

Abstract
In primary cells, overexpression of oncogenes such as RasV12 induces premature senescence rather than transformation. Senescence is an irreversible form of G1 arrest that requires the p19ARF/p53 and p16INK4a/pRB pathways and may suppress tumorigenesis in vivo. Here we show that the transcription factor C/EBPβ is required for RasV12-induced senescence. C/EBPβ−/− mouse embryo fibroblasts (MEFs) expressing RasV12 continued to proliferate despite unimpaired induction of p19ARF and p53, and lacked morphological features of senescent fibroblasts. Enforced C/EBPβ expression inhibited proliferation of wild-type MEFs and also slowed proliferation of p19Arf−/− and p53−/− cells, indicating that C/EBPβ acts downstream or independently of p19ARF/p53 to suppress growth. C/EBPβ was unable to inhibit proliferation of MEFs lacking all three RB family proteins or wild-type cells expressing dominant negative E2F-1 and, instead, stimulated their growth. C/EBPβ decreased expression of several E2F target genes and was associated with their promoters in chromatin immunoprecipitation assays, suggesting that C/EBPβ functions by repressing genes required for cell cycle progression. C/EBPβ is therefore a novel component of the RB:E2F-dependent senescence program activated by oncogenic stress in primary cells.
Keywords: C/EBPβ, cell cycle arrest, cellular senescence, oncogenic Ras, RB:E2F
Introduction
Most immortalized rodent cell lines can be transformed by the Ha-ras oncogene (RasV12), leading to neoplastic growth in culture and tumorigenicity in vivo (Weinberg, 1989). In contrast, overexpression of RasV12 or other activated oncogenes in many primary cells induces premature senescence, a permanent form of cell cycle arrest that is believed to play a role in tumor suppression (Serrano et al, 1997; Lin and Lowe, 2001). Cultured primary rodent fibroblasts also undergo ‘spontaneous' senescence after 15–30 cell doublings, possibly resulting from prolonged exposure to mitogenic signals or accumulated DNA damage upon ex vivo culture (‘culture shock'; Sherr and DePinho, 2000). The senescent state is characterized by expression of specific biochemical markers such as senescence-associated β-galactosidase and a distinctive cell morphology typified by a flattened, enlarged shape and increased focal adhesions (Goldstein, 1990; Campisi, 1996). The ‘flat cell' phenotype is due in part to the assembly of actin stress fibers, which form a highly organized network of microfilaments and associations with other cytoskeletal proteins (Pawlak and Helfman, 2001).
Both Ras-induced and spontaneous senescence in mouse embryo fibroblasts (MEFs) require activation of the p19ARF/p53 tumor suppressor pathway (Sherr and Weber, 2000). MEFs derived from p53−/− or INK4a null mice (p19ARF and p16INK4a are INK4a gene products) are intrinsically immortalized, fail to undergo spontaneous or Ras-induced senescence, and are transformed by oncogenic Ras alone, underscoring the importance of the p19ARF/p53 pathway in senescent cell cycle arrest. p19ARF stabilizes p53 by sequestering the p53 E3 ligase, Mdm2. Induction of the p53 transcription factor subsequently provokes cell cycle arrest, although the mechanism by which p53 inhibits cell growth has not been fully elucidated. One transcriptional target of p53 that contributes to cell cycle arrest is the cyclin/Cdk2 inhibitor p21Cip1, but other p53-regulated genes are likely to be involved (Groth et al, 2000).
The RB family of tumor suppressors (pRB, p107, and p130) also plays a critical role in cellular senescence by forming inhibitory complexes with E2F transcription factors that repress S-phase gene expression. pRB activity is controlled by the CDK inhibitor p16INK4a, which is induced by RasV12. p16INK4a impedes cell cycle progression by inhibiting the activity of Cdk4 and Cdk6, thus blocking phosphorylation of pRB and preventing its release from E2F. Senescence can be induced by overexpression of pRB (Xu et al, 1997; Alexander and Hinds, 2001), and MEFs doubly mutant for pRB and p107 or lacking all three RB family members are defective for Ras-induced cell cycle arrest (Dannenberg et al, 2000; Sage et al, 2000; Peeper et al, 2001). In contrast to p19ARF or p53 null cells, Rb−/−/p107−/− MEFs are not transformed by Ras alone (Peeper et al, 2001). Hence, disruption of RasV12-induced senescence does not necessarily cause oncogenic transformation. pRB-imposed cell cycle arrest occurs even in cells lacking p53, indicating that pRB acts downstream of p53 or in another pathway (Alexander and Hinds, 2001). More recently, it was shown that E2F repressor complexes are downstream targets of p19ARF/p53-induced proliferation arrest (Rowland et al, 2002), indicating a convergence of the p19ARF/p53 and p16Ink4a/RB pathways at the level of E2F-RB.
While p53 and RB are essential mediators of oncogenic Ras-induced senescence, it is likely that additional pathways or components of the p53 and RB pathways remain to be identified. In the present study, we have explored a possible role for the bZIP transcription factor C/EBPβ (CCAAT/enhancer binding protein β). C/EBPβ is broadly expressed and has diverse regulatory functions (Descombes et al, 1990; Poli et al, 1990; Cao et al, 1991; Williams et al, 1991). C/EBPβ null mice display pleiotropic phenotypes, including defects in innate immunity (Tanaka et al, 1995) and female reproduction (Sterneck et al, 1997; Robinson et al, 1998; Seagroves et al, 1998) and impaired growth or differentiation of adipocytes (Tanaka et al, 1997; Tang et al, 2003), hepatocytes (Greenbaum et al, 1998), and keratinocytes (Zhu et al, 1999). C/EBPβ activity is regulated by oncogenic Ras signaling (Nakajima et al, 1993; Kowenz-Leutz et al, 1994; Hanlon and Sealy, 1999; Zhu et al, 2002). Under basal conditions, C/EBPβ is maintained in a latent, autoinhibited state by two negative regulatory domains (Kowenz-Leutz et al, 1994; Williams et al, 1995). However, in response to receptor tyrosine kinase activation or expression of RasV12, C/EBPβ becomes derepressed and transcriptionally active (Nakajima et al, 1993; Kowenz-Leutz et al, 1994; Hanlon and Sealy, 1999). C/EBPβ activation is mediated at least partly by ERK-dependent phosphorylation of a conserved threonine residue located in one of the regulatory domains (Nakajima et al, 1993).
Members of the C/EBP family, including C/EBPβ, are implicated in regulating growth arrest of terminally differentiating cells and, when ectopically expressed, can inhibit cell proliferation (Umek et al, 1991; Hendricks-Taylor and Darlington, 1995). For example, forced expression of C/EBPβ in HepG2 hepatoma cells induces cell cycle arrest at the G1–S boundary (Buck et al, 1994), and C/EBPβ inhibits colony formation of Balb/MK2 keratinocytes (Zhu et al, 1999). Conversely, C/EBPβ knockout mice display mild epidermal hyperplasia, and C/EBPβ-deficient primary keratinocytes show defective calcium-induced proliferation arrest in vitro (Zhu et al, 1999). These growth-inhibitory functions of C/EBPβ, together with its role as a target of RasV12 signaling, prompted us to investigate whether C/EBPβ is involved in Ras-induced premature senescence.
Results
Impaired H-RasV12-induced senescence in C/EBPβ−/− MEFs
To determine if C/EBPβ has a role in oncogene-induced senescence, we examined the effect of overexpressing RasV12 in C/EBPβ+/+ and C/EBPβ−/− MEFs. Early-passage (P2/P3) MEFs were used throughout this study to avoid selecting immortalizing mutations in the p19ARF–p53 pathway. Cells were infected with control or RasV12-expressing retroviruses and analyzed for proliferation over a 6-day time course (Figure 1A). As expected, expression of RasV12 in wild-type MEFs provoked cell cycle arrest. However, RasV12-expressing C/EBPβ−/− cells continued to proliferate. This response was observed for three independent C/EBPβ−/− MEF populations, indicating that C/EBPβ null cells are defective for RasV12-induced cell cycle arrest.
Figure 1.
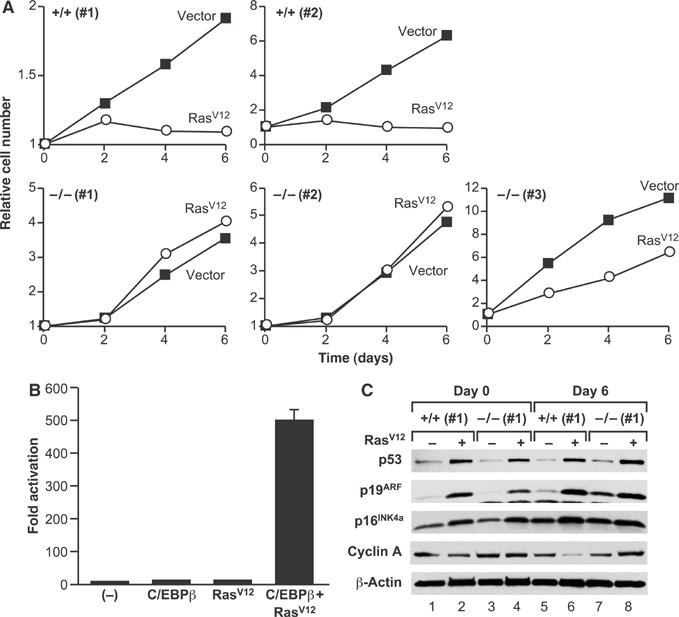
C/EBPβ−/− MEFs are defective for RasV12-induced cell cycle arrest. (A) Early-passage (P2) wild-type and C/EBPβ−/− MEFs were infected with control or RasV12-expressing pBabe-puro retroviruses, drug selected, and used for growth assays. A total of 2.5 × 104 cells/well were plated and cell numbers were measured over a 6-day period. Each value was normalized to the cell number at day 0 (post drug selection). (B) Transactivation assays. C/EBPβ−/− MEFs were transfected with a C/EBP reporter construct (2 × C/EBP-luc) either alone or with expression plasmids for C/EBPβ and/or RasV12. The cells were harvested and luciferase activity was determined; reporter activity was normalized to protein levels and the value for the reporter alone (−) was set to 1. Data are plotted as fold activation and are the means±s.e. of three experiments. (C) Induction of cell cycle regulatory proteins. Western blot analysis was performed on cell lysates prepared at days 0 and 6 from wild-type and C/EBPβ−/− MEFs transduced with RasV12 or empty vector. An 80–100 μg portion of each protein extract was analyzed by Western blotting for the indicated proteins.
Analysis of nuclear extracts from wild-type MEFs showed only minor increases in C/EBPβ expression and DNA-binding activity in RasV12-expressing cells (see Supplementary Figure 1). However, coexpression of RasV12 stimulated C/EBPβ-mediated transcription of a C/EBP-driven reporter gene nearly 500-fold in transfected MEFs, whereas C/EBPβ or RasV12 alone caused only ∼10-fold increases (Figure 1B). C/EBPβ is thus strongly activated by RasV12 signaling in MEFs, consistent with the idea that C/EBPβ functions as a Ras effector in these cells.
We next asked whether RasV12-induced activation of the p19ARF–p53 pathway is impaired in C/EBPβ null MEFs, accounting for their failure to undergo cell cycle arrest. The levels of p19ARF, p53, and other cell cycle regulators were examined on days 0 and 6 after drug selection (Figure 1C). p19ARF and p53 expression was induced by RasV12 to a similar extent in wild-type cells and C/EBPβ−/− cells. p16INK4a and p21CIP1 levels were also increased comparably in wild-type and C/EBPβ−/− cells (Figure 1C and data not shown). Therefore, the inability of C/EBPβ−/− MEFs to undergo RasV12-induced growth arrest is not due to defective engagement of the p19ARF/p53 pathway or altered expression of CDK inhibitors. Cyclin A levels were diminished in RasV12-transduced wild-type MEFs relative to control cells, signifying G1 arrest (Figure 1C, lanes 5 and 6), whereas cyclin A was unaffected or even increased by RasV12 in C/EBPβ−/− cells (lanes 7 and 8).
To determine whether proliferation of RasV12-expressing C/EBPβ−/− MEFs is stably maintained or whether the cells eventually senesce, we propagated Ras-expressing and control C/EBPβ−/− cells on a 3T3 protocol for four passages (Figure 2A). Cell numbers were comparable at each passage; moreover, the cells displayed similar growth rates before and after passaging (compare Figures 1A and 2B). The Ras-expressing cells also maintained elevated expression of p53 and p19ARF (Figure 2C). Thus, RasV12-induced premature senescence is permanently disabled in C/EBPβ−/− MEFs and proliferation remains unconstrained by induction of p19ARF/p53 over many cell doublings.
Figure 2.
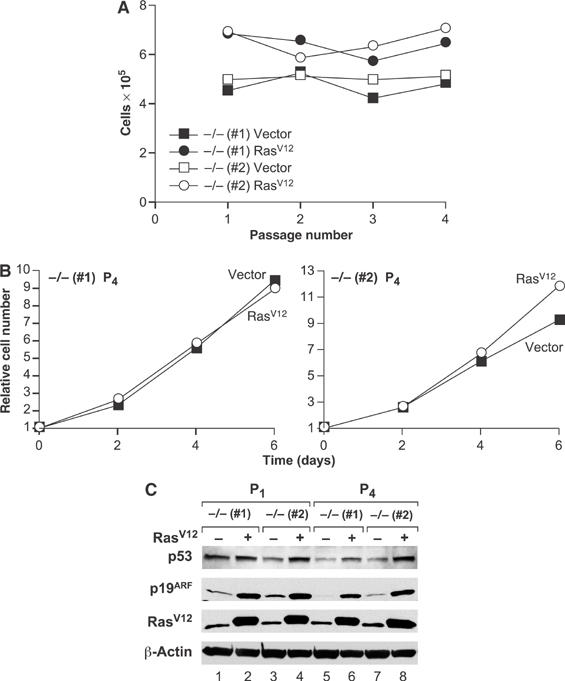
Enhanced proliferative capacity of RasV12-expressing C/EBPβ−/− MEFs. (A) Two independent populations of C/EBPβ−/− MEFs were infected and selected as described in Figure 1A and were propagated on a 3T3 protocol for four passages. (B) Growth curves of C/EBPβ−/− MEFs infected with empty vector or RasV12-expressing retrovirus, after passage 4. (C) Western blot analysis of p53, p19ARF, and Ras in lysates prepared from the indicated MEFs at P1 and P4.
We also passaged wild-type and C/EBPβ−/− primary MEFs on a 3T3 protocol to determine if the mutant cells exhibit defects in spontaneous senescence (see Supplementary Figure 2). C/EBPβ−/− cells maintained a low rate of proliferation even after numerous passages, whereas the wild-type cells eventually senesced. Moreover, the mutant cells showed an increased tendency to become immortalized, coinciding with loss of p19ARF and/or p53 expression (Supplementary Figure 2 and data not shown). MEFs lacking C/EBPβ are therefore resistant to both spontaneous and Ras-induced senescence.
Partially transformed properties of RasV12-expressing C/EBPβ−/− MEFs
In addition to inducing growth arrest, RasV12 elicits characteristic changes in fibroblast cell morphology (Serrano et al, 1997). Expression of RasV12 in C/EBPβ+/+ MEFs induced the flat, enlarged appearance typical of senescent cells (Figure 3A). By contrast, C/EBPβ-deficient cells expressing RasV12 were highly refractile, had spindle-like projections, and displayed reduced contact inhibition. Thus, RasV12-expressing C/EBPβ−/− MEFs adopt morphological features usually associated with transformed cells. We also compared the colony-forming ability of RasV12-transduced wild-type and C/EBPβ−/− MEFs plated at low density. Cells of both genotypes produced detectable colonies; however, cells displaying a transformed-like morphology were observed only with C/EBPβ−/− MEFs (Figure 3B). Many of these colonies had a star-like shape and stained intensely with crystal violet, and under higher magnification the cells showed dense growth and loss of contact inhibition (Figure 3B, right panel). In contrast, C/EBPβ+/+ colonies stained much less intensely and the cells displayed a flattened, senescent appearance.
Figure 3.
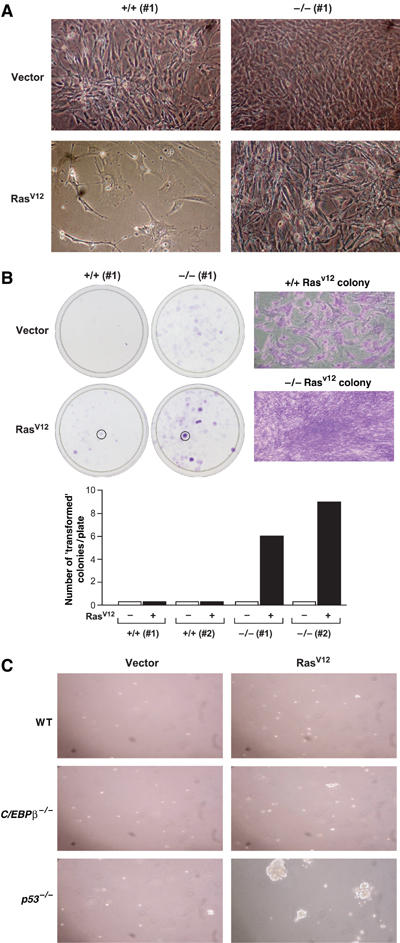
Growth properties of RasV12-expressing MEFs. (A) Cell morphology of control or RasV12-expressing wild-type and C/EBPβ−/− MEFs at day 6 postselection. (B) Colony formation assay of wild-type and C/EBPβ−/− MEFs infected with control or RasV12 retroviruses. A total of 2.5 × 104 cells were seeded into 10 cm plates, cultured for 2 weeks, and then stained with crystal violet. The right panels show higher magnification images of two selected colonies. Quantitation of intensely stained (i.e., transformed-like) colonies obtained from two MEF lines of each genotype is shown at the bottom. (C) Soft agar colony assays. RasV12- or control vector-infected MEFs of the indicated genotypes were seeded into soft agar (2.5 × 104 cells/6 cm dish). The plates were photographed after 2 weeks.
To assess the tumorigenicity of RasV12-expressing C/EBPβ−/− MEFs, we examined their ability to grow anchorage-independently and to form solid tumors in nude mice. Wild-type cells infected with the Ras virus were unable to form colonies in soft agar (Figure 3C). C/EBPβ−/− MEFs were also incapable of anchorage-independent growth despite being highly proliferative under adherent conditions. By comparison (and as previously reported; Peeper et al, 2001), RasV12-expressing p53−/− MEFs formed numerous large colonies in soft agar. When wild-type or C/EBPβ−/− MEFs expressing RasV12 were injected into athymic nude mice (3.5 × 105 cells/flank, eight mice per group), neither population gave rise to tumors, even after 10 weeks. However, RasV12-transformed NIH 3T3 cells transplanted in a parallel experiment were highly tumorigenic. Thus, by two criteria, Ras-expressing C/EBPβ−/− MEFs lack a fully transformed phenotype even though they evade cell cycle arrest.
RasV12 and C/EBPβ cooperate to induce cell cycle arrest
We next investigated the ability of C/EBPβ, when expressed alone or in combination with RasV12, to induce proliferative arrest. RasV12 and/or C/EBPβ were introduced into wild-type and C/EBPβ−/− MEFs by retroviral infection and cell proliferation was monitored over a 6-day period (Figure 4A). Expression of RasV12 or C/EBPβ alone in C/EBPβ+/+ cells significantly reduced their proliferation rate. However, cell cycle arrest was incomplete, as cell numbers increased modestly at days 2 and 4. In contrast, RasV12 and C/EBPβ together provoked rapid and efficient arrest, with no measurable increase in cell number at any time point. C/EBPβ−/− cells failed to arrest in response to RasV12 but their growth was significantly inhibited by C/EBPβ, and the combination of RasV12 and C/EBPβ completely blocked proliferation. Therefore, RasV12 and C/EBPβ act synergistically to induce cell cycle arrest.
Figure 4.
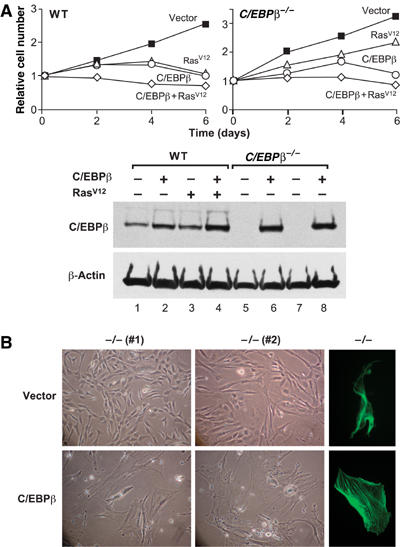
Effect of C/EBPβ and RasV12 on cell growth and cytoskeletal reorganization. (A) Growth curves of wild-type and C/EBPβ−/− MEFs transduced with C/EBPβ and/or RasV12. Each value was normalized to the cell number at day 0 (postselection). A representative example is shown from at least two independent experiments performed in duplicate. Bottom panel: Western blot analysis of C/EBPβ expression. (B) Cell morphology of control and C/EBPβ-transduced C/EBPβ−/− MEFs. Cells were photographed at day 6 postselection. The cells were also stained for stress fibers using FITC-conjugated phalloidin (right panels). A representative example from two independent experiments is shown; photographs were taken at the same magnification.
In addition to inhibiting proliferation, C/EBPβ caused enlargement and flattening of C/EBPβ−/− MEFs (Figure 4B). The cells were stained with phalloidin–FITC conjugate to examine actin-cytoskeleton rearrangements. In contrast to control cells, which were smaller, spindle-shaped, and lacked stress fibers, C/EBPβ-transduced cells displayed a highly organized network of actin stress fibers (Figure 4B, right panels). Stress fiber formation induced by RasV12 was also defective in C/EBPβ−/− MEFs (data not shown). Together, these results demonstrate that C/EBPβ regulates stress fiber formation and other morphological features associated with RasV12-induced senescence.
C/EBPβ-mediated growth suppression is largely independent of p19ARF/p53 but requires RB:E2F activity
MEFs deficient for p53, p16INK4a/p19ARF, or p19ARF do not undergo replicative arrest and are transformed by RasV12 alone (Serrano et al, 1996; Kamijo et al, 1997; Sharpless et al, 2001, 2004). To determine whether enforced C/EBPβ expression can arrest cells that lack INK4a gene products or p53, we expressed C/EBPβ and/or RasV12 in p53−/−, p16Ink4a−/−, and p19ARF−/− MEFs and analyzed cell proliferation. As expected, RasV12 suppressed the growth of wild-type and p16Ink4a−/− MEFs but not p19ARF−/− or p53−/− cells (Figure 5A). C/EBPβ decreased the proliferation of all genotypes, although to a lesser degree in p53 and p19ARF null MEFs, and its inhibitory effects were significantly enhanced by RasV12. Cell proliferation was also assessed by colony formation in low-density plating assays (Figure 5B). Wild-type or p16Ink4a−/− MEFs expressing RasV12 did not produce detectable colonies, whereas p19ARF−/−and p53−/− cells gave rise to large numbers of transformed colonies that stained intensely with crystal violet. The size and number of these colonies were reduced by C/EBPβ coexpression. Collectively, the results of Figure 5 show that C/EBPβ inhibits cell proliferation and Ras transformation in a manner that is largely independent of the p19ARF/p53 pathway.
Figure 5.
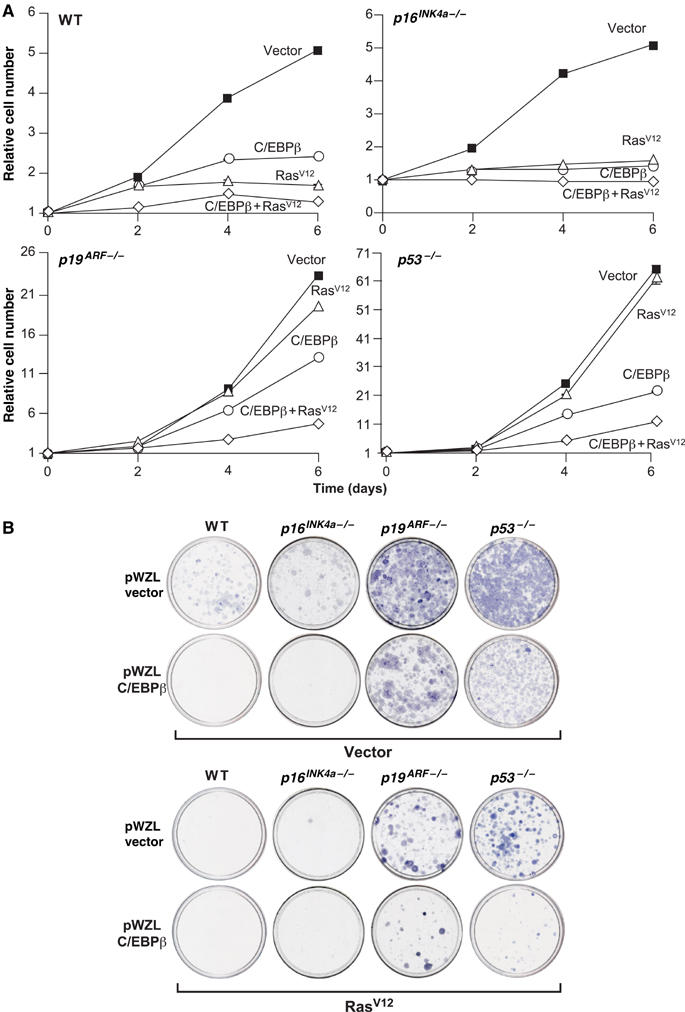
Effect of C/EBPβ and RasV12 on proliferation of p53−/−, p16Ink4a−/−, and p19Arf−/− MEFs. (A) Primary MEFs of the indicated genotypes were infected sequentially with retroviruses expressing C/EBPβ and RasV12 and cell proliferation was analyzed over a time course. Each value was normalized to the cell number at day 0 (postselection). Each experiment was performed twice with similar results; data are the mean±s.e. of triplicate time points from a representative experiment. (B) The same cells were plated at low density (2 × 104 cells/10 cm dish) and colonies were visualized 2 weeks later.
The properties of C/EBPβ−/− MEFs are similar in several respects to those of cells lacking both pRB and p107 or all three RB family members (triple knockout, ‘TKO') (Dannenberg et al, 2000; Sage et al, 2000; Peeper et al, 2001). To determine whether C/EBPβ arrests cells by an RB-dependent mechanism, we examined the effects of expressing C/EBPβ in MEFs lacking RB genes (Figure 6). C/EBPβ inhibited the growth of wild-type and p107−/−/p130−/− cells and also suppressed proliferation of Rb−/−/p130−/− and Rb−/−/p107−/− MEFs, albeit less efficiently. Remarkably, C/EBPβ not only failed to inhibit the growth of TKO cells, but also significantly increased their proliferation rate. Western blot analysis showed that C/EBPβ was overexpressed to similar levels in each cell line (lower panel). C/EBPα, which also potently inhibits proliferation of many cells (Nerlov, 2004), diminished the growth of wild-type and p107−/−/p130−/− cells and, to a lesser extent, Rb−/−/p130−/− and Rb−/−/p107−/− MEFs, yet it accelerated proliferation of TKO cells (Figure 6). Thus, both C/EBP isoforms require at least one RB family member to induce cell cycle arrest and can stimulate proliferation in the absence of all three pocket proteins.
Figure 6.
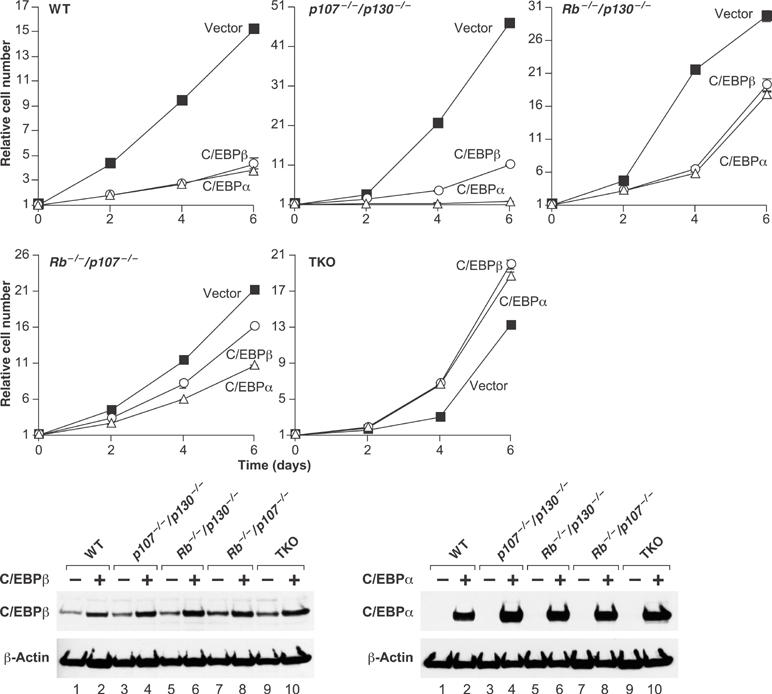
C/EBPβ requires RB family members to induce growth arrest. Primary MEFs of the indicated genotypes were infected with C/EBPβ or C/EBPα retroviruses and cell growth was analyzed over a time course. Each curve was performed at least twice, and time points were determined in triplicate. Bottom panel: Western blots of C/EBPβ and C/EBPα expression.
We next analyzed the effect of RasV12 on proliferation of C/EBPβ-expressing TKO MEFs (Figure 7A). As observed previously, C/EBPβ and/or RasV12 strongly inhibited proliferation of wild-type cells. However, RasV12 failed to suppress the growth of TKO MEFs and also enhanced the ability of C/EBPβ to stimulate proliferation (most apparent at day 8 of the time course). C/EBPβ also increased the colony-forming activity of TKO cells, as did RasV12 (Figure 7B). Cells expressing both genes generated similar numbers of colonies but many of these stained more densely with crystal violet, indicating the presence of aggressively proliferating cells. Ablation of all three Rb genes therefore disrupts the antiproliferative effects of C/EBPβ and/or RasV12.
Figure 7.
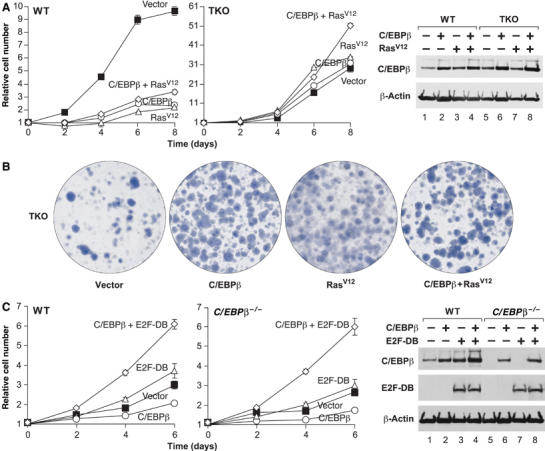
(A) Growth curves of TKO MEFs overexpressing C/EBPβ and/or RasV12. Data are the means±s.e. of triplicate assays. Right panel: Western blot for C/EBPβ expression. (B) Colony assays of the TKO MEFs from panel A. (C) Dominant negative E2F disrupts C/EBPβ-induced arrest. Wild-type or C/EBPβ−/− MEFs were infected with vectors for C/EBPβ and/or dominant negative E2F-1 (E2F-DB) and growth curves were performed. Right panel: Western blot analysis of E2F-DB and C/EBPβ expression.
RB proteins inhibit cell proliferation through their association with E2F transcription factors, which target RBs to specific growth-regulatory genes. Thus, we tested whether E2F function is required for the antiproliferative activity of C/EBPβ by transducing wild-type and C/EBPβ−/− MEFs with a dominant-negative form of E2F-1 (‘E2F-DB') that lacks the transactivation and RB protein-binding domains (Krek et al, 1995; Rowland et al, 2002). The cells were subsequently infected with C/EBPβ or control viruses and growth rates were analyzed (Figure 7C). E2F-DB alone had little effect on the proliferation rate, although there was a slight stimulation in wild-type MEFs. However, coexpression of C/EBPβ and E2F-DB caused a significant increase in proliferation, in contrast to growth inhibition by C/EBPβ alone. Therefore, disruption of the RB:E2F axis by deletion of all three Rb genes or expression of dominant negative E2F reverses the cellular growth response to C/EBPβ.
C/EBPβ downregulates expression of E2F target genes and associates with their promoters
C/EBPβ might induce G1 arrest by inhibiting expression of E2F-regulated S-phase genes, as has been proposed for C/EBPα (Timchenko et al, 1999; Slomiany et al, 2000; Porse et al, 2001). We used RT–PCR to analyze mRNA levels of several E2F-regulated genes in C/EBPβ−/− MEFs infected with C/EBPβ virus (growth-arrested), RasV12 (proliferating), or the empty vector (Figure 8A). C/EBPβ decreased the expression of E2F-1, c-Myc, DHFR, cyclin A2, and PCNA to varying degrees, while Cdc25A levels were unaffected. The C/EBPβ-induced decrease in gene expression, while modest, was greater than that observed in senescent wild-type MEFs overexpressing RasV12 (data not shown). In contrast, RasV12 generally increased expression of E2F-regulated genes in C/EBPβ−/− MEFs (except for cyclin A2), consistent with the continued proliferation of these cells. We next used chromatin immunoprecipitation (ChIP) assays to examine C/EBPβ association with the proximal promoters of the same genes, using PCR primers that amplify sequences spanning the known E2F sites (Figure 8B). Using two different C/EBPβ antibodies and an E2F4 antiserum, we detected specific binding of C/EBPβ and E2F4 to the E2F-1, c-Myc, DHFR, cyclin A2, and PCNA promoters; E2F4 also bound to Cdc25A. Promoter binding was indicated by increased PCR signals with specific antibodies versus control IgG as well as signal reduction in the presence of blocking peptides. By these criteria, C/EBPβ showed no binding to the negative controls (β-2 microglobulin and lamin B1) or to the Cdc25A promoter. Hence, C/EBPβ is associated with several E2F target genes, and binding correlates with inhibition of expression. Analysis of the promoter sequences using the TESS search tool (www.cbil.upenn.edu/tess/) revealed several potential C/EBP binding sites that could mediate repression by C/EBPβ (data not shown). In the case of DHFR, we identified a novel C/EBP site adjacent to the E2F element that binds C/EBPβ and is required for transcriptional repression in reporter assays (Supplementary Figure 3). These data support a mechanism whereby C/EBPβ elicits cell cycle arrest by repressing E2F target genes.
Figure 8.
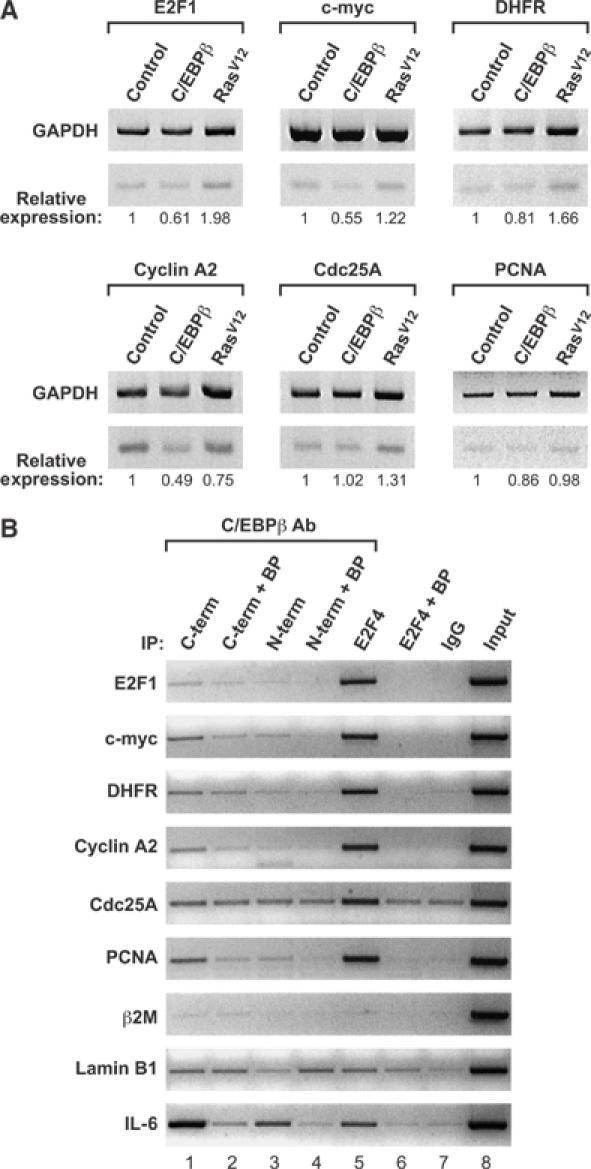
C/EBPβ downregulates and associates with E2F target genes. (A) RT–PCR analyses. Total RNA from C/EBPβ−/− MEFs infected with control, C/EBPβ, or RasV12 viruses was analyzed by RT–PCR to detect expression of E2F-1, c-myc, DHFR, cyclin A2, Cdc25A, and PCNA. GAPDH was used as an internal standard. The PCR products were quantitated and normalized to the corresponding GAPDH levels, and are expressed relative to controls. (B) ChIP assays of NIH 3T3 cells overexpressing C/EBPβ. Chromatin was immunoprecipitated with the indicated antibodies and the recovered DNA was analyzed by PCR using primers corresponding to the indicated genes. C/EBPβ COOH-terminal (C-term), C/EBPβ NH2-terminal (N-term), and E2F4 antibodies were used, as well as a normal rabbit IgG control. Specific binding of the antibodies was determined by preincubating the antibodies with their respective blocking peptides (BP) overnight before using in the immunoprecipitation reaction. Input represents 2% of the total chromatin. β2-Microglobulin (β2M) and lamin B1 are negative controls and IL-6 is a positive control for C/EBPβ binding.
Discussion
We have shown that C/EBPβ null MEFs overexpressing RasV12 fail to undergo senescence and instead display properties of partially transformed cells. C/EBPβ mutant cells also show increased resistance to spontaneous senescence. Conversely, overexpression of C/EBPβ in MEFs suppressed cell growth and, in conjunction with RasV12, induced complete cell cycle arrest. Cells transduced with C/EBPβ also acquired a senescent-like morphology, displaying a flat, extended cell shape and prominent stress fibers. These findings establish C/EBPβ as a novel regulator of the senescence program in primary fibroblasts.
C/EBPβ-mediated cell cycle arrest requires RB-E2F
Induction of p19ARF and p53 by RasV12 was normal in C/EBPβ-deficient cells, indicating that C/EBPβ is not an upstream component of the p19ARF/p53 pathway; moreover, C/EBPβ was capable of suppressing the growth of p19Arf−/− or p53−/− MEFs, particularly when coexpressed with RasV12. Thus, C/EBPβ apparently functions independently or downstream of p19ARF/p53 to implement cell cycle arrest and senescence. Because C/EBPβ did not fully arrest p19Arf−/− or p53−/− MEFs, it is possible that the p19ARF/p53 pathway also has C/EBPβ-independent targets that contribute to growth arrest.
C/EBPβ's ability to suppress cell growth was reduced in Rb−/−/p107−/− MEFs and was completely abolished in TKO cells. C/EBPβ diminished the proliferation of all combinations of double RB knockout MEFs but not TKO cells, demonstrating functional overlap among pRB, p107, and p130 in constraining cell growth (Dannenberg et al, 2000; Sage et al, 2000). C/EBPβ and pRB were previously found to associate in vitro and in vivo, and pRB can enhance the DNA-binding and transcriptional activities of C/EBPβ (Chen et al, 1996b). These two proteins were also implicated in terminal differentiation of adipocytes (Chen et al, 1996a). Thus, C/EBPβ may facilitate RB-mediated cell cycle exit in both senescent and terminally differentiating cells.
C/EBPβ increased the proliferation of TKO MEFs; moreover, an RB binding-defective form of E2F-1 also disrupted C/EBPβ-induced cell cycle arrest and rendered C/EBPβ capable of stimulating proliferation. These findings strongly implicate RB:E2F complexes as targets of C/EBPβ-dependent growth arrest. A previous study using E2F-DB demonstrated that E2F function is required for cellular senescence induced by p19ARF, p53, and RasV12 (Rowland et al, 2002). Apparently, both p53 and C/EBPβ act on or in conjunction with RB:E2F complexes to implement senescence in cells overexpressing RasV12. Whether there is a direct connection between p53 and C/EBPβ in this pathway remains to be established.
Ectopic expression of C/EBPβ decreased transcripts from several E2F target genes and ChIP assays demonstrate binding to their promoters. Hence, C/EBPβ may directly repress genes required for cell cycle progression. C/EBPβ also represses transcription from a DHFR promoter-reporter in wild-type cells but not in TKO MEFs (Supplementary Figure 3), paralleling its effects on the growth of these cells. Transcriptional repression requires a C/EBP-like element adjacent to the E2F site in the DHFR promoter. The related C/EBPα protein can repress transcription from the DHFR promoter and other E2F-regulated genes such as Myc (Timchenko et al, 1999; Slomiany et al, 2000; Johansen et al, 2001; Porse et al, 2001). It was proposed that C/EBPα repression involves an indirect (i.e., non-DNA-binding) mechanism requiring the E2F sites in these promoters (Timchenko et al, 1999; Slomiany et al, 2000; Johansen et al, 2001; Porse et al, 2001). However, a more recent report suggests that C/EBPα can bind directly to the DHFR E2F site (Iakova et al, 2003). We detected weak binding of C/EBPβ to the DHFR E2F motif but much stronger interaction with the adjacent C/EBP-like sequence (Supplementary Figure 3), further suggesting that the latter is the functional C/EBPβ response element. Additional studies are required to determine whether other E2F target genes contain C/EBP sites that mediate transcriptional responses to C/EBPs.
C/EBPβ functions as an effector of Ras signaling
Our studies examined C/EBPβ function in primary fibroblasts overexpressing RasV12. These cells display constitutive high-level Ras signaling and sustained activation of the MEK/ERK cascade and are irreversibly growth-arrested (Serrano et al, 1997; Lin et al, 1998). In this context, C/EBPβ is a key effector of Ras-induced senescence. Since C/EBPβ activity is strongly potentiated by RasV12 (Figure 1B) (Nakajima et al, 1993; Kowenz-Leutz et al, 1994; Zhu et al, 2002), its antiproliferative effects may also require post-translational activation by downstream Ras effectors. RasV12 expression induces phosphorylation on at least two C/EBPβ residues, Thr188 (Nakajima et al, 1993) and Ser64 (Shuman et al, 2004), and C/EBPβ mutants lacking these phosphoacceptor residues are functionally impaired in certain Ras-dependent activity assays (Nakajima et al, 1993; Kowenz-Leutz et al, 1994; Hanlon and Sealy, 1999; Zhu et al, 2002; Mo et al, 2004; Shuman et al, 2004). However, alanine substitutions at residues 188 and/or 64 do not disrupt C/EBPβ's ability to inhibit proliferation of C/EBPβ−/− MEFs, even in the presence of RasV12 (T Sebastian and PF Johnson, unpublished data). Hence, phosphorylation of these two sites is not essential for cell cycle arrest. We are currently investigating whether other Ras-induced modifications on C/EBPβ may be involved.
Studies using conditional activation of an endogenous mutant K-Ras allele (K-rasG12D) indicate that oncogenic Ras does not always cause growth arrest in normal cells (Tuveson et al, 2004). Activation of endogenous K-rasG12D stimulates proliferation of primary MEFs, and in the lung and intestine it induces preneoplastic hyperplasias. Thus, it was suggested that senescence is elicited specifically by sustained, high-level Ras signaling, such as when oncogenic Ras is overexpressed (Tuveson et al, 2004). Conceivably, C/EBPβ is part of the sensing mechanism for high-intensity Ras signaling, instructing cells to senesce under conditions of oncogenic stress instead of undergoing normal responses to Ras signals. In this regard, it will be important to examine the effect of C/EBPβ deficiency on proliferation of cells expressing endogenous levels of oncogenic Ras.
Pro- and antiproliferative functions for C/EBPβ
Our study demonstrates that C/EBPβ is required for RasV12-induced growth arrest in primary fibroblasts and therefore might act as a tumor suppressor. Paradoxically, C/EBPβ also promotes the growth and survival of some cells and tumors. For example, C/EBPβ−/− mice are completely resistant to the development of papillomas in the two-stage model of skin carcinogenesis, which produces Ras-dependent tumors (Zhu et al, 2002). The protumorigenic effect of C/EBPβ in skin cancer may involve its ability to suppress apoptosis. In addition, Myc/Raf-transformed murine macrophages are dependent on C/EBPβ for survival; this antiapoptotic activity is mediated by autocrine production of IGF-I, whose gene is regulated by C/EBPβ (Wessells et al, 2004). Forced expression of C/EBPβ in transformed macrophages also greatly increases colony size, suggesting that C/EBPβ promotes proliferation as well as survival of these cells. Similar promitogenic effects of C/EBPβ were observed in mammary epithelial cells (Bundy and Sealy, 2003) and hepatic cells (Buck et al, 1999). Thus, depending on the cell type, C/EBPβ can either augment or inhibit growth. Such opposite activities are reminiscent of E2F, which mediates the antiproliferative and tumor suppressor effects of pRB but can also stimulate growth by positively regulating transcription of S-phase genes when pRB is mutated or phosphorylated (Johnson, 2000). These parallels may indicate that C/EBPβ, like E2F, interacts with RB proteins to promote cell cycle arrest in specific cells. Future studies should illuminate the molecular basis for cell-specific growth responses to C/EBPβ.
Materials and methods
Plasmid constructs
The pcDNA3.1-C/EBPβ vector was described previously (Shuman et al, 2004).
Cell culture and preparation of MEFs
Wild-type and C/EBPβ−/− MEFs were prepared from day 13.5 embryos derived from mating C/EBPβ+/− mice (Sterneck et al, 1997) on pure 129/Sv and C57BL/6 genetic strain backgrounds. Cells were cultured in DMEM (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Hyclone), 200 mM L-glutamine, and 100 U/ml penicillin–streptomycin (Gibco). Phoenix ecotropic packaging cells (provided by H Young), p53−/− MEFs (P3) (Harvey et al, 1993), p16Ink4a−/− (P5) and p19Arf−/− (P3) MEFs (Sharpless et al, 2001, 2004), and Rb−/−/p130−/−, Rb−/−/p107−/−, p107−/−/p130−/−, and Rb−/−/p107−/−/p130−/− (TKO) MEFs (Dannenberg et al, 2000; Sage et al, 2000) were propagated in the same medium. For 3T3 experiments, MEFs were maintained on a 3-day serial passaging protocol (Todaro and Green, 1963). Cells (3 × 105) were plated in 6 cm dishes, and 3 days later the cell number was determined and 3 × 105 cells were replated.
Retroviral vectors and viral infection
MEFs were infected with a pBabe-puro vector expressing human H-RasV12 cDNA (pBabe-H-RasV12) provided by S Lowe; expression of H-RasV12 using pLXSP3 or pWZL-hygro gave similar results. pWZL-hygro and pBabe-puro were used to generate retroviruses expressing the p35 form of murine C/EBPβ (Descombes and Schibler, 1991) or the p42 form of rat C/EBPα. pWZL-hygro and pWZL-H-RasV12-hygro were obtained from K Vousden. pBabe-puro expressing truncated E2F-1 (residues 1–368; ‘E2F-DB') was kindly provided by B Rowland (Krek et al, 1995; Rowland et al, 2002). Retroviral plasmids were transfected into the Phoenix packaging line using a standard CaPO4 method. At 24–72 h after transfection, viral supernatants were collected every 5–6 h, pooled, filtered (0.45 μm), supplemented with 5 μg/ml polybrene, and used to infect P2-P3 MEFs. Three infections were performed and the cells were selected for 3 days in 2 μg/ml puromycin or for 5 days in 100 μg/ml hygromycin. Multiple genes were introduced by sequential infection and drug selection.
Growth curves
Retrovirally infected MEFs were seeded at 2.5 × 104 cells/well in six-well plates. At the indicated times, cells were washed with phosphate-buffered saline (PBS), fixed in 10% formalin, rinsed with water, stained with 0.1% crystal violet (Sigma) for 30 min, rinsed extensively, and dried. The dye was extracted with 10% acetic acid and absorbance measured at 590 nm. All values were normalized to day 0.
Colony assays
MEFs were plated at 2 × 104 cells/10 cm dish in DMEM containing 10% FBS, and after 2 weeks colonies were visualized by crystal violet staining. For soft agar colony assays, cells were placed into 0.35% agar in DMEM containing 10% FBS at 2.5 × 104 cells/6 cm plate and seeded onto solidified 0.7% agar containing culture medium. The cells were fed weekly and colonies were evaluated after 2 weeks.
Reporter assays
C/EBPβ−/− MEFs were plated 12 h prior to transfection (7 × 104 cells/well in six-well plates). A 1 μg portion of 2 × C/EBP Luc reporter plasmid was cotransfected with 100 ng of murine C/EBPβ expression vector (pcDNA3.1-C/EBPβ) and/or H-RasV12 (pcDNA3.1-Ras) using Fugene6 (Roche Molecular Biochemicals). At 16 h prior to harvesting, the cells were placed into medium containing 0.5% serum. At 48 h after transfection, the cells were lysed and analyzed using the Luciferase assay system (Promega). Luciferase values were normalized to protein levels; the data represent the average of three independent determinations and are graphed as the mean±s.e.
Immunoblotting
Lysates from retrovirally transduced MEFs were prepared in NP-40 lysis buffer (50 mM Tris–HCl (pH 8.0), 400 mM NaCl, 1% NP-40, 1 mM EDTA) containing protease inhibitors and cleared by high-speed centrifugation. Nuclear extracts were prepared as described (Baer et al, 1998). A 25 μg portion of nuclear extract or 80–100 μg of whole cell lysate was resolved by 12% SDS–PAGE and blotted to nitrocellulose membranes. Primary antibodies used were as follows: C/EBPβ (Santa Cruz, C-19, 1:1000), C/EBPα (Santa Cruz, 14AA, 1:1000), p53 (Novacastra, CM5, 1:1000), p19ARF (Abcam, ab80, 1:500), p16INK4a (Santa Cruz, M-156, 1:1000), cyclin A (Santa Cruz, C-19, 1:1000), E2F-1 (Santa Cruz, KH-95, 1:1000), and actin (Santa Cruz, C-11, 1:1000). Secondary antibodies conjugated to horseradish peroxidase were used to detect antigen–antibody by chemiluminescence (ECL detection system; Pierce).
Immunofluorescence staining of actin stress fibers
MEFs grown on 18 mm glass coverslips were fixed with 3.7% formaldehyde in PBS for 10 min, permeabilized with 0.2% Triton X-100 in PBS for 5 min, and blocked with 1% bovine serum albumin in PBS for 30 min. To visualize actin stress fibers, samples were incubated with FITC-conjugated phalloidin (Molecular Probes) for 20 min at room temperature. The cells were washed three times with PBS and mounted on glass slides using Vectashield (Vector Laboratories). Fluorescence images were obtained using a Zeiss microscope.
RT–PCR analysis
Total cellular RNA was prepared using Trizol reagent (Invitrogen). A 1 μg portion of total RNA was reverse transcribed using the first strand cDNA synthesis kit (Super Array). After reverse transcription, cDNAs were amplified using primers for GAPDH (control), E2F-1, c-myc, DHFR, cyclin A2, Cdc25A, and PCNA. PCR products were separated on agarose gels and visualized by ethidium bromide staining. The gene-specific primers and Hot-start PCR reagents were purchased from SuperArray Bioscience.
Chromatin immunoprecipitation
ChIP assay was performed according to Spencer et al (2003) with minor modifications. In brief, subconfluent cell cultures (15 cm dish per one ChIP reaction) were crosslinked by addition of 1% formaldehyde and incubated for 10 min at room temperature. Cells were washed with PBS and resuspended in lysis buffer (0.1% SDS, 0.5% Triton X-100, 150 mM NaCl, 20 mM Tris–HCl, pH 8.1) and sonicated to obtain DNA fragments of 500–1000 bp. Immunoprecipitation was performed using 5 μg of the following antibodies: C/EBPβ C-terminal (C-19, Santa-Cruz), C/EBPβ N-terminal (Williams et al, 1991), and E2F-4 (C-20, Santa-Cruz). In control reactions, antibodies were preincubated overnight with their respective blocking peptides. Samples were incubated with antibodies overnight at 4°C and then StaphA cells (Calbiochem) were added and incubated for 30 min at 4°C. Precipitates were washed and processed for DNA purification. DNA was amplified by PCR using sequence-specific primers (30–35 cycles). Primers sequences are available upon request.
Supplementary Material
Supplemental Material
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Acknowledgments
We thank S Lowe and K Vousden for RasV12 retroviral vectors, L Hernandez and C Stewart for p53−/− MEFs, N Sharpless for p16Ink4a−/− and p19Arf−/− MEFs, P Farnham for DHFR reporter constructs, B Rowland for the E2F-DB vector, L Warg and B Shankle for maintaining the mouse colony and excellent technical assistance, and J Wessells and C McCauslin for critical reading of the manuscript. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- Alexander K, Hinds PW (2001) Requirement for p27(KIP1) in retinoblastoma protein-mediated senescence. Mol Cell Biol 21: 3616–3631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer M, Williams SC, Dillner A, Schwartz RC, Johnson PF (1998) Autocrine signals control CCAAT/enhancer binding protein beta expression, localization, and activity in macrophages. Blood 92: 4353–4365 [PubMed] [Google Scholar]
- Buck M, Poli V, van der Geer P, Chojkier M, Hunter T (1999) Phosphorylation of rat serine 105 or mouse threonine 217 in C/EBP beta is required for hepatocyte proliferation induced by TGF alpha. Mol Cell 4: 1087–1092 [DOI] [PubMed] [Google Scholar]
- Buck M, Turler H, Chojkier M (1994) LAP (NF-IL-6), a tissue-specific transcriptional activator, is an inhibitor of hepatoma cell proliferation. EMBO J 13: 851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundy LM, Sealy L (2003) CCAAT/enhancer binding protein beta (C/EBPbeta)-2 transforms normal mammary epithelial cells and induces epithelial to mesenchymal transition in culture. Oncogene 22: 869–883 [DOI] [PubMed] [Google Scholar]
- Campisi J (1996) Replicative senescence: an old lives' tale? Cell 84: 497–500 [DOI] [PubMed] [Google Scholar]
- Cao Z, Umek RM, McKnight SL (1991) Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev 5: 1538–1552 [DOI] [PubMed] [Google Scholar]
- Chen PL, Riley DJ, Chen Y, Lee WH (1996a) Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes Dev 10: 2794–2804 [DOI] [PubMed] [Google Scholar]
- Chen PL, Riley DJ, Chen-Kiang S, Lee WH (1996b) Retinoblastoma protein directly interacts with and activates the transcription factor NF-IL6. Proc Natl Acad Sci USA 93: 465–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg JH, van Rossum A, Schuijff L, te Riele H (2000) Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev 14: 3051–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descombes P, Chojkier M, Lichtsteiner S, Falvey E, Schibler U (1990) LAP, a novel member of the C/EBP gene family, encodes a liver-enriched transcriptional activator protein. Genes Dev 4: 1541–1551 [DOI] [PubMed] [Google Scholar]
- Descombes P, Schibler U (1991) A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 67: 569–579 [DOI] [PubMed] [Google Scholar]
- Goldstein S (1990) Replicative senescence: the human fibroblast comes of age. Science 249: 1129–1133 [DOI] [PubMed] [Google Scholar]
- Greenbaum LE, Li W, Cressman DE, Peng Y, Ciliberto G, Poli V, Taub R (1998) CCAAT enhancer-binding protein beta is required for normal hepatocyte proliferation in mice after partial hepatectomy. J Clin Invest 102: 996–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth A, Weber JD, Willumsen BM, Sherr CJ, Roussel MF (2000) Oncogenic Ras induces p19ARF and growth arrest in mouse embryo fibroblasts lacking p21Cip1 and p27Kip1 without activating cyclin D-dependent kinases. J Biol Chem 275: 27473–27480 [DOI] [PubMed] [Google Scholar]
- Hanlon M, Sealy L (1999) Ras regulates the association of serum response factor and CCAAT/enhancer-binding protein beta. J Biol Chem 274: 14224–14228 [DOI] [PubMed] [Google Scholar]
- Harvey M, Sands AT, Weiss RS, Hegi ME, Wiseman RW, Pantazis P, Giovanella BC, Tainsky MA, Bradley A, Donehower LA (1993) In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene 8: 2457–2467 [PubMed] [Google Scholar]
- Hendricks-Taylor LR, Darlington GJ (1995) Inhibition of cell proliferation by C/EBP alpha occurs in many cell types, does not require the presence of p53 or Rb, and is not affected by large T-antigen. Nucleic Acids Res 23: 4726–4733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iakova P, Awad SS, Timchenko NA (2003) Aging reduces proliferative capacities of liver by switching pathways of C/EBPalpha growth arrest. Cell 113: 495–506 [DOI] [PubMed] [Google Scholar]
- Johansen LM, Iwama A, Lodie TA, Sasaki K, Felsher DW, Golub TR, Tenen DG (2001) c-Myc is a critical target for c/EBPalpha in granulopoiesis. Mol Cell Biol 21: 3789–3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DG (2000) The paradox of E2F1: oncogene and tumor suppressor gene. Mol Carcinog 27: 151–157 [DOI] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ (1997) Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 91: 649–659 [DOI] [PubMed] [Google Scholar]
- Kowenz-Leutz E, Twamley G, Ansieau S, Leutz A (1994) Novel mechanism of C/EBP β (NF-M) transcriptional control: activation through derepression. Genes Dev 8: 2781–2791 [DOI] [PubMed] [Google Scholar]
- Krek W, Xu G, Livingston DM (1995) Cyclin A-kinase regulation of E2F-1 DNA binding function underlies suppression of an S phase checkpoint. Cell 83: 1149–1158 [DOI] [PubMed] [Google Scholar]
- Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW (1998) Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 12: 3008–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AW, Lowe SW (2001) Oncogenic ras activates the ARF–p53 pathway to suppress epithelial cell transformation. Proc Natl Acad Sci USA 98: 5025–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo X, Kowenz-Leutz E, Xu H, Leutz A (2004) Ras induces mediator complex exchange on C/EBP beta. Mol Cell 13: 241–250 [DOI] [PubMed] [Google Scholar]
- Nakajima T, Kinoshita S, Sasagawa T, Sasaki K, Naruto M, Kishimoto T, Akira S (1993) Phosphorylation at threonine-235 by a ras-dependent mitogen-activated protein kinase cascade is essential for transcription factor NF-IL6. Proc Natl Acad Sci USA 90: 2207–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerlov C (2004) C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer 4: 394–400 [DOI] [PubMed] [Google Scholar]
- Pawlak G, Helfman DM (2001) Cytoskeletal changes in cell transformation and tumorigenesis. Curr Opin Genet Dev 11: 41–47 [DOI] [PubMed] [Google Scholar]
- Peeper DS, Dannenberg JH, Douma S, te Riele H, Bernards R (2001) Escape from premature senescence is not sufficient for oncogenic transformation by Ras. Nat Cell Biol 3: 198–203 [DOI] [PubMed] [Google Scholar]
- Poli V, Mancini FP, Cortese R (1990) IL-6DBP, a nuclear protein involved in interleukin-6 signal transduction, defines a new family of leucine zipper proteins related to C/EBP. Cell 63: 643–653 [DOI] [PubMed] [Google Scholar]
- Porse BT, Pedersen TA, Xu X, Lindberg B, Wewer UM, Friis-Hansen L, Nerlov C (2001) E2F repression by C/EBPalpha is required for adipogenesis and granulopoiesis in vivo. Cell 107: 247–258 [DOI] [PubMed] [Google Scholar]
- Robinson GW, Johnson PF, Hennighausen L, Sterneck E (1998) The C/EBPbeta transcription factor regulates epithelial cell proliferation and differentiation in the mammary gland. Genes Dev 12: 1907–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland BD, Denissov SG, Douma S, Stunnenberg HG, Bernards R, Peeper DS (2002) E2F transcriptional repressor complexes are critical downstream targets of p19(ARF)/p53-induced proliferative arrest. Cancer Cell 2: 55–65 [DOI] [PubMed] [Google Scholar]
- Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T (2000) Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev 14: 3037–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seagroves TN, Krnacik S, Raught B, Gay J, Burgess-Beusse B, Darlington GJ, Rosen JM (1998) C/EBPbeta, but not C/EBPalpha, is essential for ductal morphogenesis, lobuloalveolar proliferation, and functional differentiation in the mouse mammary gland. Genes Dev 12: 1917–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA (1996) Role of the INK4a locus in tumor suppression and cell mortality. Cell 85: 27–37 [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602 [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA (2001) Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 413: 86–91 [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Ramsey MR, Balasubramanian P, Castrillon DH, DePinho RA (2004) The differential impact of p16(INK4a) or p19(ARF) deficiency on cell growth and tumorigenesis. Oncogene 23: 379–385 [DOI] [PubMed] [Google Scholar]
- Sherr CJ, DePinho RA (2000) Cellular senescence: mitotic clock or culture shock? Cell 102: 407–410 [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Weber JD (2000) The ARF/p53 pathway. Curr Opin Genet Dev 10: 94–99 [DOI] [PubMed] [Google Scholar]
- Shuman JD, Sebastian T, Kaldis P, Copeland TD, Zhu S, Smart RC, Johnson PF (2004) Cell cycle-dependent phosphorylation of C/EBPbeta mediates oncogenic cooperativity between C/EBPbeta and H-RasV12. Mol Cell Biol 24: 7380–7391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomiany BA, D'Arigo KL, Kelly MM, Kurtz DT (2000) C/EBPalpha inhibits cell growth via direct repression of E2F-DP-mediated transcription. Mol Cell Biol 20: 5986–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer VA, Sun JM, Li L, Davie JR (2003) Chromatin immunoprecipitation: a tool for studying histone acetylation and transcription factor binding. Methods 31: 67–75 [DOI] [PubMed] [Google Scholar]
- Sterneck E, Tessarollo L, Johnson PF (1997) An essential role for C/EBPβ in female reproduction. Genes Dev 11: 2153–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Akira S, Yoshida K, Umemoto M, Yoneda Y, Shirafuji N, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T (1995) Targeted disruption of the NF-IL6 gene discloses its essential role in bacteria killing and tumor cytotoxicity by macrophages. Cell 80: 353–361 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Yoshida N, Kishimoto T, Akira S (1997) Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J 16: 7432–7443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QQ, Otto TC, Lane MD (2003) CCAAT/enhancer-binding protein beta is required for mitotic clonal expansion during adipogenesis. Proc Natl Acad Sci USA 100: 850–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko NA, Wilde M, Darlington GJ (1999) C/EBPalpha regulates formation of S-phase-specific E2F–p107 complexes in livers of newborn mice. Mol Cell Biol 19: 2936–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todaro GJ, Green H (1963) Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol 17: 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, Hingorani SR, Zaks T, King C, Jacobetz MA, Wang L, Bronson RT, Orkin SH, DePinho RA, Jacks T (2004) Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5: 375–387 [DOI] [PubMed] [Google Scholar]
- Umek RM, Friedman AD, McKnight SL (1991) CCAAT-enhancer binding protein: a component of a differentiation switch. Science 251: 288–292 [DOI] [PubMed] [Google Scholar]
- Weinberg RA (1989) Oncogenes, antioncogenes, and the molecular bases of multistep carcinogenesis. Cancer Res 49: 3713–3721 [PubMed] [Google Scholar]
- Wessells J, Yakar S, Johnson PF (2004) Critical prosurvival roles for C/EBP beta and insulin-like growth factor I in macrophage tumor cells. Mol Cell Biol 24: 3238–3250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SC, Baer M, Dillner AJ, Johnson PF (1995) CRP2 (C/EBPβ) contains a bipartite regulatory domain that controls transcriptional activation, DNA binding and cell specificity. EMBO J 14: 3170–3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SC, Cantwell CA, Johnson PF (1991) A family of C/EBP-related proteins capable of forming covalently linked leucine zipper dimers in vitro. Genes Dev 5: 1553–1567 [DOI] [PubMed] [Google Scholar]
- Xu HJ, Zhou Y, Ji W, Perng GS, Kruzelock R, Kong CT, Bast RC, Mills GB, Li J, Hu SX (1997) Reexpression of the retinoblastoma protein in tumor cells induces senescence and telomerase inhibition. Oncogene 15: 2589–2596 [DOI] [PubMed] [Google Scholar]
- Zhu S, Oh HS, Shim M, Sterneck E, Johnson PF, Smart RC (1999) C/EBPbeta modulates the early events of keratinocyte differentiation involving growth arrest and keratin 1 and keratin 10 expression. Mol Cell Biol 19: 7181–7190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Yoon K, Sterneck E, Johnson PF, Smart RC (2002) CCAAT/enhancer binding protein-beta is a mediator of keratinocyte survival and skin tumorigenesis involving oncogenic Ras signaling. Proc Natl Acad Sci USA 99: 207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3