

Abstract
Signaling from the activin/transforming growth factor β (TGFβ) family of cytokines is a tightly regulated process. Disregulation of TGFβ signaling is often the underlying basis for various cancers, tumor metastasis, inflammatory and autoimmune diseases. In this study, we identify the protein G-coupled receptor kinase 2 (GRK2), a kinase involved in the desensitization of G protein-coupled receptors (GPCR), as a downstream target and regulator of the TGFβ-signaling cascade. TGFβ-induced expression of GRK2 acts in a negative feedback loop to control TGFβ biological responses. Upon TGFβ stimulation, GRK2 associates with the receptor-regulated Smads (R-Smads) through their MH1 and MH2 domains and phosphorylates their linker region. GRK2 phosphorylation of the R-Smads inhibits their carboxyl-terminal, activating phosphorylation by the type I receptor kinase, thus preventing nuclear translocation of the Smad complex, leading to the inhibition of TGFβ-mediated target gene expression, cell growth inhibition and apoptosis. Furthermore, we demonstrate that GRK2 antagonizes TGFβ-induced target gene expression and apoptosis ex vivo in primary hepatocytes, establishing a new role for GRK2 in modulating single-transmembrane serine/threonine kinase receptor-mediated signal transduction.
Keywords: activin/TGFβ, cancer, GRK2, growth inhibition, Smad phosphorylation
Introduction
The transforming growth factor β (TGFβ) superfamily of growth factors represents a large group of pluripotent polypeptides, comprised of activins, TGFβs and bone morphogenetic proteins (BMPs) among others, that regulate cell growth, differentiation and apoptosis, in nearly all cell types (Shi and Massague, 2003). TGFβ, activin and their receptors are widely expressed in all tissues, and the regulatory role played by these growth factors is of central importance to human diseases. The involvement of these growth factors in human cancer is multifaceted. While they initially contribute to tumor suppression by efficiently inhibiting the proliferation of cells derived from epithelial, endothelial and hemaetopoietic origins, the TGFβ growth-inhibitory responses in cancer cells are often replaced by invasive and prometastatic responses as tumors progress, highlighting the dual role of TGFβ as both a tumor suppressor and a tumor-promoting agent (Derynck et al, 2001; Wakefield and Roberts, 2002).
The current paradigm of TGFβ/activin signal transduction begins with ligand binding to a single-transmembrane-spanning, constitutively auto-phosphorylated serine/threonine kinase, type II receptor. Following ligand binding, the type II receptor recruits and transphosphorylates the type I receptor within a juxtamembrane glycine and serine-rich region, thereby activating the kinase activity of the type I receptor. The activated type I receptor then phosphorylates intracellular mediators known as receptor-regulated Smads (R-Smads), Smad2 and 3, on their carboxy-terminal serine residues (SxS motif). This phosphorylation event results in the release of auto-inhibitory intramolecular interactions between the Mad-homology 1 (MH1) and Mad-homology 2 (MH2) domains of Smads 2 and 3, allowing for their subsequent heterodimerization with their common partner, Smad4. Once activated, the Smad complex translocates into the nucleus, where it associates with coactivators or corepressors of transcription to regulate the expression of various target genes (Shi and Massague, 2003).
Although mechanistically simple, the activin/TGFβ signaling cascade has numerous auto-regulatory mechanisms that exist to maintain a tightly regulated ligand-induced message. The inhibitory Smad7 functions through a negative feedback loop mechanism to terminate signaling by sterically preventing access of Smad2/3 to the kinase domain of the type I receptor (Hayashi et al, 1997; Nakao et al, 1997) and by recruiting protein phosphatases and ubiquitin ligases to the activated TGFβ receptor (Kavsak et al, 2000; Shi et al, 2004). Receptor internalization and/or downregulation are also now emerging as predominant means of regulating signaling from the TGFβ pathways (Di Guglielmo et al, 2003). These receptors can be constitutively internalized through clathrin-independent or -dependent mechanisms, which may involve the recruitment to the TGFβ receptor of endocytic adaptors like AP-2 and βarrestins (Yao et al, 2002; Chen et al, 2003). At the level of the R-Smads, nuclear translocated Smad2 and 3 can be subject to ubiquitin–proteasome-mediated degradation (Lo and Massague, 1999) or dephosphorylation (Xu et al, 2002), leading to termination of signaling. Crosstalk from other signaling pathways, which interferes with the nuclear translocation of the Smads, can further act to negatively regulate Smad-dependent signal transduction (Kretzschmar et al, 1997, 1999). In fact, nonreceptor kinases have been shown to utilize the R-Smads as substrates to regulate TGFβ signaling (Wicks et al, 2000; Matsuura et al, 2004; Waddell et al, 2004).
In the present study, we describe a novel inhibitory mechanism downstream of activin/TGFβ. We have identified GRK2 to act in a negative feedback loop downstream of the activin/TGFβ receptors. GRK2 is known to phosphorylate a large number of G protein-coupled receptors (GPCRs), leading to the uncoupling of the GPCRs from their heterotrimeric G proteins, subsequent receptor desensitization and downregulation to terminate signaling (Pitcher et al, 1998). Although GRK2 is critical to GPCR signaling and has been shown to be involved in receptor tyrosine kinase desensitization (Freedman et al, 2002), to date no implications of GRK2 acting downstream of single-transmembrane serine/threonine kinase growth factor receptors have been established. We show here that GRK2 expression levels are upregulated in response to activin/TGFβ signaling, and that GRK2 physically interacts with the MH1 and MH2 domains of the receptor-regulated Smads and phosphorylates their linker region on a specific single serine/threonine residue. GRK2-induced Smad phosphorylation blocks activin/TGFβ-induced Smad activation, nuclear translocation and target gene expression. The net effect of GRK2 on activin/TGFβ responses leads to an inhibition of their antiproliferative and proapoptotic functions. We further demonstrate that GRK2 antagonizes activin/TGFβ responses ex vivo and potently inhibits activin-mediated cell death in primary hepatocytes from liver perfused animals. Thus, GRK2 appears as a novel TGFβ antagonist that strongly inhibits activin/TGFβ-mediated cell growth arrest and apoptosis in both normal and cancer liver cells.
Results and discussion
Activin/TGFβ induces cell growth arrest and apoptosis in human hepatocarcinoma cells
Human hepatocellular carcinoma (HuH7) and hepatoblastoma (HepG2) cells, treated with activin or TGFβ for 72 h, led to a clear inhibition of cell growth (Figure 1A). Using flow cytometry (FACS) analysis, we found that activin/TGFβ regulates cell growth of these two hepatocarcinoma cell lines by inhibiting cell proliferation (G1 arrest) and inducing apoptosis (Figure 1B). The strong proapoptotic effect of these growth factors was confirmed by Annexin V/propidium iodide (PI) staining (Figure 1C). Collectively, these results indicate that the two human hepatoma cell lines, HepG2 and HuH7, respond in a highly similar manner to activin and TGFβ. These findings are consistent with previous studies demonstrating that activin and TGFβ play a major role in regulating liver function by modulating growth arrest and apoptosis in normal and cancer liver cells (Oberhammer et al, 1992; Yasuda et al, 1993; Ho et al, 2004).
Figure 1.
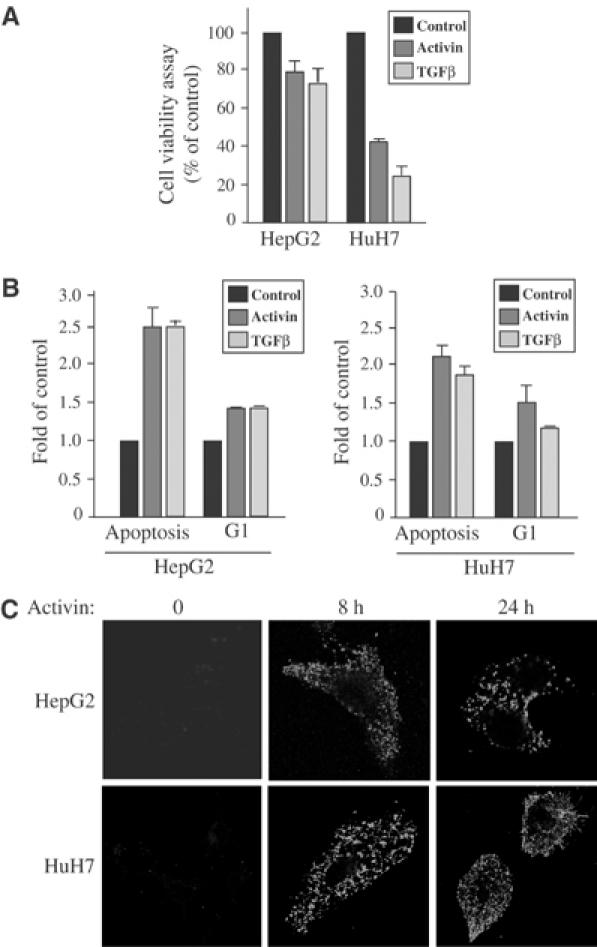
Activin/TGFβ's biological effect in human heptocarcinoma cells. (A) Normal HepG2 and HuH7 cells were stimulated or not with either activin or TGFβ for 72 h before cell growth was measured by cell viability colorimetric (MTT) assay. Values are representative of three independent experiments performed in triplicate, and are expressed in arbitrary units. P<0.05 compared with no activin/TGFβ treatment. (B) HepG2 and HuH7 cells were stimulated or not with either activin or TGFβ for 48 h. The distribution of cells in the cell cycle was quantified by analysis of PI-stained cells using flow cytometry, and the data are representative of three independent experiments. (C) HepG2 and HuH7 cells either treated or not with activin for 0, 8 and 24 h were labeled for Annexin V-FITC and PI to ensure that necrotic cells were not included in the analysis (data not shown) and analyzed by confocal microscopy.
Activin/TGFβ selectively induces GRK2 expression in human hepatocarcinoma cells
To identify novel activin/TGFβ target genes which may be responsible for mediating their growth-inhibitory effects, we performed Affymetrix human Gene Chip U95A microarray experiments using activin or TGFβ-treated human hepatocarcinoma (HuH7) cells. From our microarray experiments, we found the mRNA level of the GPCR kinase-2 (GRK2) to be significantly increased in HuH7 cells treated for 8 h with activin or TGFβ (3.5 and 3, respectively). Our initial microarray findings were verified by Northern blot analysis (Figure 2A).
Figure 2.
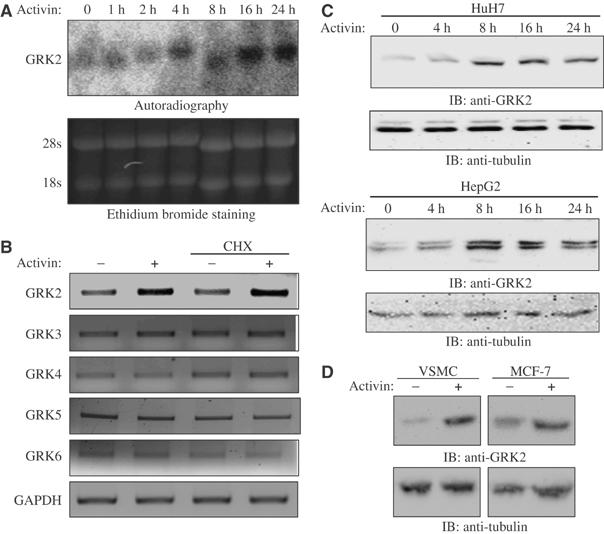
Activin/TGFβ induces upregulation of GRK2 in human hepatocarcinoma cells. (A) HuH7 cells were stimulated with activin for 0, 1, 2, 4, 8, 16 and 24 h, and total RNA was analyzed by Northern blot using specific probes for GRK2 (upper panel). Equal loading was assessed by ethidium bromide staining (lower panel). (B) Reverse transcription reactions were performed using oligo-dT and cDNAs were amplified for 30 cycles using specific oligonucleotides to GRKs 2–6 and GAPDH as a control. The samples were treated or not with the translation inhibitor cycloheximide and stimulated or not with activin for 16 h. (C) HuH7 and HepG2 cells were stimulated with activin for 0, 4, 8, 16 and 24 h and total cell lysates were analyzed by Western blot using a specific monoclonal antibody against GRK2 (upper panels) or tubulin (lower panels). (D) Vascular smooth muscle and MCF7 breast cancer cells were stimulated or not with activin for 24 h and total cell lysates were analyzed by Western blot using a specific monoclonal antibody against GRK2 (upper panels) or tubulin (lower panels).
The activin-induced increase in GRK2 mRNA levels was further confirmed by RT–PCR using primers specific for GRK2, and appeared to occur through a direct transcriptional regulatory mechanism, as it was not affected by treatment with the translational inhibitor cycloheximide (Figure 2B). To determine whether activin/TGFβ could also induce expression of other GRK family members, semiquantitative RT–PCR experiments were performed in HuH7 cells treated or not with activin, and, as shown in Figure 2B, only GRK2 levels were affected by activin treatment. Thus, this suggests that activin selectively regulates GRK2 mRNA levels in these cells.
Consistent with the increase in the mRNA levels of GRK2, we also observed an increase in GRK2 protein levels in response to activin in the two hepatocarcinoma cell lines, HuH7 and HepG2 (Figure 2C). This effect is not liver specific, as activin was also able to induce GRK2 protein expression levels in breast cancer cells (MCF7) and vascular smooth muscle cells (VSMC), two distinct activin-responsive cell lineages (Figure 2D). Thus, our findings identify activin/TGFβ to be key modulators of the expression levels of GRK2 in both normal and cancer cells.
GRK2 inhibits activin/TGFβ-mediated gene expression and cell growth arrest
To determine the role of an increase in GRK2 expression on activin/TGFβ-mediated signal transduction, we cotransfected HuH7 liver cells with an activin/TGFβ-responsive gene promoter construct (3TP-luc) fused to the luciferase gene. As shown in Figure 3A, in the presence of GRK2, a 75% decrease in 3TP-luc activity was observed upon activin stimulation, as compared to cell expressing 3TP-luc alone. Moreover, transfection of a GRK2 mutant in which the kinase domain was inactivated by point mutation (K220M) reversed this inhibitory effect, indicating that the kinase domain of GRK2 is required for inhibition of activin-induced target gene expression (Figure 3A).
Figure 3.

GRK2 inhibits activin-mediated Smad2 phosphorylation, gene transcription, cell growth arrest, apoptosis and target gene expression. (A) HuH7 cells were cotransfected with pcDNA3, GRK2-WT or GRK2-K220M and the 3TP-luc reporter construct, together with a β-galactosidase expression plasmid. Cells were treated with activin for 18 h and luciferase assays were performed. *P<0.05 compared with no activin treatment. (B) HuH7 cells were cotransfected with pcDNA3, GRK2-WT, GRK3-WT, GRK4-WT, GRK5-WT or GRK6-WT and the 3TP-luc reporter construct, together with a β-galactosidase expression plasmid. Cells were treated with activin or TGFβ for 18 h and luciferase assays were performed. *P<0.05 compared with no activin treatment. (C) Magnetically enriched HuH7 GFP-expressing cells and HuH7 GFP-GRK2-expressing cells were stimulated or not with activin for 72 h before cell growth was measured by cell viability MTT assay. Values are expressed in arbitrary units. P<0.05 compared with no activin treatment. (D) WT CHO cells, CHO cells stably overexpressing either GRK2 or GRK2-K220M were stimulated or not with TGFβ for 48 h. The distribution of cells in the cell cycle was quantified by analysis of PI-stained cells using flow cytometry, and the data are representative of three independent experiments. (E) WT CHO cells, CHO cells stably overexpressing either GRK2 or GRK2-K220M, either treated or not with activin for 8 h and labeled with Annexin V-FITC and PI (data not shown), and analyzed by confocal microscopy. (F) HuH7 cells were mock transfected, or transfected with a scrambled siRNA oligonucleotide sequence, or with a specific siRNA sequence to GRK2. Cells were treated with activin for 24 h and total cell lysates were analyzed by Western blot using either specific polyclonal antibodies against Bax (first panel), p15 (second panel) or c-myc (third panel), or using monoclonal antibodies against GRK2 (fourth panel) or tubulin (fifth panel).
Given the high homology of the kinase domains of the GRK family members, we examined whether any of the other GRKs could have potential effects upon activin/TGFβ-induced promoter activity. Similar to GRK2, overexpression of any of GRKs 3, 4, 5 or 6 resulted in an inhibition of activin/TGFβ-induced 3TP-luc promoter activity (Figure 3B). This ability of the other GRK family members to block activin/TGFβ-induced 3TP-luc promoter activity emphasizes the importance of the highly homologous kinase domain of the GRKs in mediating their inhibitory effect upon activin/TGFβ signaling.
Having established that activin/TGFβ treatment induces an upregulation of GRK2 protein levels in liver cells and that overexpression of GRK2 inhibits activin/TGFβ-mediated gene transcription, we next examined the effect of GRK2 on activin/TGFβ-induced cell growth inhibition. For this, HuH7 hepatocarcinoma cells were cotransfected with a GFP-tagged GRK2 (GFP-GRK2) expression construct and a bicistronic pMACS Kk II vector encoding the truncated mouse H-2Kk surface marker. MACselect Kk microbeads conjugated to a monoclonal antibody directed against the surface marker H-2Kk were then used to magnetically select, enrich and purify GFP-GRK2/H-2Kk-overexpressing cells. Control GFP/pMACS-transfected cells, which went through a similar purification process, and GFP-GRK2-positive cells were subsequently treated or not with activin, and cell viability was assessed by MTT assay. In control HuH7 cells, activin stimulation resulted in a 42% cell growth inhibition, as compared to cells that had been left untreated (Figure 3C). In contrast, in an enriched population of overexpressing GFP-GRK2 HuH7 cells, activin-mediated cell growth inhibition was reversed. Furthermore, we performed FACS analysis on either wild-type (WT) Chinese hamster ovary (CHO) cells, CHO cells stably overexpressing GRK2 or the kinase-inactive (K220M) mutant. As seen in Figure 3D, TGFβ treatment of WT-CHO cells clearly induced an increase in apoptosis, and, to a more modest extent, G1 arrest. However, stable overexpression of GRK2 reversed these TGFβ-mediated effects, while overexpression of the kinase-inactive mutant GRK2 had no effect. GRK2's inhibitory effect upon activin-induced apoptosis was further confirmed by Annexin V staining. As shown in Figure 3E, the activin-induced onset of apoptosis, detectable following activin treatment of WT cells, was clearly inhibited in GRK2 stable cells, while the K220M stable cells behaved in a manner similar to control cells. These findings indicate that elevated levels of GRK2 serve to antagonize activin/TGFβ regulation of cell growth and apoptosis.
Activin/TGFβ-induced apoptosis and G1 arrest are mediated through upregulation of proapoptotic factors, such as Bax (Kanzler and Galle, 2000), and cyclin-dependent kinase inhibitors, such as p15 (Beach, 1994; Reynisdottir and Massague, 1997; Ho et al, 2004), and downregulation of proto-oncogenic factors, such as c-myc (Frederick et al, 2004). To examine the effect of GRK2 on the expression levels of these known activin/TGFβ target genes, we used siRNA to specifically block GRK2 expression in liver cancer cells. In both normal HuH7 cells and cells transfected with a scrambled siRNA, activin stimulation led to a clear increase in both endogenous Bax and p15 expression levels, as well as a decrease in c-myc protein levels (Figure 3F). However, decreasing endogenous GRK2 levels using a specific GRK2 siRNA resulted in a potentiated activin-induced upregulation of Bax and p15, coupled with a robust activin-induced downregulation of c-myc (Figure 3F). Thus, removal of GRK2 activity in liver cancer cells leads to increased activin/TGFβ regulation of target genes, which may contribute to GRK2's antagonistic effect upon activin/TGFβ-induced growth inhibition and apoptosis.
Collectively, these findings indicate that GRK2 overexpression antagonizes activin/TGFβ-mediated transcriptional activity, target gene expression, G1 arrest and apoptosis in human hepatocarcinoma cells. Taken together, these results also suggest that GRK2 acts in a negative feedback loop manner to block activin/TGFβ-mediated physiological effects on cell growth inhibition, thus defining the kinase GRK2 as a multipotent inhibitor of activin/TGFβ signaling.
Norepinephrine (NE) induces GRK2 expression and antagonizes activin-induced cell growth inhibition
Previous studies have demonstrated that NE, which signals through the GPCR α1-adrenergic receptor (α1-AR), can increase GRK2 gene promoter activity (Ramos-Ruiz et al, 2000). As such, we decided to examine if NE could increase GRK2 protein expression levels in human hepatocarcinoma cells. As shown in Figure 4A, NE treatment resulted in a clear increase in GRK2 protein expression levels. We then tested whether utilizing NE to upregulate GRK2 levels would have an antagonistic effect upon activin-induced cell growth inhibition. As shown in Figure 4B, activin strongly inhibited cell growth, while NE by itself had no effect. Interestingly, a 24-h pretreatment of HuH7 cells with NE, prior to activin stimulation, led to a partial reversal of the activin-inhibitory effect, indicating that these two growth factors exert antagonistic effects in human liver cells, and further supports a role for GRK2 as an antagonist to activin-mediated signaling.
Figure 4.
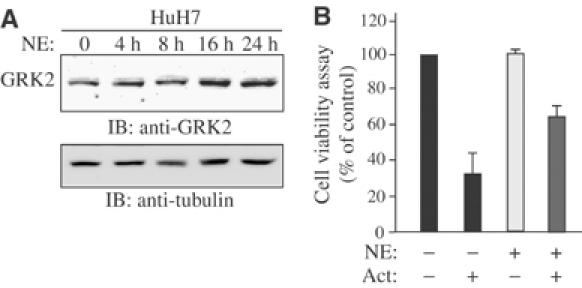
NE induces upregulation of GRK2 and antagonizes activin growth-inhibitory effect. (A) HuH7 cells were stimulated with NE for 0, 4, 8, 16 and 24 h. Total cell lysates were analyzed by Western blot using a specific monoclonal antibody to GRK2 (upper panels) or tubulin (lower panels). (B) HuH7 cells were pretreated or not with NE for 24 h before being stimulated or not with activin for 72 h. Cell growth was assessed by cell viability MTT assay. Values are expressed in arbitrary units. P<0.05 compared with no activin treatment.
GRK2 blocks activin/TGFβ-induced Smad phosphorylation and nuclear translocation
To further elucidate the mechanism by which GRK2 inhibits activin/TGFβ-induced gene expression, we examined the effect of overexpressing GRK2 on Smad phosphorylation and nuclear accumulation. For this, CHO cells were transfected or not with cDNA encoding Flag-GRK2 or a Flag-K220M kinase-inactive mutant, and stimulated with activin for varying time periods. As shown in Figure 5A, in total cell lysates analyzed by Western blot, using a specific antibody recognizing C-terminal phosphorylated serine residues of Smad2, activin-induced Smad2 phosphorylation was inhibited in the presence of GRK2, but not by the kinase-inactive mutant. Nuclear extracts from CHO cells transfected or not with GRK2 were analyzed using anti-phospho-Smad2 and anti-Smad2/3 antibodies and, as shown in Figure 5B, while activin strongly induced Smad2 phosphorylation and nuclear translocation in control cells, these effects were robustly diminished in cells overexpressing GRK2.
Figure 5.
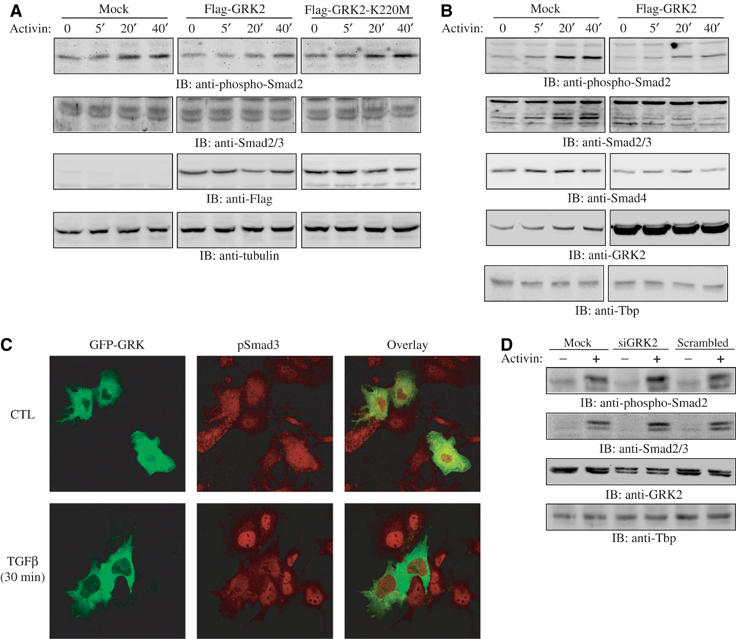
GRK2 inhibits Smad2 carboxyl-terminal phosphorylation. (A) Whole-cell lysates from CHO cells transfected with empty vector, Flag-GRK2 or Flag-GRK2-K220M and stimulated with activin were analyzed by Western blot using anti-phospho-Smad2, anti-Smad2/3, anti-Flag and anti-tubulin antibodies. (B) Nuclear extracts from CHO cells transfected or not with Flag-GRK2 and stimulated with activin for the indicated periods of time were analyzed by Western blot using anti-phospho-Smad2 antibody, anti-Smad2/3, anti-Smad4 and anti-GRK2 antibodies. (C) An immunofluorescence field of view of HuH7 cells containing both transfected GFP-GRK2 cells and nontransfected cells, stimulated or not for 30 min with 0.1 nM TGFβ and labeled for endogenous phospho-Smad3 in rhodamine. (D) HuH7 nuclear extracts from cells either mock transfected or transfected with a scrambled siRNA oligonucleotide sequence, or with a specific siRNA sequence to GRK2, that were either treated or not with activin for 30 min and analyzed by Western blot using anti-phospho-Smad2 antibody, anti-Smad2/3, anti-GRK2 and anti-Tbp antibodies.
In Figure 5C, using confocal microscopy, we further demonstrate that, while normal HuH7 liver cancer cells (left panels, nonfluorescent cells) exhibited a strong nuclear accumulation of phosphorylated Smad3 (middle panels, control nonstimulated as compared to TGFβ-stimulated cells), cells overexpressing GFP-GRK2 (left panel, green cells) failed to exhibit nuclear accumulation and phosphorylation of Smad3 upon 30 min TGFβ treatment as compared to cells not overexpressing GFP-GRK2. Taken together, our results indicate that the kinase GRK2 prevents Smad phosphorylation and nuclear translocation, thus inhibiting Smad signaling.
We next examined the effect of blocking GRK2 expression on activin-mediated Smad activation. As shown in Figure 5D, transfection of HuH7 liver cells with siRNA to GRK2 significantly enhanced Smad2 phosphorylation and nuclear translocation. Together, these results clearly indicate that induced expression of the kinase GRK2 inhibits activin/TGFβ signaling, while knocking down the GRK2 gene potently enhances their signaling effects.
GRK2 physically interacts with the Smads in an activin-dependent manner
CHO cells were stimulated or not with activin for either 4 or 24 h, and cell lysates were immunoprecipitated using an anti-Smad2/3 antibody and proteins in the complex were revealed by immunoblot analysis with an antibody directed against GRK2. Results indicate that ligand stimulation induces complex formation between endogenous GRK2 and Smad2/3 (Figure 6A), an association that is markedly increased after 24 h, possibly due to the activin-induced increase in GRK2 expression at the later time point.
Figure 6.

Activin induces complex formation between GRK2 and the Smads. (A) Cell lysates from CHO cells stimulated with activin for 0, 4 or 24 h were immunoprecipitated with an anti-Smad2/3 antibody and analyzed by Western blot with an anti-GRK2 antibody (top panel). (B) Total cell lysates representing 10% of the immunoprecipitating input were analyzed by Western blot using antibodies to Smad2/3 and tubulin for loading controls. (C) Cell lysates from CHO cells overexpressing Flag-GRK2 or Flag-PRLR were incubated with either GST–Smad2 (C), GST–Smad3 (D) or GST–Smad4 (E) fusion constructs, and analyzed by Western blot using anti-Flag antibody (top panels). Ponceau staining of membranes used in the pulldown experiment, to illustrate relative levels of the fusion proteins used (bottom panels).
Distinct modular domains within the Smads mediate their interactions with various partner proteins. As such, we set out to map the domains of the Smads required to mediate their interaction with GRK2. GST-fusion constructs encompassing the MH1, linker or MH2 domains of Smad2 were used in GST pulldown experiments using lysates from cells overexpressing either Flag-GRK2 or a nonrelated Flag-cDNA (prolactin receptor) as a negative control. The results indicate that Flag-GRK2 interacts with both MH1 and MH2, but not the linker region of Smad2 and 3 (Figure 6C and D). Interestingly, all domains of Smad4 were found to interact with the Flag-GRK2 (Figure 6E). Taken together, these results indicate that GRK2 can interact with all activin/TGFβ-specific Smads, and that this complex formation is ligand regulated.
The linker domains of Smad2/3 are substrates for the kinase GRK2
As GRK2 kinase domain is required to block activin/TGFβ biological effects, we investigated whether Smads were substrates for GRK2. GST-fusion constructs containing the MH1, linker and MH2 domains of Smad2 and 3 were used in an in vitro kinase assay with purified GRK2. A representation of the relative amounts of fusion protein used for the kinase assays is shown in Figure 7A. Our results indicate that the GST-linker domain, but not the MH1 and MH2 domains of Smad2 and Smad3, are highly phosphorylated by GRK2 in vitro, and suggest that the Smads are substrates for the kinase GRK2 (Figure 7B).
Figure 7.
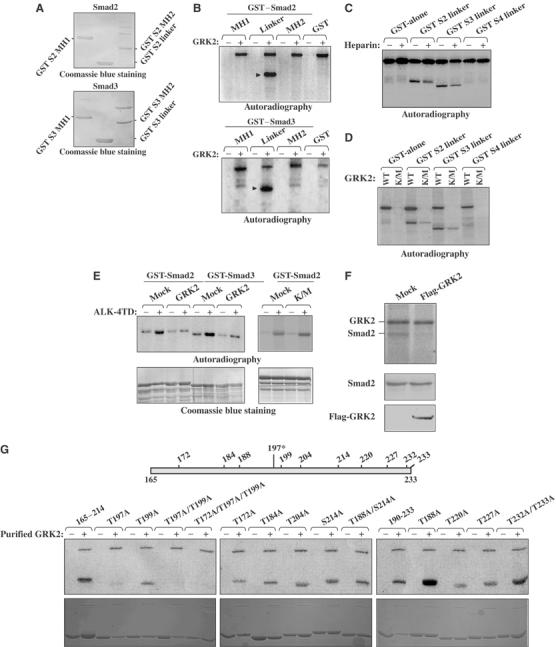
The linker region of Smad2/3 is phosphorylated by GRK2 in an in vitro kinase assay. (A) Coomassie blue staining demonstrating the relative amounts of GST–Smad2 (top panel) and GST–Smad3 (top panel) fusion protein used for the kinase assays. (B) Purified GRK2 was used in a kinase assay with the various GST–Smad constructs (top: Smad2, bottom: Smad3). (C) Purified GRK2 was used in a kinase assay in the absence or presence of heparin with the various GST–Smad fusion proteins. (D) Purified GRK2 or GRK2-K220M was used in a kinase assay with the various GST–Smad constructs. (E) Kinase assay in which GST–full-length Smad2/3 were first incubated with either in the presence or absence of either purified GRK2 or purified GRK2-K220M and unlabeled ATP (or not), the GRKs removed and the GST fusion constructed subsequently incubated with (or not) immunoprecipitated constitutively active activin type I receptor (ALK4-TD) in the presence of [γ-32P]ATP (top panels). Coomassie blue staining demonstrating relative amounts of GST-fusion protein used (bottom panels). (F) Kinase assay in which Myc–Smad2 complexes immunoprecipitated from CHO cells overexpressing either Myc–Smad2 or Myc–Smad2 and Flag-GRK2 were incubated in the presence of purified GRK2 and [γ-32P]ATP. Total cell lysates representing 10% of the immunoprecipitation input were analyzed by Western blot using antibodies to c-myc and flag (bottom). (G) Kinase assay to map the residue of phosphorylation by GRK2. The top is a schematic representation of the location of the potential residues on Smad2, and the top bottom panel is the kinase assay and bottom is the corresponding Coomassie blue staining demonstrating relative amounts of GST-fusion protein used.
To eliminate the possibility that the phosphorylation of the Smad linker domain resulted from the copurification of another kinase, such as the p42/p44 MAP kinase which can coimmunoprecipitate with GRK2 (Elorza et al, 2000), we used heparin, a known inhibitor of GRK2 kinase activity. As shown in Figure 7C, in the presence of heparin, the phosphorylation of the linker region of Smad2 and 3 was markedly reduced. Furthermore, using purified GRK2-K220M, the kinase-inactive mutant, we also observed a marked decrease in the levels of phosphorylation of the linker domain of Smad2 and 3 (Figure 7D). These results indicate that the phosphorylation of Smad2/3 linker domain in our assay is mediated primarily through the kinase GRK2. The slight residual phosphorylation of GST–Smads observed in the presence of the purified kinase-inactive GRK2 could be possibly due to a low amount of copurified MAP kinase.
Then, to determine if phosphorylation of Smad2/3 by GRK2 prevents C-terminal phosphorylation of Smad2/3 by the activin type I receptor, we performed a kinase assay in which GST-full-length Smad2/3 was first incubated in the presence or the absence of either purified GRK2 or GRK2-K220M and unlabeled ATP. The GRKs were then removed and the GST–Smads were subsequently incubated (or not) with purified constitutively active activin type I receptor (ALK-4TD) in the presence of [γ-32P]ATP. As shown in Figure 7E, both GST–Smad2 and GST–Smad3 were phosphorylated following incubation with ALK-4TD. However, when GST–Smads were first incubated with purified GRK2, ALK4-TD-mediated phosphorylation of the Smads was strongly decreased. In contrast, GST–Smad2 that had been incubated with purified GRK2-K220M did not display a decreased level of ALK4-TD-mediated phosphorylation. Together, these findings indicate that phosphorylation of the Smads within their linker region by GRK2 prevents their phosphorylation on their C-terminal residues by the type I activin receptor.
To assess the role of GRK2 phosphorylation of native Smads, CHO cells were transfected with either myc–Smad2 alone, or both myc–Smad2 and Flag-GRK2. Myc–Smad2 complexes were then immunoprecipitated and subsequently subjected to an in vitro kinase assay with purified GRK2 and [γ-32P]ATP. As seen in Figure 7F, there was a clear reduction in the observed level of Smad2 phosphorylation in cells overexpressing Flag-GRK2, as compared to control cells. This is presumably due to the fact that the immunoprecipitated Smad2 in the GRK2-overexpressing cells has already been phosphorylated on its linker domain by the expressed Flag-GRK2, prior to the in vitro kinase assay. Thus, our results indicate that the kinase GRK2 potently phosphorylates GST-fusion Smads in vitro, and also native Smads in living cells.
Mapping of the GRK2 phosphorylation site(s) within the Smad2 linker domain
To then specifically map which serine/threonine (S/T) residue(s) within this region serves as the phosphorylation site(s) for GRK2, we generated various GST point-mutant constructs containing either single, double or triple serine/threonine to alanine (A) mutations (Figure 7G). These point-mutant constructs were then tested in in vitro kinase assays using purified GRK2. Interestingly, we found that a single point mutation of threonine 197 to alanine significantly prevented GRK2 phosphorylation of this region (Figure 7G). Moreover, combining the T197A mutation with any other S/T to A mutation did not appear to have a synergistic effect upon inhibition of GRK2 phosphorylation within this domain. In summary, we have identified threonine 197 in Smad2 as the main site for phosphorylation by GRK2. Assuringly, the amino-acid environment surrounding threonine 197 matches the preferred GRK2 phosphorylation motif, which usually contains acidic residues surrounding the target serine/threonine residues.
Interestingly, the Smad linker domain is also a target for phosphorylation by other protein kinases, including MAP kinase, CAM kinase and cyclin-dependent kinases (Wicks et al, 2000; Matsuura et al, 2004; Waddell et al, 2004). Although the phosphorylation of the Smad linker domain by these kinases also leads to an inhibition of TGFβ signaling, the serine/threonine target residues affected by these kinases differ from threonine 197 identified here. Thus, in this study, we have identified a novel Smad-inhibitory phosphorylation site.
GRK2 antagonizes activin signaling in primary rat hepatocytes
To further translate our findings to a physiological context, we examined the effects of GRK2 upon activin/TGFβ signal transduction in rat primary hepatocytes. Primary hepatocytes were isolated by in situ liver perfusion with collagenase, harvested and infected with either a control recombinant β-galactosidase adenovirus (Ad-LacZ) or an adenovirus containing a cDNA encoding GRK2 (Ad-GRK2). As seen in Figure 8A, while activin stimulation led to an increase in both endogenous Bax and p15 protein levels in Ad-LacZ-infected primary hepatocytes, Ad-GRK2 infection significantly inhibited the ability of activin to induce Bax and p15 expression. Furthermore, Annexin V staining of the cells clearly indicates that Ad-GRK2 infection of primary hepatocytes blocked activin-induced apoptosis, as compared to Ad-LacZ-infected hepatocytes (Figure 8B). These findings demonstrate that GRK2 inhibits activin-induced target gene expression and apoptosis in primary hepatocytes from in situ liver perfused animals.
Figure 8.
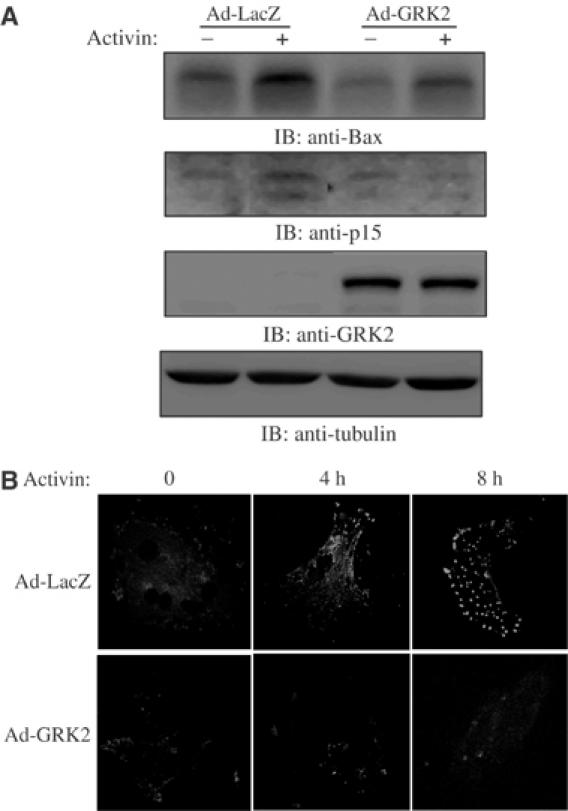
GRK2 antagonizes activin signaling in vivo. (A) Whole-cell lysates from rat primary hepatocytes infected with either control β-galactosidase adenovirus (Ad-LacZ) or GRK2 adenovirus (Ad-GRK2) and stimulated with activin for 24 h were analyzed by Western blot using anti-Bax, anti-p15, anti-GRK2 and anti-tubulin antibodies. (B) Primary hepatocytes infected with either Ad-LacZ or Ad-GRK2 were treated or not with activin for 0, 4 and 8 h, and were labeled for Annexin V-FITC and PI to ensure that necrotic cells were not included in the analysis (data not shown) and analyzed by confocal microscopy.
Conclusions
Here we demonstrate that GRK2 regulates activin/TGFβ receptor responses, extending the central role played by this kinase in modulating signaling from multiple classes of membrane receptors to control a wide range of biological processes. A previous study has recently highlighted the importance of βarrestin, another regulator of GPCR signaling, in the internalization and downregulation of TGFβ receptors (Chen et al, 2003). We have identified the kinase GRK2 as a key player in activin/TGFβ signal transduction, which acts in a negative feedback loop manner to phosphorylate and inhibit Smad signaling. Our results are consistent with a model in which, in a normal physiological and cellular context, where activin/TGFβ levels are low, the endogenous GRK2 levels that are expressed may be insufficient to significantly inhibit activin/TGFβ-induced signaling. After prolonged exposure to activin/TGFβ, there is an upregulation of GRK2 expression levels, which then serves to terminate or slow down the activin/TGFβ-induced receptor-mediated signaling. This method of regulation is reminiscent of the mechanism of action of the inhibitory Smad7 (Hayashi et al, 1997; Nakao et al, 1997), and suggests that negative feedback loop regulation may represent a universal method of inhibiting signaling downstream of growth factor receptors. Unlike Smad7, which acts at the receptor level by binding to the type I receptor, GRK2 acts directly at the Smad level by binding to and phosphorylating the R-Smads, thus preventing their type I receptor carboxyl-terminal phosphorylation, an event required for their nuclear accumulation and subsequent action upon target gene expression. This is of particular novelty, as, unlike Smad7, which may inhibit all activin/TGFβ signaling pathways, GRK2 specifically blocks Smad-dependent signaling cascades.
Our study unveils a novel role played by GRK2 in phosphorylating the Smad linker domain within a specific S/T residue to inhibit activin/TGFβ-mediated signaling. Interestingly, phosphorylation of the Smad linker region by several intracellular protein kinases, such as MAP kinase, Cam kinase II and cyclin-dependent kinase 4/6, also leads to inhibition of Smad signaling (Wicks et al, 2000; Matsuura et al, 2004; Waddell et al, 2004). Thus, this highlights the predominant role played by the linker domain of the receptor-regulated Smads as a primary site for negative regulation of activin/TGFβ signal transduction.
The identification of a novel antagonist to TGFβ/activin-Smad-mediated signaling is of high significance to the development of anticancer therapies. In recent years, many studies have highlighted the therapeutic potential of various TGFβ antagonists as antimetastatic drugs (Bandyopadhyay et al, 2002; Muraoka et al, 2002; Yang et al, 2002). As such, modulation of GRK2 expression and GRK2 kinase activity may provide to be a useful method of specifically regulating activin/TGFβ proinvasive and metastatic responses.
GRK2 controls the activity of many different families of growth factor receptors, including GPCRs, single-transmembrane tyrosine kinases and, from our study, serine/threonine kinases. Regulation of GRK2 expression by multiple growth factors from distinct families may have important physiological consequences towards crosstalk mechanisms acting downstream of these growth factor receptors. Finally, our findings highlight the preponderant role played by GRK2 in maintaining cellular homeostasis and growth control in both normal and tumor cells, and will open new avenues to human cancer therapy.
Materials and methods
Cell culture, cell viability MTT assays, flow cytometry and Annexin V staining
All cell lines were cultured in Dulbecco's modified Eagles medium (DMEM) in the presence of 10% fetal clone III serum (Hyclone). For cell viability assays, cells were plated in triplicates in 96-well dishes, at 10 000 cells/100 μl density in 2% serum, treated or not with 40 μM NE 0.5 nM activin or 0.2 nM TGFβ. After 3 days, cell growth was assessed using the nonradioactive MTT cell proliferation assay for eukaryotic cells (Cell Titer 96, Promega G 4000). Absorbance was measured at 570 nm using a Bio-tek Microplate reader. For flow cytometry, cells were plated in triplicate at 200 000 cells/ml in DMEM, 2% serum, and stimulated or not with activin (0.5 nM) or TGF-β (0.2 nM). After 48 h, cellular DNA was labeled with 50 ng ml−1 PI. Cells were analyzed in a Becton Dickinson FACSCalibur flow cytometer. At least 10 000 gated events were recorded for each sample and the data were analyzed by WinList2.0 software for Windows (Verify Software House Inc., Topsham, ME). For Annexin V staining, cells were plated at a density of 40 000 cells/22 × 22 mm coverslip in 2% serum for an overnight period, and, subsequently, cells were stimulated (or not) with 0.5 nM activin A for 0, 8 or 24 h. Annexin V staining was then assessed according to an Annexin V apoptosis detection kit (Santa Cruz) and analyzed using an LSM-510 Zeiss confocal microscope.
Northern blot analysis
HuH7 cells were plated at a density of 2 × 106 cells/10 cm dish in DMEM containing 2% FBS and stimulated with 0.5 nM activin for different periods of time. Total RNA was extracted using Trizol (Invitrogen). In all, 40 μg of each sample was then separated on agarose gels and transferred to nylon membranes. Probes for GRK2 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were labeled using the Random Priming Kit (Roche), and added to the hybridization solution (0.5 M NaPO4, pH 7.2, 1 mM EDTA, pH 8.0, 7% SDS, 1% BSA, and 200 μg/ml salmon sperm DNA). Results were revealed using a phosphorimager Cyclone Storage Phosphorscreen (Packard Canberra).
Reverse-transcription PCR
HuH7 cells were treated with or without 0.5 nM activin in the presence or absence of cycloheximide (20 μg/ml) for 16 h, and total RNA was extracted using Trizol reagents (Invitrogen). Reverse transcription of total cellular RNA using oligo-dT primers was carried out using Stratacript Reverse Transcriptase (Stratagene) as per the manufacturer's instruction. Subsequently, amplification of cDNA to obtain products for GRK2, GRK3, GRK4, GRK5, GRK6 and GAPDH was performed. The PCR conditions were as follows: 30 cycles (94°C for 30 s, 60°C for 30 s, 72°C for 1 min 30 s).
Purification of GFP-GRK2-overexpressing cells
HuH7 cells were transiently transfected with cDNAs encoding pMACS Kk II and either GFP-GRK2 or GFP alone using lipofectin reagent (Invitrogen). Following overnight recovery, cells were incubated with MACSelect Kk MicroBeads, magnetically selected and eluted according to the manufacturer's specifications (Miltenyi Biotech). The efficiency of enrichment was then assessed using both light and fluorescence microscopy to determine the percent of GFP- and GFP-GRK2-positive contained within 10 fields of view, for four independent experiments.
Luciferase assays
HuH7 cells were transiently cotransfected with empty vector or 0.4 μg of cDNAs encoding either Flag-GRK2, GRK3, GRK4, GRK5, GRK6 or Flag-GRK2-K220M, and the β-galactosidase and 3TP-Luc gene reporter construct using lipofectin reagent (Invitrogen), and treated or not with 0.5 nM activin or 0.2 nM TGFβ for an overnight period. Luciferase activity was measured (EG&G Berthold Luminometer) and normalized to the relative β-galactosidase activity.
siRNA design, generation and transfection
Three siRNAs corresponding to the human GRK2 gene (GenBank accession no. NM_001619) were designed using the ‘siRNA Selection Program' (Whitehead Institute for Biomedical Research). All siRNA sequences were Blast searched in the NCBI's search for short nearly exact matches mode against all human sequences of the GenBank database, and were not found to have significant homology to genes other than the human GRK2 (ADRBK1) gene. siRNAs were synthesized using the Silencer siRNA Construction Kit (Ambion) according to the manufacturer's instructions. SiRNAs sequences were as follows: siGRK2 5′-AAGCAGGTGCCTCCGGATCTCTTCCT GTCTC-3′. 1.6 × 106 cells were transfected using the Lipofectamine 2000 transfection agent (Invitrogen) according to the manufacturer's instructions. Expression levels of GRK2 were analyzed by Western blot.
Coimmunoprecipitation analysis
Cell lysates from cells stimulated or not with 0.5 nM activin for 4 and 24 h were incubated with 0.6 μg of rabbit anti-Smad2/3 (Fl-425) antibody (Santa Cruz) for an overnight period. The following day, 30 μl of a 50% protein A sepharose beads slurry solution (Santa Cruz) was added for 2 h at 4°C. After washing, the immunoprecipitates were eluted with 2 × SDS loading buffer, boiled for 10 min and resolved by 7.5% SDS–PAGE, transferred onto nitrocellulose and incubated with the indicated specific antibodies overnight at 4°C.
Confocal immunofluorescence
HuH7 cells transfected with GFP-GRK were stimulated or not with TGFβ for 30 min, fixed in 4% paraformaldehyde/PBS at room temperature for 15 min, permeabilized in (0.2% Triton-X100, 2% BSA dissolved in PBS) for 30 min, incubated for 1 h with 1:200 dilution of rabbit anti-phospho-Smad3 (Biosource), washed three times for 10 min in PBS, incubated for 1 h with 1:250 dilution of goat anti-rabbit rhodamine (Molecular Probes), washed three times for 10 min in PBS and mounted. Slides were analyzed using an LSM-510 Zeiss confocal microscope.
Western analysis
Cells were cultured in DMEM, 2% fetal clone serum and stimulated or not with 0.5 nM activin for different periods of time as indicated in the figures. Total cell extracts were separated on a 10% polyacrylamide gel, transferred onto nitrocellulose and incubated with the indicated specific antibodies (anti-phospho-Smad2, Chemicon; anti-Flag M2, Sigma; anti-Bax (N-20), Santa-Cruz; anti-p15 (C-20), Santa-Cruz; anti-c-myc (N-262), Santa-Cruz; anti-tubulin, Sigma). Immunoreactivity was revealed by chemiluminescence (Lumi-Light Plus Western Blotting substrate, Roche) according to the manufacturer's instructions and measured using an Alpha Innotech Fluorochem Imaging system (Packard Canberra).
GST pull-down assays
GST–Smad fusion proteins were bacterially expressed and incubated overnight with cell lysates from CHO cells overexpressing Flag-GRK2 or Flag-PRLR. The bound complexes were eluted with 2 × Laemmli buffer and analyzed by Western analysis.
In vitro kinase assay
Reactions were performed (30°C, 20 min) in kinase buffer (20 mM Tris–HCl, pH 8.0, 2 mM EDTA; 10 mM MgCl2, 1 mM DTT), with 0.2 μM of GRK2 or K220M, each of the GST-fusion constructs and 0.1 mM ATP containing 10 μCi of [γ-32P]ATP, in the presence or absence of 5 μM heparin. Reactions were terminated by the addition of 30 μl of 2 × Laemmli buffer, heated to 65°C for 20 min, separated on SDS–PAGE gels, dried and revealed using a phosphor imager. For the double-kinase assay, GST-full-length Smad2/3 fusion constructs were incubated or not with either 0.2 μM purified GRK2 or GRK2-K220M, in the presence of cold 0.1 mM ATP in kinase reaction buffer for 20 min at 30°C. The GST-fusion proteins were then washed five times with cold 1 × PBS and resuspended in a final volume of 6 μl. Using these fusion constructs, a radioactive kinase assay was then carried out using HA-immunoprecipitated ALK-4TD, in the presence of 0.1 mM ATP containing 10 μCi of [γ-32P]ATP, and proceeded as described above.
Rat primary hepatocyte isolation and adenoviral infection
Rat primary hepatocytes were isolated as described previously (Kong et al, 2000). Briefly, primary hepatocytes were isolated from 120–140 g male Harlan Sprague–Dawley rats (Charles River, St Constant, Quebec, Canada) by in situ liver perfusion with collagenase (protocol 4110, approved by McGill), and were plated on a collagen matrix (Vitrogen-100). Cultures were prepared by seeding 1 × 106 cells onto 9.6-cm2 × six-well plates in the absence (for Western blotting) or presence of 22 × 22 mm coverslips (for Annexin V labeling). Cells were bathed for 24 h in seeding medium (DMEM/Ham's F-12 containing 10% FBS, 10 mM Hepes, 20 mM NaHCO3, 500 IU/ml penicillin and 500 μg/ml streptomycin) and then for 48 h in a serum-free medium that differed from the seeding medium in that it lacked FBS and contained 1.25 μg/ml fungizone, 0.4 mM ornithine, 2.25 μg/ml L-lactic acid, 2.5 × 10−8 M selenium and 1 × 10−8 M ethanolamine. For adenovirus infection, cells were infected with either recombinant β-galactosidase adenovirus or GRK2 adenovirus for 4 h at 37°C. After viral exposure, infected cells were serum-starved for 24 h in serum-free medium and subsequently stimulated with 0.5 nM activin A for the indicated periods of time. Western blotting analysis and Annexin V labeling were carried out as described previously.
Acknowledgments
We are thankful to Dr Y Eto and Ajinomoto Co., Inc. for activin A, Dr J Benovic for his generous gift of the purified GRK2 kinase, Dr S Ferguson for the GRK3, GRK4, GRK5 and GRK6 constructs, Dr Ross Feldmann for the GRK2 adenovirus and Dr J Massague for the 3TP-luc construct. JJL is Research Scientist of the National Cancer Institute of Canada, supported with funds provided by the Canadian Cancer Society, SAL holds a Canadian research chair in Molecular Endocrinology, EC is a recipient of a Fonds de Recherche de Sante du Quebec scholarship and JH is a recipient of a McGill University Health Center scholarship. This work was supported by grants from the CIHR (MOP-53141 to JJL and MOP-49447 to SAL).
References
- Bandyopadhyay A, Lopez-Casillas F, Malik SN, Montiel JL, Mendoza V, Yang J, Sun LZ (2002) Antitumor activity of a recombinant soluble betaglycan in human breast cancer xenograft. Cancer Res 62: 4690–4695 [PubMed] [Google Scholar]
- Beach GJHD (1994) p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 371: 257–261 [DOI] [PubMed] [Google Scholar]
- Chen W, Kirkbride KC, How T, Nelson CD, Mo J, Frederick JP, Wang XF, Lefkowitz RJ, Blobe GC (2003) Beta-arrestin 2 mediates endocytosis of type III TGF-beta receptor and down-regulation of its signaling. Science 301: 1394–1397 [DOI] [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ, Balmain A (2001) TGF-beta signaling in tumor suppression and cancer progression. Nat Genet 29: 117–129 [DOI] [PubMed] [Google Scholar]
- Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL (2003) Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol 5: 410–421 [DOI] [PubMed] [Google Scholar]
- Elorza A, Sarnago S, Mayor F Jr (2000) Agonist-dependent modulation of G protein-coupled receptor kinase 2 by mitogen-activated protein kinases. Mol Pharmacol 57: 778–783 [DOI] [PubMed] [Google Scholar]
- Frederick JP, Liberati NT, Waddell DS, Shi Y, Wang X-F (2004) Transforming growth factor β-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol Cell Biol 24: 2546–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman NJ, Kim LK, Murray JP, Exum ST, Brian L, Wu JH, Peppel K (2002) Phosphorylation of the platelet-derived growth factor receptor-beta and epidermal growth factor receptor by G protein-coupled receptor kinase-2. Mechanisms for selectivity of desensitization. J Biol Chem 277: 48261–48269 [DOI] [PubMed] [Google Scholar]
- Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA Jr, Wrana JL, Falb D (1997) The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 89: 1165–1173 [DOI] [PubMed] [Google Scholar]
- Ho J, de Guise C, Kim C, Lemay S, Wang XF, Lebrun JJ (2004) Activin induces hepatocyte cell growth arrest through induction of the cyclin-dependent kinase inhibitor p15INK4B and Sp1. Cell Signal 16: 693–701 [DOI] [PubMed] [Google Scholar]
- Kanzler S, Galle PR (2000) Apoptosis and the liver. Semin Cancer Biol 10: 173–184 [DOI] [PubMed] [Google Scholar]
- Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell 6: 1365–1375 [DOI] [PubMed] [Google Scholar]
- Kong M, Mounier C, Wu J, Posner BI (2000) Epidermal growth factor-induced phosphatidylinositol 3-kinase activation and DNA synthesis. Identification of Grb2-associated binder 2 as the major mediator in rat hepatocytes. J. Biol. Chem. 275: 36035–36042 [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Massague J (1997) Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 389: 618–622 [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Timokhina I, Massague J (1999) A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev 13: 804–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo RS, Massague J (1999) Ubiquitin-dependent degradation of TGF-beta-activated smad2. Nat Cell Biol 1: 472–478 [DOI] [PubMed] [Google Scholar]
- Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F (2004) Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 430: 226–231 [DOI] [PubMed] [Google Scholar]
- Muraoka RS, Dumont N, Ritter CA, Dugger TC, Brantley DM, Chen J, Easterly E, Roebuck LR, Ryan S, Gotwals PJ, Koteliansky V, Arteaga CL (2002) Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J Clin Invest 109: 1551–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P (1997) Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 389: 631–635 [DOI] [PubMed] [Google Scholar]
- Oberhammer FA, Pavelka M, Sharma S, Tiefenbacher R, Purchio AF, Bursch W, Schulte-Hermann R (1992) Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc Natl Acad Sci USA 89: 5408–5412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher JA, Freedman NJ, Lefkowitz RJ (1998) G protein-coupled receptor kinases. Annu Rev Biochem 67: 653–692 [DOI] [PubMed] [Google Scholar]
- Ramos-Ruiz R, Penela P, Penn RB, Mayor F Jr (2000) Analysis of the human G protein-coupled receptor kinase 2 (GRK2) gene promoter: regulation by signal transduction systems in aortic smooth muscle cells. Circulation 101: 2083–2089 [DOI] [PubMed] [Google Scholar]
- Reynisdottir I, Massague J (1997) The subcellular locations of p15(Ink4b) and p27(Kip1) coordinate their inhibitory interactions with cdk4 and cdk2. Genes Dev 11: 492–503 [DOI] [PubMed] [Google Scholar]
- Shi W, Sun C, He B, Xiong W, Shi X, Yao D, Cao X (2004) GADD34-PP1c recruited by Smad7 dephosphorylates TGFβ type I receptor. J Cell Biol 164: 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113: 685–700 [DOI] [PubMed] [Google Scholar]
- Waddell DS, Liberati NT, Guo X, Frederick JP, Wang XF (2004) Casein kinase Iepsilon plays a functional role in the transforming growth factor-beta signaling pathway. J Biol Chem 279: 29236–29246 [DOI] [PubMed] [Google Scholar]
- Wakefield LM, Roberts AB (2002) TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev 12: 22–29 [DOI] [PubMed] [Google Scholar]
- Wicks SJ, Lui S, Abdel-Wahab N, Mason RM, Chantry A (2000) Inactivation of smad-transforming growth factor beta signaling by Ca(2+)-calmodulin-dependent protein kinase II. Mol Cell Biol 20: 8103–811111027280 [Google Scholar]
- Xu L, Kang Y, Col S, Massague J (2002) Smad2 nucleocytoplasmic shuttling by nucleoporins CAN/Nup214 and Nup153 feeds TGFbeta signaling complexes in the cytoplasm and nucleus. Mol Cell 10: 271–282 [DOI] [PubMed] [Google Scholar]
- Yang YA, Dukhanina O, Tang B, Mamura M, Letterio JJ, MacGregor J, Patel SC, Khozin S, Liu ZY, Green J, Anver MR, Merlino G, Wakefield LM (2002) Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J Clin Invest 109: 1607–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D, Ehrlich M, Henis YI, Leof EB (2002) Transforming growth factor-beta receptors interact with AP2 by direct binding to beta2 subunit. Mol Biol Cell 13: 4001–4012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Mine T, Shibata H, Eto Y, Hasegawa Y, Takeuchi T, Asano S, Kojima I (1993) Activin A: an autocrine inhibitor of initiation of DNA synthesis in rat hepatocytes. J Clin Invest 92: 1491–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]