boat antibodies do not crossreact with ataxin-1, and that the ataxin-1 antibodies do not crossreact with Boat (Supplementary Result 5, not shown for ataxin-1(4–197) antibody).
Drosophila stocks, salivary gland dissection, and genetic analysis
Constructs for UAS-FLAG-Boat, UAS-FLAG-ataxin-1(0Q), and UAS-HA-Boat(NBA) fly lines were generated by shuffling the DNA fragments from the CMX-FLAG-Boat, CMX-FLAG-ataxin-1(0Q), and CMX-HA-Boat(4–197) plasmid to the UAST vector. In the case of HA-Boat(4–197), we additionally tagged the fusion protein with a nucleus localization signal. These constructs were injected into w1118 embryos to generate the corresponding stable fly lines. Multiple lines were obtained and tested for each of the mentioned experiments.
Hsp70-Gal4, GMR-Gal4, w1118, UAS-GFP, and UAS-RedStinger were obtained from the Bloomington Stock Center. UAS-ataxin-1(82Q) was a gift of M Feany (Feany and Bender, 2000); UAS-MJDtr(78Q) was a gift of N Bonini (Warrick et al, 1998); UAS-HA-Q127 was a gift of P Kazemi-Esfarjani (Kazemi-Esfarjani and Benzer, 2000). The GMR-Gal4, UAS-ataxin-1(82Q) line, the GMR-Gal4; UAS-MJDtr(78Q) line, and the GMR-Gal4; UAS-HA-Q127 line were generated by chromosomal recombination. All genetic experiments were carried out at 23°C.
Salivary glands were dissected from third instar larvae in 1 × PBS with 0.1% Tween 20 for morphological analysis. For immunostaining purposes, the salivary glands were pressed on Superfrost Plus slides (VWR) using silicone-coated coverglass prior to proceeding with the immunostaining steps with either ataxin-1, Boat, SMRTER, FLAG, or/and HA antibodies.
Additional materials and methods are described in Supplementary data.
Supplementary Material
Supplementary Result 1
Supplementary Result 2
Supplementary Result 3
Supplementary Result 4
Supplementary Result 5
Supplementary Materials and Methods
Supplementary Figure Legends
Acknowledgments
We thank Ron Evans for his support of early stages of this work; R Head, J Fondell, K Madura, J Lenard, and P Casaccia-Bonnefil for comments on the manuscript; H Orr for ataxin-1[77]Δ plasmid; H Zoghbi for SCA1 mouse; N Bonini, M Feany, P Kazemi-Esfarjani, and the Bloomington Stock Center for Drosophila stocks; J Fondell for HeLa nuclear extracts; and Qiufu Ma for in situ hybridization protocol. This work is supported by the UMDNJ-RWJMS start-up fund, UMDNJ Foundation, the National Ataxia Foundation, and the Hereditary Disease Foundation to C-CT.
References
- Banfi S, Servadio A, Chung MY, Kwiatkowski TJ Jr, McCall AE, Duvick LA, Shen Y, Roth EJ, Orr HT, Zoghbi HY (1994) Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat Genet 7: 513–520 [DOI] [PubMed] [Google Scholar]
- Burright EN, Clark HB, Servadio A, Matilla T, Feddersen RM, Yunis WS, Duvick LA, Zoghbi HY, Orr HT (1995) SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 82: 937–948 [DOI] [PubMed] [Google Scholar]
- Burright EN, Davidson JD, Duvick LA, Koshy B, Zoghbi HY, Orr HT (1997) Identification of a self-association region within the SCA1 gene product, ataxin-1. Hum Mol Genet 6: 513–518 [DOI] [PubMed] [Google Scholar]
- Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EM, Orr HT, Botas J, Zoghbi HY (2003) Interaction of akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 113: 457–468 [DOI] [PubMed] [Google Scholar]
- Chen JD, Evans RM (1995) A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 377: 454–457 [DOI] [PubMed] [Google Scholar]
- Chen YW, Allen MD, Veprintsev DB, Lowe J, Bycroft M (2004) The structure of the AXH domain of spinocerebellar ataxin-1. J Biol Chem 279: 3758–3765 [DOI] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, Matilla A, Zoghbi HY, Orr HT (1997) Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci 17: 7385–7395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Chiara C, Giannini C, Adinolfi S, de Boer J, Guida S, Ramos A, Jodice C, Kioussis D, Pastore A (2003) The AXH module: an independently folded domain common to ataxin-1 and HBP1. FEBS Lett 551: 107–112 [DOI] [PubMed] [Google Scholar]
- Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, Clark HB, Orr HT (2003) Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38: 375–387 [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW (2000) A Drosophila model of Parkinson's disease. Nature 404: 394–398 [DOI] [PubMed] [Google Scholar]
- Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, McCall A, Canal I, Orr HT, Zoghbi HY, Botas J (2000) Identification of genes that modify ataxin-1-induced neurodegeneration. Nature 408: 101–106 [DOI] [PubMed] [Google Scholar]
- Fields S, Song O (1989) A novel genetic system to detect protein–protein interactions. Nature 340: 245–246 [DOI] [PubMed] [Google Scholar]
- Hong S, Ka S, Kim S, Park Y, Kang S (2003) p80 coilin, a coiled body-specific protein, interacts with ataxin-1, the SCA1 gene product. Biochim Biophys Acta 1638: 35–42 [DOI] [PubMed] [Google Scholar]
- Hong S, Kim SJ, Ka S, Choi I, Kang S (2002) USP7, a ubiquitin-specific protease, interacts with ataxin-1, the SCA1 gene product. Mol Cell Neurosci 20: 298–306 [DOI] [PubMed] [Google Scholar]
- Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, Rosenfeld MG (1995) Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 377: 397–404 [DOI] [PubMed] [Google Scholar]
- Kazemi-Esfarjani P, Benzer S (2000) Genetic suppression of polyglutamine toxicity in Drosophila. Science 287: 1837–1840 [DOI] [PubMed] [Google Scholar]
- Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. (1998) Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95: 41–53 [DOI] [PubMed] [Google Scholar]
- Matilla A, Koshy BT, Cummings CJ, Isobe T, Orr HT, Zoghbi HY (1997) The cerebellar leucine-rich acidic nuclear protein interacts with ataxin-1. Nature 389: 974–978 [DOI] [PubMed] [Google Scholar]
- Ordentlich P, Downes M, Xie W, Genin A, Spinner NB, Evans RM (1999) Unique forms of human and mouse nuclear receptor corepressor SMRT. Proc Natl Acad Sci USA 96: 2639–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT, Chung MY, Banfi S, Kwiatkowski TJ Jr, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LP, Zoghbi HY (1993) Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 4: 221–226 [DOI] [PubMed] [Google Scholar]
- Park EJ, Schroen DJ, Yang M, Li H, Li L, Chen JD (1999) SMRTe, a silencing mediator for retinoid and thyroid hormone receptors-extended isoform that is more related to the nuclear receptor corepressor. Proc Natl Acad Sci USA 96: 3519–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privalsky ML (2004) The role of corepressors in transcriptional regulation by nuclear hormone receptors. Annu Rev Physiol 66: 315–360 [DOI] [PubMed] [Google Scholar]
- Skinner PJ, Koshy BT, Cummings CJ, Klement IA, Helin K, Servadio A, Zoghbi HY, Orr HT (1997) Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature 389: 971–974 [DOI] [PubMed] [Google Scholar]
- Tevosian SG, Shih HH, Mendelson KG, Sheppard KA, Paulson KE, Yee AS (1997) HBP1: a HMG box transcriptional repressor that is targeted by the retinoblastoma family. Genes Dev 11: 383–396 [DOI] [PubMed] [Google Scholar]
- Tsai CC, Fondell JD (2004) Nuclear receptor recruitment of histone-modifying enzymes to target gene promoters. Vitam Horm 68: 93–122 [DOI] [PubMed] [Google Scholar]
- Tsai CC, Kao HY, Mitzutani A, Banayo E, Rajan H, McKeown M, Evans RM (2004) Ataxin 1, a SCA1 neurodegenerative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc Natl Acad Sci USA 101: 4047–4052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CC, Kao HY, Yao TP, McKeown M, Evans RM (1999) SMRTER, a Drosophila nuclear receptor coregulator, reveals that EcR-mediated repression is critical for development. Mol Cell 4: 175–186 [DOI] [PubMed] [Google Scholar]
- Warrick JM, Paulson HL, Gray-Board GL, Bui QT, Fischbeck KH, Pittman RN, Bonini NM (1998) Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 93: 939–949 [DOI] [PubMed] [Google Scholar]
- Yue S, Serra HG, Zoghbi HY, Orr HT (2001) The spinocerebellar ataxia type 1 protein, ataxin-1, has RNA-binding activity that is inversely affected by the length of its polyglutamine tract. Hum Mol Genet 10: 25–30 [DOI] [PubMed] [Google Scholar]
- Zoghbi HY, Botas J (2002) Mouse and fly models of neurodegeneration. Trends Genet 18: 463–471 [DOI] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT (2000) Glutamine repeats and neurodegeneration. Annu Rev Neurosci 23: 217–247 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Result 1
Supplementary Result 2
Supplementary Result 3
Supplementary Result 4
Supplementary Result 5
Supplementary Materials and Methods
Supplementary Figure Legends