

Abstract
MDM2 is a RING domain ubiquitin E3 ligase and a major regulator of the p53 tumor suppressor. MDM2 binds to p53, inactivates p53 transcription function, inhibits p53 acetylation, and promotes p53 degradation. Here, we present evidence that MDM2 interacts with the nuclear corepressor KAP1. The binding is mediated by the N-terminal coiled-coil domain of KAP1 and the central acidic domain of MDM2. KAP1 stimulates formation of p53–HDAC1 complex and inhibits p53 acetylation by interacting with MDM2. Expression of KAP1 cooperates with MDM2 to promote p53 ubiquitination and degradation. The tumor suppressor ARF competes with KAP1 in MDM2 binding; oncogene induction of ARF expression reduces MDM2–KAP1 interaction. Depletion of endogenous KAP1 expression by RNAi stimulates p53 transcriptional activity, sensitizes p53 response to DNA damage, and increases apoptosis. Therefore, MDM2 interaction with KAP1 contributes to p53 functional regulation. ARF may regulate p53 acetylation and stability in part by inhibiting KAP1–MDM2 binding.
Keywords: acetylation, ARF, KAP1, MDM2, p53
Introduction
The p53 tumor suppressor is critical for maintenance of genome stability and protection against malignant transformation. p53 is regulated by multiple signal pathways and mechanisms, allowing it to respond to a wide range of stress conditions and act as a tumor suppressor in many cell types (Vousden, 2000). p53 turnover is regulated by MDM2, which binds p53 and functions as an ubiquitin E3 ligase to promote p53 ubiquitination and degradation by the proteasomes (Zhang and Xiong, 2001). Stress signals such as DNA damage induce p53 accumulation by phosphorylation (Prives and Hall, 1999). Mitogenic signals activate p53 by induction of the ARF tumor suppressor encoded by an alternative open reading frame in the p16INK4a locus, which inhibits the ability of MDM2 to ubiquitinate p53 (Zhang and Xiong, 2001).
MDM2 is an ubiquitin E3 ligase that promotes ubiquitination of itself, p53, and several other cellular proteins, including androgen receptor, Tip60, glucocorticoid receptor, and the MDM2 homolog MDMX (De Graaf et al, 2003; Kawai et al, 2003a; Pan and Chen, 2003). Ubiquitination of p53 and MDM2 requires the C-terminal RING domain of MDM2, which is involved in recruitment of the ubiquitin conjugating enzyme E2 (Joazeiro and Weissman, 2002). Efficient ubiquitination of p53 also requires the central acidic domain of MDM2, which is an important regulatory domain targeted by ARF and possibly other signals (Midgley et al, 2001; Blattner et al, 2002; Meulmeester et al, 2003). The acidic domain also has important functions in targeting the degradation of ubiquitinated p53 (Blattner et al, 2002).
In addition to regulating p53 ubiquitination, MDM2 has been shown to inhibit p53 acetylation by coactivators p300/CBP and PCAF (Ito et al, 2001; Kobet et al, 2002). Competition with p300 for p53 binding may in part account for the inhibitory effect of MDM2 on p53 acetylation. However, MDM2 can also directly interact with p300 and appears to be able to inhibit the acetylation of p53 directly (Ito et al, 2001). Additionally, MDM2 may also promote p53 deacetylation by recruiting histone deacetylases, mainly HDAC1 (Ito et al, 2002). Acetylation of p53 stimulates its DNA-binding activity in vitro and may directly contribute to enhanced transcriptional function in vivo (Gu and Roeder, 1997; Ito et al, 2001). Because p53 ubiquitination and acetylation occur on similar lysine residues in the C terminus, acetylation can inhibit p53 ubiquitination (Li et al, 2002; Rodriguez et al, 2002). More recently, MDM2 itself has also been found to be acetylated by p300 and CBP in the RING domain, possibly inhibiting the ubiquitin E3 ligase function of MDM2 (Wang et al, 2004).
In addition to ARF, MDM2 activity is also regulated by interaction with other proteins. The p300 coactivator interacts with MDM2 and stimulates p53 polyubiquitination by MDM2 (Grossman et al, 2003). Ribosomal proteins L5 and L11 are significant binding partners of MDM2 (Marechal et al, 1994; Lohrum et al, 2003; Zhang et al, 2003). L11 has been shown to inhibit p53 ubiquitination by binding to the central acidic region of MDM2 and behaves very similar to ARF, suggesting that it may play a role in p53 signaling by sensing a different type of cellular stress that affects ribosomal biogenesis or function (Zhang et al, 2003). Recent studies showed that the multifunctional transcription regulator YY1 interacts with MDM2 through the acidic region, stimulating MDM2–p53 complex formation and p53 ubiquitination (Sui et al, 2004). YY1 also directly binds to p53 transactivation domain and blocks p53–p300 binding (Gronroos et al, 2004). Elimination of YY1 by RNAi or genetic knockout leads to p53 accumulation and activation, suggesting that YY1 is critical for maintaining normal levels of p53. YY1–MDM2 interaction is competitively inhibited by ARF (Sui et al, 2004), suggesting that a mechanism of p53 activation by ARF is disruption of YY1–MDM2 binding.
In this report, we describe the identification of nuclear corepressor KAP1 as a novel MDM2-binding protein. KAP1 was first identified as a factor important for the transcription repression function of Kruppel-associated box (KRAB) domain (Friedman et al, 1996). KAP1 contains conserved domains for RING finger, B boxes, leucine zipper alpha helical coiled-coil region, PHD (plant homodomain) finger, and bromodomain. The N-terminal RING, B boxes, and coiled-coil sequences (RBCC) are necessary and sufficient for interaction with KRAB and formation of KAP1 oligomers (Peng et al, 2000). The RBCC tripartite motif (also called TRIM) is found in a large family of proteins including PML, TIF1, and Rfp (Jensen et al, 2001). The C-terminal PHD finger and bromodomain of KAP1 cooperatively function as a transcription repression domain by recruiting the histone deacetylase complex NuRD and histone H3 lysine 9-specific methyltransferase SETDB1 (Schultz et al, 2001, 2002). Therefore, KAP1 may function as a corepressor in part by being targeted to DNA by other transcription factors and promotes modification of chromatin structure. KAP1 expression is important for normal development in mice. Germline homozygous knockout of KAP1 results in embryonic lethality at E5.5 (Cammas et al, 2000).
Results described below show that MDM2 and KAP1 interact through the coiled-coil domain of KAP1 and the central acidic domain of MDM2. KAP1 stimulates p53–HDAC1 complex formation and inhibits p53 acetylation by interacting with MDM2. Overexpression of KAP1 cooperates with MDM2 to promote p53 ubiquitination and degradation. ARF competes with KAP1 for MDM2 binding during mitogenic stress response. Reduction of endogenous KAP1 level sensitizes cells to p53 activation and apoptosis after DNA damage. Therefore, KAP1 is a novel MDM2-binding protein that contributes to p53 functional regulation.
Results
KAP1 is a novel MDM2-interacting protein
To identify novel MDM2-binding proteins, human MDM2 expression plasmid was transiently transfected into 293T cells and MDM2 was affinity purified. MDM2 is a protein with very short half-life due to self-ubiquitination and rapid degradation. To increase the yield of MDM2, the transfected cells were also treated with proteasome inhibitor MG132 for 4 h before harvest to block protein degradation. Mass spectrometric sequencing of multiple MDM2-copurifying bands revealed the presence of multiple ribosomal proteins including L5 and L11 as observed previously (Figure 1A) (Marechal et al, 1994; Lohrum et al, 2003; Zhang et al, 2003). However, the analysis also revealed that the transcription corepressor KAP1 was copurified with MDM2 (Figure 1A), migrating as a ∼120 kDa band.
Figure 1.
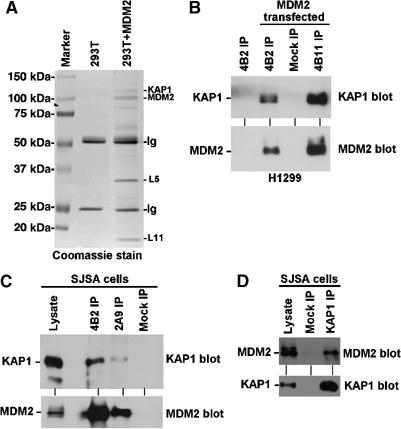
KAP1 forms a complex with MDM2. (A) 293T cells were transiently transfected with MDM2 expression plasmid for 48 h and treated for 4 h with 30 μM MG132. MDM2 was immunoprecipitated using 2A9 antibody. The MDM2-binding proteins were detected by Coomassie blue staining and identified by mass spectrometry. (B) H1299 cells were transiently transfected with MDM2 and immunoprecipitated using MDM2 antibodies 4B2 and 4B11. Co-precipitation of endogenous KAP1 was detected by Western blot. (C) Endogenous MDM2 in SJSA cells was precipitated using MDM2 antibodies 4B2 and 2A9, and co-precipitation of endogenous KAP1 was detected by Western blot. (D) Endogenous KAP1 was immunoprecipitated from SJSA cells and co-precipitation of endogenous MDM2 was detected by Western blot.
In order to confirm the interaction between MDM2 and KAP1, 293T and H1299 cells were transiently transfected with MDM2. MDM2 was immunoprecipitated using two monoclonal antibodies (4B2, 4B11) (Chen et al, 1993). Western blot of MDM2 complexes using KAP1 antibody revealed the presence of KAP1, which correlated with the amount of MDM2 in the immunoprecipitate (Figure 1B). To confirm binding between endogenous MDM2 and KAP1, the osteosarcoma cell line SJSA (overexpressing MDM2) was immunoprecipitated with MDM2 antibodies 4B2 and 2A9. KAP1 was also detected in the MDM2 complex, which correlated with the level of MDM2 recovery by each antibody (Figure 1C). In a reverse immunoprecipitation (IP)–Western blot, endogenous MDM2 in SJSA cells was also co-precipitated by the KAP1 antibody (Figure 1D). The binding between endogenous MDM2 and KAP1 was not affected by ionizing radiation (data not shown). These results demonstrated that KAP1 is a novel MDM2-binding protein.
The MDM2 homolog MDMX also shares extensive sequence similarity to MDM2 and the ability to bind p53 (Shvarts et al, 1996). Although no KAP1 band was detected in affinity purification of MDMX-binding proteins in unrelated experiments, cotransfection of MDMX with KAP1 also resulted in their co-precipitation in sensitive IP–Western blot assay (data not shown). Although we will focus on MDM2–KAP1 binding in this study, it is possible that MDMX-KAP1 interaction also plays a role in regulating p53 function.
Mapping of KAP1–MDM2 interaction domains
To identify the regions on MDM2 and KAP1 involved in complex formation, a panel of MDM2 deletion mutants were transfected into 293T cells. Co-precipitation of endogenous KAP1 was determined by MDM2 IP–KAP1 Western blot. The effects of the deletions on p53 binding were also confirmed by MDM2 IP–p53 Western blot. The initial results revealed that the central region of MDM2 from 150 to 325 was required for binding KAP1 (Figure 2A). Further mapping using an additional set of MDM2 deletion mutants showed that 1–230 of MDM2 was sufficient for KAP1 binding, whereas 1–200 was defective (Figure 2B). Collectively, these results indicated that 150–230 of MDM2 was involved in KAP1 binding, with 200–230 being a critical region. This region contains several important elements, including the nuclear import signal (NLS, 179–185), nuclear export signal (NES, 191–202), and part of the central acidic domain (200–300). The KAP1-interacting region also partially overlaps with the binding site for ARF (212–244) that is important for regulating the ability of MDM2 to ubiquitinate and degrade p53 (Midgley et al, 2001; Kawai et al, 2003b).
Figure 2.
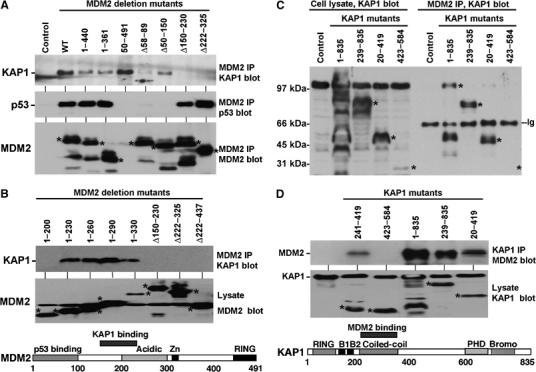
Mapping of MDM2–KAP1 interaction. (A) The region of MDM2 required for KAP1 binding was determined by expression of MDM2 mutants in 293T cells followed by MDM2 IP and Western blot for endogenous KAP1. Expression of MDM2 mutants was confirmed by Western blot using 4B2 (recognizing an epitope between 19 and 50), which detects all except the 50–491 mutant. The p53-binding activity MDM2 mutants was confirmed by MDM2 IP and p53 Western blot. (B) Mapping of MDM2–KAP1 binding using additional MDM2 deletion mutants and summary of results in the MDM2 diagram. (C) The region of KAP1 critical for MDM2 binding was determined by cotransfection of KAP1 deletion mutants with full-length MDM2 into 293T cells, followed by MDM2 IP and KAP1 Western blot. Endogenous and exogenous KAP1 expression was confirmed by direct Western blot on the left half of the panel. (D) The coiled-coil fragment (241–419) of KAP1 was expressed as a miniprotein and its ability to bind MDM2 was confirmed as in panel C. The results are summarized in the diagram for KAP1. * indicates the expected positions of mutant proteins.
To map the MDM2-binding domain on KAP1, a panel of KAP1 deletion mutants were cotransfected with MDM2 into 293T cells. MDM2 was precipitated with 2A9 antibody and bound KAP1 mutants were detected by Western blot using antibodies against the N- and C-terminal regions of the protein. The results showed that the coiled-coil region (239–419) was required for binding to MDM2 (Figure 2C). Furthermore, the 241–419 fragment of KAP1 was sufficient for MDM2 binding when expressed as a miniprotein (Figure 2D). The C-terminal PHD and bromodomains of KAP1 are involved in recruitment of histone deacetylase-containing complex and methyltransferases in transcription repression (Schultz et al, 2001, 2002). Therefore, MDM2 interaction with the coiled-coil region may recruit KAP1 and its C-terminal-associated cofactors to modify p53 acetylation level, which is further tested below.
KAP1 stimulates p53–HDAC1 interaction and p53 deacetylation
Since the KAP1-binding site on MDM2 is separate from the N-terminal p53-binding domain, MDM2 may mediate interaction between p53 and KAP1. Formation of such a ternary complex would lead to modification of p53 by histone deacetylases recruited by KAP1. IP of endogenous or transfected p53 from 293T cells did not result in significant co-precipitation of endogenous KAP1, possibly due to SV40 T-antigen inactivation of p53 and low MDM2 expression level. However, transfection of exogenous MDM2 resulted in formation of a complex between endogenous p53 and KAP1. Furthermore, MDM2 mutants deficient for KAP1 binding did not stimulate endogenous p53–KAP1 interaction, whereas a point mutation that inactivates the RING domain (457S) had no effect (Figure 3A). Therefore, MDM2 mediates the formation of p53–MDM2–KAP1 ternary complex by acting as an adapter molecule.
Figure 3.
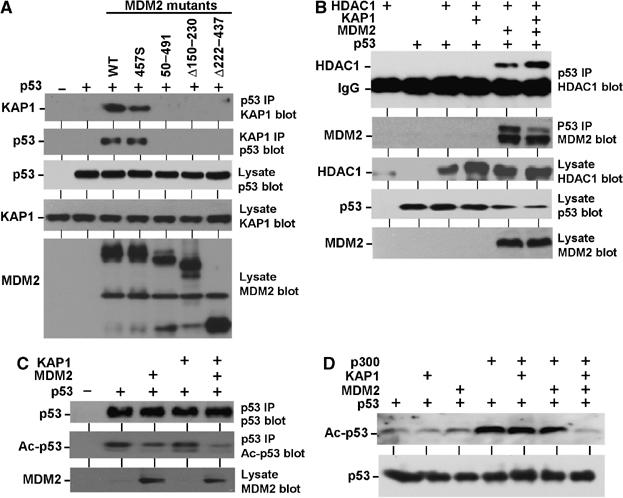
KAP1 and MDM2 cooperatively regulate p53 acetylation. (A) Formation of p53–MDM2–KAP1 ternary complex. MDM2 mutants and p53 were transfected into H1299 cells, p53 was immunoprecipitated using Pab1801 antibody, and KAP1 co-precipitation was detected by Western blot. The result was also confirmed by KAP1 IP using polyclonal antibody followed by p53 Western blot using DO-1. (B) KAP1 promotes p53–HDAC1 binding. H1299 cells were transfected with indicated plasmids and p53 was immunoprecipitated by Pab1801. The co-precipitated FLAG-HDAC1 was detected by Western blot using the M2 antibody. (C) KAP1 and MDM2 cooperate in inhibiting p53 acetylation. H1299 cells were transfected with indicated plasmids and the level of acetylated p53 was determined by Western blot using an antibody specifically recognizing acetylated lysine 382. The total level of p53 was detected by Western blot using DO-1 antibody. (D) H1299 cells were transfected with p53 and p300 and the level of acetylated p53 was determined by Western blot using an antibody specifically recognizing acetylated lysine 382.
MDM2 and p53 have been shown to bind HDAC1 (Juan et al, 2000; Luo et al, 2000; Ito et al, 2002). In order to determine whether KAP1 promotes interaction of p53 with HDAC1, H1299 cells were transiently cotransfected with expression plasmids for each protein and analyzed by IP–Western blot. In this experiment, p53 co-precipitated with HDAC1 in an MDM2-dependent fashion, and the interaction was further enhanced by coexpression of KAP1 (Figure 3B). These results showed that MDM2 and KAP1 cooperate to recruit HDAC1 into a complex containing p53.
MDM2 has been shown to inhibit the acetylation of p53 by p300 (Ito et al, 2002). To test the effect of KAP1 on p53 acetylation level, H1299 cells were transfected with p53, MDM2, and KAP1. Acetylation of p53 on lysines 373 and 382 was detected by Western blot using acetylation-specific antibodies. As expected, p53 acetylation level was reduced by expression of MDM2 but not by KAP1. However, MDM2 and KAP1 coexpression led to further reduction of p53 acetylation level (Figure 3C). In a second assay, p300 was cotransfected to further stimulate p53 acetylation. KAP1 expression also led to reduction of p53 acetylation level in an MDM2-dependent fashion (Figure 3D). Therefore, KAP1 and MDM2 cooperate in inhibiting the acetylation of p53, possibly due to recruitment of HDAC1 to the p53–MDM2 complex by KAP1.
KAP1 cooperates with MDM2 to promote p53 ubiquitination and degradation
An important function of MDM2 is to promote ubiquitination and degradation of p53. Ubiquitination and acetylation of p53 occur on the same lysine residues on the C terminus of p53 and are mutually exclusive events (Li et al, 2002). To determine whether KAP1 cooperates with MDM2 to ubiquitinate p53, H1299 cells were transfected with p53 and His6-ubiquitin expression plasmids. The in vivo ubiquitination level of p53 was determined by Ni-NTA purification of proteins conjugated to His6-ubiquitin and p53 Western blot. As expected, ubiquitination of p53 was stimulated by MDM2, and expression of KAP1 further enhanced p53 ubiquitination level in an MDM2-dependent fashion (Figure 4A). The ability of KAP1 to stimulate p53 ubiquitination required the MDM2 RING domain. The stable MDM2-457S E3 ligase mutant did not cooperate with KAP1, and possibly acted in a dominant-negative fashion over the endogenous MDM2 due to high-level expression (Figure 4A).
Figure 4.
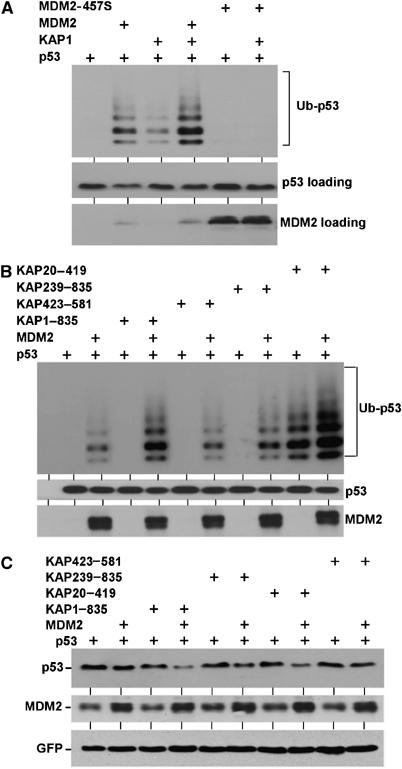
KAP1 promotes p53 ubiquitination by MDM2. (A) H1299 cells were transfected with His6-ubiquitin and indicated plasmids. The level of p53 ubiquitination was determined by Ni-NTA purification and p53 Western blot. KAP1 stimulated p53 ubiquitination in the presence of full-length MDM2. The MDM2-457S mutant contains an inactivating point mutation in the RING domain. (B) The ability of KAP1 mutants to promote p53 ubiquitination in the presence or absence of MDM2 was determined in MDM2-null 174.1 MEFs using the same assay as in panel A. The KAP20–419 RBCC fragment promoted p53 ubiquitination independent of MDM2. (C) H1299 cells were transfected with p53 in combination with indicated plasmids and p53 expression level was determined by Western blot after 48 h. KAP1 promoted degradation of p53 in the presence of MDM2.
KAP1 has a RING domain and a PHD domain, which are features of ubiquitin E3 ligases (Joazeiro and Weissman, 2002). Figure 4A suggested that KAP1 alone does not function as E3 for p53. To determine which domain of KAP1 is important for cooperation with MDM2, KAP deletion mutants were tested in MDM2-null MEFs. The results showed that KAP1 239–835 mutant without the N-terminal RING domain showed reduced cooperation with MDM2 in p53 ubiquitination (Figure 4B). Interestingly, the N-terminal fragment 20–419 (RBCC) showed strong stimulation of p53 ubiquitination independent of MDM2 (Figure 4B). Since full-length KAP1 did not exhibit such activity, we introduced point mutations into the RBCC fragment to target conserved cysteine residues that may be important for ubiquitin E3 ligase function (C68S, C88S, C156S, C209S) or to cause disruption of the coiled-coil region (L306P). Surprisingly, these single point mutations failed to block the ability of RBCC to stimulate p53 ubiquitination level in MDM2-null cells (data not shown). It is noteworthy that another RBCC protein Efp has recently been shown to have RING domain-dependent ubiquitin E3 activity against 14-3-3 sigma (Urano et al, 2002). The effect of KAP1 RBCC on p53 ubiquitination suggested a cryptic activity that may regulate p53 or other KAP1-binding proteins under certain conditions.
Next, we examined the effect of KAP1 on p53 degradation by MDM2. H1299 cells were cotransfected with p53 and MDM2, and p53 expression level was determined by Western blot. In this assay, KAP1 expression stimulated p53 degradation when MDM2 dose was limiting. Moderate activities were also observed for KAP239–835 and KAP20–419 mutants that can cooperate with MDM2 in the ubiquitination assay, but not for the KAP423–581 mutant (Figure 4C). These results showed that KAP1 cooperates with MDM2 to stimulate p53 ubiquitination and degradation, which contributes to p53 inactivation.
KAP1 cooperates with MDM2 to inhibit p53 functions
To investigate whether KAP1–MDM2 cooperate to inhibit p53, a luciferase reporter gene assay was used. Transfection of the p53-responsive BP100-luciferase reporter (containing the p53-binding site from the MDM2 promoter) into U2OS cells resulted in activation of luciferase expression by the endogenous p53. Coexpression of MDM2 inhibited p53 transcriptional activity, and expression of KAP1 further inhibited p53 in cooperation with a suboptimal dose of MDM2 (Figure 5A). Treatment with the histone deacetylase inhibitor TSA alleviated the cooperation between MDM2 and KAP1, suggesting that HDAC activity was required for the effect of KAP1. In a different assay, a GAL4-p53N fusion containing only the N-terminal 1–82-residue transactivation domain of p53 was used to activate the luciferase reporter G5BTK-luc containing five GAL4-binding sites and the thymidine kinase minimal promoter. KAP1 also cooperated with MDM2 to inactivate the p53 transactivation domain (Figure 5B). Since the GAL4-p53N fusion does not contain the lysine residues required for MDM2-mediated ubiquitination, this result suggested that KAP1 recruited by MDM2 may also directly repress transcription, possibly through modification of promoter chromatin structure.
Figure 5.

Regulation of p53 function by KAP1. (A) U2OS cells were transiently transfected with p53-responsive BP100-luc reporter. Luciferase expression was determined after 24 h and normalized to cotransfected CMV-lacZ expression level. TSA (500 nM) was added to indicated samples 8 h before analysis. (B) GAL4-p53N (1–82) fusion protein was used to activate the G5BTK-luc GAL4 reporter in H1299 cells and luciferase activity was determined 24 h after transfection. Coexpression of MDM2 and KAP1 cooperatively inhibits the p53 transactivation domain. (C) H1299 cells were transiently transfected with p53 and CD20 expression plasmids, and apoptosis (sub-2N population) in CD20-positive cells was quantified by FACS analysis after 48 h. Examples of FACS histograms are shown.
Next, the effect of KAP1 on p53 apoptosis induction was examined. p53-deficient H1299 cells were transiently transfected with p53 and CD20 marker and the level of cell death was quantified by measuring the population of CD20-positive sub-2N apoptotic cells in FACS. Expression of p53 induced efficient apoptosis in H1299 cells. Coexpression of MDM2 plasmid at 2:1 ratio only caused a small reduction in apoptosis. When KAP1 was cotransfected with this suboptimal amount of MDM2, significant inhibition of apoptosis was observed (Figure 5C). Therefore, KAP1 has the potential to regulate p53 transcription and apoptosis functions in cooperation with MDM2.
ARF inhibits KAP1–MDM2 interaction
The tumor suppressor ARF binds to the acidic region of MDM2 and inhibits p53 ubiquitination (Zhang and Xiong, 2001). ARF has also been shown to abrogate MDM2 inhibition of p53 acetylation (Ito et al, 2002). Since ARF- and KAP1-binding sites on MDM2 partially overlap, we tested whether ARF and KAP1 binding to MDM2 is mutually exclusive. H1299 cells were transfected with MDM2, KAP1, and ARF expression plasmids and MDM2–KAP1 binding was determined by IP–Western blot. Cotransfection of ARF strongly inhibited MDM2–KAP1 co-precipitation (Figure 6A), suggesting that ARF competes with KAP1 for binding to MDM2. Furthermore, overexpression of KAP1 also inhibited MDM2–ARF binding in a reciprocal experiment (Figure 6B). KAP1 deletion mutant retaining MDM2 binding (20–419) also blocked ARF–MDM2 binding, whereas MDM2-binding-deficient mutant (423–584) had no effect. ARF expression is known to target MDM2 into the nucleolus. As expected, KAP1 overexpression effectively blocked nucleolar sequestration of MDM2 by ARF in U2OS cells (Figure 6E). Therefore, ARF and KAP1 competitively interact with MDM2 and have opposing effects on MDM2 localization and p53 activity.
Figure 6.
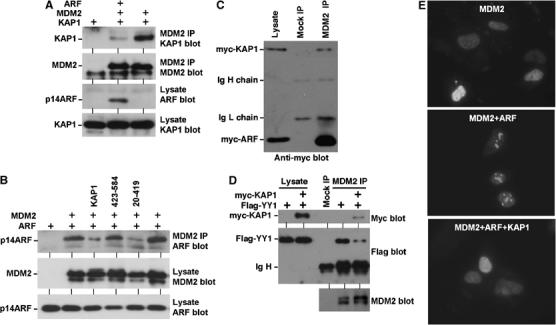
Competitive binding of ARF and KAP1 to MDM2. (A) ARF blocks MDM2–KAP1 binding. H1299 cells were transiently transfected with indicated plasmids and MDM2 was immunoprecipitated using 2A9. The amount of co-precipitated KAP1 was determined by Western blot. (B) KAP1 overexpression blocks ARF–MDM2 binding. H1299 cells were transiently transfected with indicated plasmids and MDM2 was immunoprecipitated using 2A9. Co-precipitation of p14ARF was determined by Western blot. (C) Relative binding affinity of MDM2 to ARF and KAP1. H1299 cells were transfected with MDM2, myc-KAP1, and myc-ARF. MDM2 2A9 immunoprecipitate was blotted with anti-myc antibody to detect the relative amounts of co-precipitated myc-ARF and myc-KAP1. Comparison to the ratio of myc-ARF/myc-KAP1 in the lysate suggested stronger binding to ARF than KAP1. (D) KAP1 overexpression blocks YY1–MDM2 binding. H1299 cells were transfected with indicated plasmids and MDM2 was immunoprecipitated using 2A9. Co-precipitation of YY1 was determined by Western blot. (E) KAP1 blocks MDM2 nucleolar targeting by ARF. U2OS cells were transfected with indicated plasmids for 24 h and MDM2 localization was determined by immunofluorescence staining using 2A9.
To determine the relative binding affinity of MDM2 to ARF and KAP1, H1299 cells were transfected with MDM2, myc-KAP1, and myc-ARF. Western blot against the common myc epitope tag allowed us to compare the expression levels of KAP1 and ARF. When myc-KAP1 and myc-ARF were expressed at similar levels, MDM2 IP co-precipitated significantly more ARF than KAP1 (Figure 6C). Therefore, ARF binds to MDM2 with higher affinity than KAP1 in vivo, suggesting that fluctuations in the ARF level during stress response should be able to modulate MDM2–KAP1 complex formation.
Recently, the transcription factor YY1 has been shown to bind to MDM2 in a competitive manner with ARF and regulates p53 ubiquitination by MDM2 (Sui et al, 2004). The similarity to KAP1–MDM2 binding led to the question whether YY1 and KAP1 also compete for interaction with MDM2. We found that MDM2 co-precipitated with YY1 in a cotransfection assay as reported, and expression of KAP1 reduced YY1–MDM2 binding (Figure 6D). Therefore, KAP1 and YY1 interact with MDM2 in a mutually exclusive manner and may compensate for each other. The effect of YY1 on p53 ubiquitination is believed to be due to enhanced MDM2–p53 binding (Sui et al, 2004). We examined the effect of KAP1 expression on p53–MDM2 binding and found that KAP1 did not alter their binding efficiency in IP–Western blot (data not shown).
Next, the effect of oncogene-induced endogenous ARF expression on MDM2–KAP1 binding was examined. To mimic mitogenic stress, H1299 cells stably expressing an E2F1-ER fusion construct were treated with 4-hydroxytamoxifen, which led to activation of the E2F1 fusion protein (Ma et al, 2003). Activation of E2F1-ER resulted in the induction of ARF expression (Figure 7A), which is a direct transcription target for E2F1 (Aslanian et al, 2004). MDM2 levels in the activated cells were also increased, possibly due to stabilization by ARF. IP–Western blot showed that after E2F1 activation, the amount of KAP1 co-precipitated with MDM2 was decreased, indicating a reduced binding efficiency (Figure 7B). This result showed that MDM2–KAP1 interaction can be modulated by ARF induction during mitogenic stress response.
Figure 7.
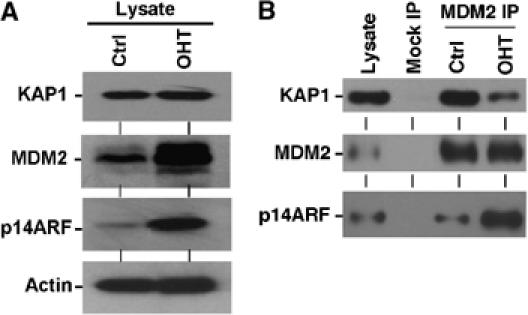
Regulation of MDM2–KAP1 interaction by oncogene activation. (A) H1299 cells expressing E2F1-ER were treated with 100 nM 4-hydroxytamoxifen (OHT) for 18 h to activate the E2F1-ER fusion protein. Expression levels of p14ARF, MDM2, and KAP1 were analyzed by Western blot. (B) MDM2 in control and OHT-treated H1299-E2F1-ER cells were immunoprecipitated with 3G9 antibody and the co-precipitated KAP1 was determined by Western blot. Sample loading was adjusted to obtain similar amounts of MDM2. Induction of p14ARF expression by E2F1 activation was associated with reduced KAP1 binding.
Regulation of p53 function by endogenous KAP1
To investigate the function of endogenous KAP1 in regulating p53, HT1080 (wild-type (wt) p53) and H1299 (p53-null) cells were infected with lentiviral vectors expressing control siRNA (GT) or KAP1 siRNA (K2). Stable expression of KAP1 siRNA resulted in ∼95% reduction of endogenous KAP1 level in HT1080 and ∼80% reduction in H1299 (Figure 8A). In HT1080 cells, knockdown of endogenous KAP1 was associated with increased expression of p53 target genes, including p21WAF1, Bax, and MDM2. No changes were observed in p53-null H1299 cells, suggesting that the induction in HT1080 was p53 mediated. Contrary to the expectation that knockdown of KAP1 would lead to increased p53 accumulation based on its ability to stimulate p53 ubiquitination, increased MDM2 level coupled with relatively unchanged p53 level was observed (Figure 8A). This suggests that increased MDM2 expression in the KAP1 knockdown cells compensated for the reduced ability to degrade p53, maintaining p53 level and activity within a survivable range.
Figure 8.
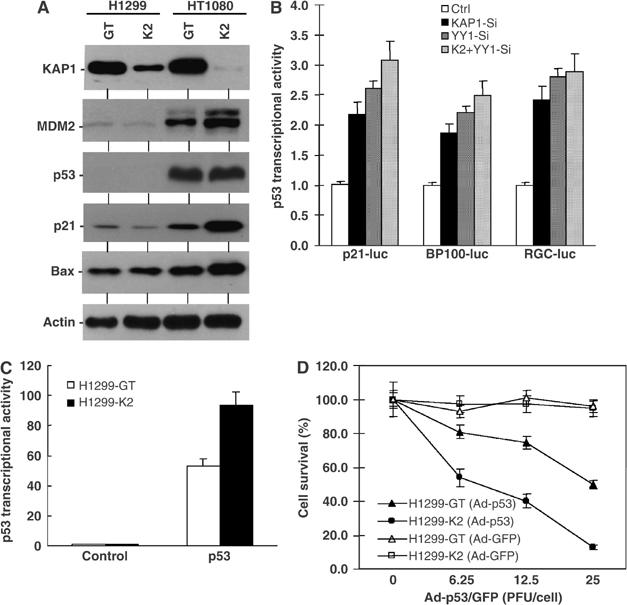
Endogenous KAP1 contributes to p53 regulation. (A) HT1080 and H1299 cells stably infected with lentiviral vectors expressing control (GT) or KAP1 (K2) siRNA were analyzed by Western blot for KAP1 levels and markers of the p53 pathway. (B) HT1080 cells were transiently transfected with p53-responsive luciferase reporters and siRNA expression plasmids against KAP1 or YY1. Luciferase expression was determined 24 h later and normalized to cotransfected CMV-lacZ expression level. (C) H1299-GT and H1299-K2 cells were transiently transfected with p53-responsive BP100-luciferase reporter. Luciferase expression was determined 24 h later and normalized to cotransfected CMV-lacZ expression level. (D) H1299-GT and H1299-K2 cells were infected with p53 adenovirus or GFP adenovirus at the indicated titers for 60 h and cell survival was determined by MTT assay.
To further confirm that knockdown of KAP1 increased p53 transcriptional activity, p53-responsive reporters (BP100-luc, p21-luc, RGC-luc) were transiently transfected into HT1080 cells and luciferase expression was compared after normalizing to cotransfected CMV-lacZ. Cotransfection with KAP1 siRNA plasmid showed more than two-fold stimulation of p53 reporter readout (Figure 8B). Consistent with recently reported role of YY1 in regulating p53 function (Gronroos et al, 2004; Sui et al, 2004), cotransfection of YY1 siRNA plasmid also resulted in p53 activation of similar magnitude (Figure 8B). Cotransfection with both KAP1 and YY1 siRNA plasmids did not have significant cooperative or additive effect, possibly because of inefficient double knockdown.
Knockdown of KAP1 in the p53-null H1299 cells also increased cellular response to ectopic p53 expression. When the H1299-K2 cells were transfected with p53 and the BP100-luc reporter, an approximately two-fold increase in p53 transcriptional activity was observed (Figure 8C). H1299 cells undergo apoptosis when infected with recombinant adenovirus expressing wt p53 (Lu et al, 2002). As expected, H1299-K2 cells showed increased cell death after infection with p53 adenovirus compared to control H1299-GT cells (Figure 8D). Therefore, endogenous KAP1 expression protects cells from the apoptotic effects of p53.
To examine the role of KAP1 in regulating p53 response to DNA damage, HT1080-GT and HT1080-K2 cells were treated with ionizing radiation. The levels of MDM2, p21, and Bax expression were also significantly higher in HT1080-K2 cells compared to control (Figure 9A). Consistent with the ability of KAP1 to regulate p53 acetylation in transfection assays, KAP1 knockdown also resulted in a higher level of p53 acetylation after irradiation (Figure 9B). HT1080 cells undergo apoptosis after ionizing radiation, which was associated with cleavage of the 117 kDa caspase 3 substrate PARP to a 87 kDa fragment (Figure 9D). Knockdown of KAP1 also significantly sensitized the cells to apoptosis after radiation, consistent with increased p53 activation and expression of Bax (Figure 9C). Collectively, these results demonstrated that KAP1 plays a role in regulating p53 activity and response to DNA damage.
Figure 9.
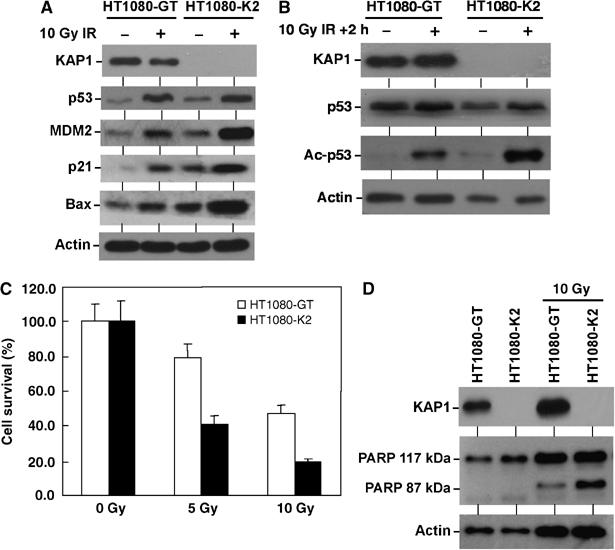
Endogenous KAP1 regulates p53 response to DNA damage. (A) HT1080-GT and HT1080-K2 cells were treated with 10 Gy gamma radiation for 6 h and analyzed by Western blot for the indicated protein markers. (B) HT1080-GT and HT1080-K2 cells were treated with 10 Gy gamma radiation for 2 h before significant p53 accumulation occurs and p53 acetylation was analyzed by IP followed by Western blot. (C) HT1080 cells were treated with gamma radiation and cell survival were analyzed by MTT assay after 60 h. Knockdown of KAP1 increased p53 target gene induction after radiation and correlated with increased apoptosis. (D) HT1080 cells were treated with 10 Gy gamma radiation and cleavage of the apoptotic substrate PARP as indicated by the generation of the 87 kDa fragment was analyzed by Western blot after 48 h.
Discussion
MDM2 is a critical regulator of p53 that functions through several mechanisms. Ubiquitination of p53 by MDM2 is a major mechanism by which p53 level is regulated in the cell. Additionally, MDM2 can inactivate p53 by concealing the p53 transactivation domain, inhibiting p53 acetylation by coactivators, and promoting p53 deacetylation by recruiting histone deacetylases (Ito et al, 2001, 2002; Kobet et al, 2002). Although some aspects of these functions can be carried out by MDM2 alone, cooperation with other MDM2-interacting proteins is expected to play important roles. Results described in this report identified the nuclear corepressor KAP1 as a novel MDM2-interacting protein. KAP1 cooperates with MDM2 to inhibit p53 acetylation, stimulate p53 ubiquitination, and inhibit p53 transcription and apoptosis functions.
Our results showed that KAP1 alone does not interact with p53, but it can be targeted to p53 by interacting with MDM2. KAP1 and MDM2 coexpression stimulates p53–HDAC1 complex formation and reduces the level of p53 acetylation. This suggested that one mechanism of KAP1 function is to promote p53 deacetylation by recruitment of deacetylases through its C-terminal PHD and bromodomains. This effect is also similar to a recent finding that MDM2 recruits HDAC1 and promotes p53 deacetylation (Ito et al, 2002). It is possible that KAP1 is one of the mediators of MDM2–HDAC1 interaction. Because acetylation and ubiquitination of p53 use common lysine residues and are mutually exclusive events, the ability of MDM2 to recruit KAP1 would cooperatively deacetylate and then ubiquitinate p53. KAP1 can directly repress transcription if targeted to the promoter (Friedman et al, 1996). Presumably, KAP1 may also be targeted to p53-responsive promoters by forming ternary complex with p53 and MDM2, directly inhibiting transcription of p53 target genes.
The KAP1 coiled-coil domain is involved in binding to the central region of MDM2. The tumor suppressor ARF binds to MDM2 central acidic domain and causes p53 stabilization and activation. ARF inhibits the ability of MDM2 to promote p53 ubiquitination, and overcomes MDM2 inhibition of p53 acetylation (Honda and Yasuda, 1999; Ito et al, 2001; Midgley et al, 2001). The ARF–MDM2 complex is still competent in ubiquitination of MDMX (Pan and Chen, 2003), suggesting that ARF does not inactivate the MDM2 RING domain. ARF may target other MDM2 functions encoded by the acidic domain that is necessary for efficient p53 ubiquitination and degradation. Our results show that ARF and KAP1 interact with MDM2 in a mutually exclusion fashion. Therefore, ARF may induce p53 stabilization and activation in part by blocking KAP1–MDM2 interaction. Our results also showed that induction of ARF by activated E2F1 was associated with reduced KAP1–MDM2 binding, consistent with this mechanism playing a role in signaling to p53 during mitogenic stress response.
RING domain-containing proteins often have ubiquitin E3 ligase function (Lorick et al, 1999). The RBCC motif protein Efp has been shown to promote 14-3-3 sigma ubiquitination and degradation by functioning as a RING-dependent E3 ligase (Urano et al, 2002). The RING domain of KAP1 is important for interaction with KRAB and oligomerization of KAP1 (Peng et al, 2000), but has not been reported to have E3 function. We show here that KAP1 overexpression cooperates with MDM2 to promote p53 ubiquitination in vivo. RNAi knockdown of endogenous KAP1 leads to p53 activation and increased MDM2 expression. However, a higher level of MDM2 was not associated with degradation of p53, suggesting a reduced efficiency in p53 degradation. One interpretation of our results is that knockdown of KAP1 leads to partial activation of p53 and a shift in the MDM2 negative feedback loop. The increased MDM2 expression compensates for its reduced p53 degradation function, preventing significant accumulation of p53, which would be detrimental for cell survival. Therefore, stable cell lines with knockdown of KAP1 were viable but more sensitive to stress that disrupts the MDM2 feedback loop.
Recent studies showed that YY1 also plays a role in promoting MDM2 ubiquitination of p53 (Gronroos et al, 2004; Sui et al, 2004). Therefore, the MDM2 acidic domain interacts with multiple corepressors, both being displaced by binding of ARF. KAP1 and YY1 also compete for binding to MDM2 and may function as backup for each other. The contributions of different corepressors in the regulation of MDM2 and p53 remain to be further elucidated. However, current data suggested key differences in the effects of KAP1 and YY1. YY1 stimulates p53 ubiquitination by enhancing MDM2–p53 binding, whereas KAP1 does not affect MDM2–p53 binding affinity. Furthermore, knockdown of YY1 leads to significant accumulation of p53 and was incompatible with long-term cell survival. In contrast, cells with knockdown of KAP1 are still viable with moderately increased p53 activity. It is possible that the effect of KAP1 knockdown is compensated by other MDM2 cofactors, including YY1. Alternatively, because KAP1 is an abundant protein, the level of knockdown achievable by RNAi may not be sufficient to uncover the extent of its involvement in p53 regulation. Development of inducible genetic knockout system may reveal additional functions of KAP1–MDM2 interaction.
The ability of KAP1 RBCC fragment to promote p53 ubiquitination in vivo suggests that this region may encode a cryptic ubiquitin E3 ligase activity that contributes to p53 ubiquitination when complexed with MDM2. Recent experiments also suggested that KAP1 RBCC can stimulate ubiquitination of MDM2 RING deletion mutants that normally cannot undergo self-ubiquitination (unpublished observations). Therefore, the biochemical activity of this fragment and its role in the context of full-length KAP1 protein remain to be further investigated. Several general transcription coactivators (p300, CBP, TAF250) have been shown to have ubiquitin E3 ligase function (Pham and Sauer, 2000; Grossman et al, 2003). It will be interesting to determine whether KAP1 regulates other factors in part by promoting their ubiquitination or degradation. It will also be important to investigate the possibility of KAP1 playing a role in cancer due to its inhibitory effect on p53.
Materials and methods
Cell lines, plasmids and reagents
H1299 (p53-null), U2OS (wt p53), SJSA (wt p53, amplified MDM2), and 293T were maintained in DMEM medium with 10% fetal bovine serum. MDM2/p53 double-null MEF 174.1 was a kind gift from Dr Guillermina Lozano (McMasters et al, 1996). H1299 cell line expressing hormone-regulated E2F1-ER™ fusion protein was kindly provided by Dr Douglas Cress (Ma et al, 2003). KAP1 constructs were described previously (Friedman et al, 1996; Ryan et al, 1999). MDM2 monoclonal antibodies (2A9, 4B2, 4B11, 3G9), and MDM2 deletion and point mutation constructs were described in previous studies (Chen et al, 1993, 1995; Chen and Chen, 2003; Pan and Chen, 2003). Additional FLAG-KAP1 and MDM2 deletion mutants were constructed by PCR amplification and cloning into pcDNA3 (KAP1) and pCMV-neo-Bam (MDM2) vectors, respectively. YY1 siRNA expression plasmid was kindly provided by Dr Yang Shi (Sui et al, 2004). All p53, MDM2, p14ARF, and KAP1 constructs used in this study were of human origin. Inhibition of KAP1 expression was achieved by hairpin RNA expression using lentiviral vector pLSL-GFP (gift from Dr Peter Chumakov). The following oligos (only 23-mer sense strand is shown) were used: K2 5′-ccagccaaccagcggaaatgtga-3′ (specific against KAP1); GT 5′-tcctctttcttatcctcgtatgt-3′ (control, against alternatively spliced isoform of SETDB1). Purified adenovirus expressing p53 was prepared as described previously (Lu et al, 2002).
Western blot
Cells were lysed in lysis buffer (50 mM Tris–HCl (pH 8.0), 5 mM EDTA, 150 mM NaCl, 0.5% NP-40, 1 mM PMSF), centrifuged for 5 min at 10 000 g, and the insoluble debris was discarded. Cell lysate (10–50 μg protein) was fractionated by SDS–PAGE and transferred to Immobilon P filters (Millipore). The filter was blocked for 1 h with phosphate-buffered saline (PBS) containing 5% nonfat dry milk and 0.1% Tween 20. The following antibodies were used: 3G9 or polyclonal rabbit serum for MDM2 (Chen et al, 1993), DO-1 (Pharmingen) or FL393 (Santa Cruz Biotechnology) for p53, rabbit anti-KAP1 antibody (BETHYL), 9B11 for myc tag (Cell Signaling), and rabbit anti-acetyl-p53 antibody (lysines 373 and 382; Upstate). The filter was developed using HRP-conjugated secondary antibodies and ECL-plus reagent (Amersham).
Purification of MDM2-associated protein
Human MDM2 cDNA expression plasmid was transiently transfected into 293T cells. At 2 days after transfection, cells (∼2 × 108) were treated with 30 μM MG132 for 4 h, lysed in a total of 10 ml lysis buffer, centrifuged for 5 min at 10 000 g, and the insoluble debris was discarded. The lysate was precleared with protein A-Sepharose beads for 30 min, and then incubated with 40 μl bed volume of protein A-Sepharose beads and 0.5 ml of 2A9 hybridoma supernatant for 4 h at 4°C. The beads were washed with SNNTE buffer (5% sucrose, 5 mM Tris–HCl (pH 7.5), 5 mM EDTA, 500 mM NaCl, 1% NP-40) and boiled in SDS sample buffer. The eluted proteins were fractionated on SDS–PAGE and stained with Coomassie blue. Proteins copurified with MDM2 were excised from the gel and subjected to protease digestion and peptide sequencing by mass spectrometry at the Harvard Microchemistry Laboratory.
Immunoprecipitation–Western blot assay
Cells were lysed in lysis buffer, centrifuged for 5 min at 10 000 g, and the insoluble debris was discarded. Cell lysate (200–500 μg protein) was immunoprecipitated using 100 μl of 4B2 or 4B11 hybridoma supernatant against MDM2 and protein A-agarose beads for 4 h at 4°C. The beads were washed extensively with lysis buffer, boiled in SDS sample buffer, fractionated by SDS–PAGE, and analyzed by anti-KAP1 or p53 Western blot using rabbit polyclonal antibodies to reduce background from Ig heavy chain.
In vivo ubiquitination assay
H1299 cells in 10 cm plates were transfected with combinations of 5 μg His6-ubiquitin expression plasmid, 1–5 μg human MDM2, 5 μg p53, and 5 μg KAP1 expression plasmids. At 32 h after transfection, cells were lysed in buffer A (6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris–HCl pH 8.0, 5 mM imidazole, 10 mM β-mercaptoethanol) and incubated with Ni2+-NTA beads (Qiagen) for 4 h at room temperature. The beads were washed with buffer A, B (8 M urea, 0.1 M Na2PO4/NaH2PO4, 0.01 M Tris–HCl pH 8.0, 10 mM β-mercaptoethanol), C (8 M urea, 0.1 M Na2PO4/NaH2PO4, 0.01 M Tris–HCl pH 6.3, 10 mM β-mercaptoethanol), and bound proteins were eluted with buffer D (200 mM imidazole, 0.15 M Tris–HCl pH 6.7, 30% glycerol, 0.72 M β-mercaptoethanol, 5% SDS). The eluted proteins were analyzed by Western blot for the presence of conjugated p53 by DO1 antibody.
Luciferase reporter assay
Cells (50 000/well) were plated in 24-well plates and transfected with a mixture containing 10 ng p53-responsive BP100-luciferase reporter plasmid (Freedman et al, 1997), 5 ng CMV-lacZ plasmid, 0.1–1.0 ng p53 expression plasmid, and 20 ng KAP1 plasmid. Transfection was achieved using Lipofectamine PLUS reagents (Invitrogen) and cells were analyzed for luciferase and beta galactosidase expression after 24 h. The ratios of luciferase/beta galactosidase activity were used as indicators of p53 transcriptional activity.
Acknowledgments
We thank Dr William Lane for help on protein identification by mass spectrometry, Dr Yang Shi for YY1 siRNA vector, Dr Douglas Cress for the E2F1 inducible cell line, the Moffitt Molecular Biology Core for DNA sequence analysis, and the Moffitt Flow Cytometry Core for FACS analysis. This work was supported by grants from the American Cancer Society and National Institutes of Health to J Chen.
References
- Aslanian A, Iaquinta PJ, Verona R, Lees JA (2004) Repression of the Arf tumor suppressor by E2F3 is required for normal cell cycle kinetics. Genes Dev 18: 1413–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner C, Hay T, Meek DW, Lane DP (2002) Hypophosphorylation of Mdm2 augments p53 stability. Mol Cell Biol 22: 6170–6182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammas F, Mark M, Dolle P, Dierich A, Chambon P, Losson R (2000) Mice lacking the transcriptional corepressor TIF1beta are defective in early postimplantation development. Development 127: 2955–2963 [DOI] [PubMed] [Google Scholar]
- Chen J, Lin J, Levine AJ (1995) Regulation of transcription functions of the p53 tumor suppressor by the mdm-2 oncogene. Mol Med 1: 142–152 [PMC free article] [PubMed] [Google Scholar]
- Chen J, Marechal V, Levine AJ (1993) Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol 13: 4107–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen J (2003) MDM2–ARF complex regulates p53 sumoylation. Oncogene 22: 5348–5357 [DOI] [PubMed] [Google Scholar]
- De Graaf P, Little NA, Ramos YF, Meulmeester E, Letteboer SJ, Jochemsen AG (2003) Hdmx protein stability is regulated by the ubiquitin ligase activity of mdm2. J Biol Chem 278: 38315–38324 [DOI] [PubMed] [Google Scholar]
- Freedman DA, Epstein CB, Roth JC, Levine AJ (1997) A genetic approach to mapping the p53 binding site in the MDM2 protein. Mol Med 3: 248–259 [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ (1996) KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev 10: 2067–2078 [DOI] [PubMed] [Google Scholar]
- Gronroos E, Terentiev AA, Punga T, Ericsson J (2004) YY1 inhibits the activation of the p53 tumor suppressor in response to genotoxic stress. Proc Natl Acad Sci USA 101: 12165–12170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Deato ME, Brignone C, Chan HM, Kung AL, Tagami H, Nakatani Y, Livingston DM (2003) Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 300: 342–344 [DOI] [PubMed] [Google Scholar]
- Gu W, Roeder RG (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90: 595–606 [DOI] [PubMed] [Google Scholar]
- Honda R, Yasuda H (1999) Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J 18: 22–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP (2002) MDM2–HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J 21: 6236–6245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, Yao TP (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J 20: 1331–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Shiels C, Freemont PS (2001) PML protein isoforms and the RBCC/TRIM motif. Oncogene 20: 7223–7233 [DOI] [PubMed] [Google Scholar]
- Joazeiro CA, Weissman AM (2002) RING finger proteins: mediators of ubiquitin ligase activity. Cell 102: 549–552 [DOI] [PubMed] [Google Scholar]
- Juan LJ, Shia WJ, Chen MH, Yang WM, Seto E, Lin YS, Wu CW (2000) Histone deacetylases specifically down-regulate p53-dependent gene activation. J Biol Chem 275: 20436–20443 [DOI] [PubMed] [Google Scholar]
- Kawai H, Wiederschain D, Kitao H, Stuart J, Tsai KK, Yuan ZM (2003a) DNA damage-induced MDMX degradation is mediated by MDM2. J Biol Chem 278: 45946–45953 [DOI] [PubMed] [Google Scholar]
- Kawai H, Wiederschain D, Yuan ZM (2003b) Critical contribution of the MDM2 acidic domain to p53 ubiquitination. Mol Cell Biol 23: 4939–4947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobet E, Zeng X, Zhu Y, Keller D, Lu H (2002) MDM2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proc Natl Acad Sci USA 97: 12547–12552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Luo J, Brooks CL, Gu W (2002) Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem 277: 50607–50611 [DOI] [PubMed] [Google Scholar]
- Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH (2003) Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3: 577–587 [DOI] [PubMed] [Google Scholar]
- Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM (1999) RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci USA 96: 11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Lin J, Chen J (2002) Expression of p14ARF overcomes tumor resistance to p53. Cancer Res 62: 1305–1310 [PubMed] [Google Scholar]
- Luo J, Su F, Chen D, Shiloh A, Gu W (2000) Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408: 377–381 [DOI] [PubMed] [Google Scholar]
- Ma Y, Yuan J, Huang M, Jove R, Cress WD (2003) Regulation of the cyclin D3 promoter by E2F1. J Biol Chem 278: 16770–16776 [DOI] [PubMed] [Google Scholar]
- Marechal V, Elenbaas B, Piette J, Nicolas J, Levine AJ (1994) The ribosomal L5 protein is associated with mdm-2 and mdm-2–p53 complexes. Mol Cell Biol 14: 7414–7420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMasters KM, Montes de Oca Luna R, Pena JR, Lozano G (1996) mdm2 deletion does not alter growth characteristics of p53-deficient embryo fibroblasts. Oncogene 13: 1731–1736 [PubMed] [Google Scholar]
- Meulmeester E, Frenk R, Stad R, De Graaf P, Marine JC, Vousden KH, Jochemsen AG (2003) Critical role for a central part of Mdm2 in the ubiquitylation of p53. Mol Cell Biol 23: 4929–4938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley CA, Desterro JM, Saville MK, Howard S, Sparks A, Hay RT, Lane DP (2001) An N-terminal p14ARF peptide blocks Mdm2-dependent ubiquitination in vitro and can activate p53 in vivo. Oncogene 19: 2312–2323 [DOI] [PubMed] [Google Scholar]
- Pan Y, Chen J (2003) MDM2 promotes ubiquitination and degradation of MDMX. Mol Cell Biol 23: 5113–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Begg GE, Schultz DC, Friedman JR, Jensen DE, Speicher DW, Rauscher FJ (2000) Reconstitution of the KRAB–KAP-1 repressor complex: a model system for defining the molecular anatomy of RING-B box-coiled-coil domain-mediated protein–protein interactions. J Mol Biol 295: 1139–1162 [DOI] [PubMed] [Google Scholar]
- Pham AD, Sauer F (2000) Ubiquitin-activating/conjugating activity of TAFII250, a mediator of activation of gene expression in Drosophila. Science 289: 2357–2360 [DOI] [PubMed] [Google Scholar]
- Prives C, Hall PA (1999) The p53 pathway. J Pathol 187: 112–126 [DOI] [PubMed] [Google Scholar]
- Rodriguez MS, Desterro JM, Lain S, Lane DP, Hay RT (2002) Multiple C-terminal lysine residues target p53 for ubiquitin–proteasome-mediated degradation. Mol Cell Biol 20: 8458–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan RF, Schultz DC, Ayyanathan K, Singh PB, Friedman JR, Fredericks WJ, Rauscher FJ (1999) KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: a potential role for Kruppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol Cell Biol 19: 4366–4378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ (2002) SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 16: 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DC, Friedman JR, Rauscher FJ (2001) Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev 15: 428–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shvarts A, Steegenga WT, Riteco N, van Larr T, Dekker P, Bazuine M, van Ham RCA, van Oordt WVDH, Hateboer G, van der Eb AJ, Jochemsen AG (1996) MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J 15: 5349–5357 [PMC free article] [PubMed] [Google Scholar]
- Sui G, Affar el B, Shi Y, Brignone C, Wall NR, Yin P, Donohoe M, Luke MP, Calvo D, Grossman SR, Shi Y (2004) Yin Yang 1 is a negative regulator of p53. Cell 117: 859–872 [DOI] [PubMed] [Google Scholar]
- Urano T, Saito T, Tsukui T, Fujita M, Hosoi T, Muramatsu M, Ouchi Y, Inoue S (2002) Efp targets 14-3-3 sigma for proteolysis and promotes breast tumour growth. Nature 417: 871–875 [DOI] [PubMed] [Google Scholar]
- Vousden KH (2000) p53: death star. Cell 103: 691–694 [DOI] [PubMed] [Google Scholar]
- Wang X, Taplick J, Geva N, Oren M (2004) Inhibition of p53 degradation by Mdm2 acetylation. FEBS Lett 561: 195–201 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y (2003) Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol 23: 8902–8912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y (2001) Control of p53 ubiquitination and nuclear export by MDM2 and ARF. Cell Growth Differ 12: 175–186 [PubMed] [Google Scholar]