

Abstract
Vascular occlusion is the major cause of morbidity and mortality in sickle cell disease but its mechanisms are poorly understood. We demonstrate by using intravital microscopy in mice expressing human sickle hemoglobin (SS) that SS red blood cells (RBCs) bind to adherent leukocytes in inflamed venules, producing vasoocclusion of cremasteric venules. SS mice deficient in P- and E-selectins, which display defective leukocyte recruitment to the vessel wall, are protected from vasoocclusion. These data uncover a previously unsuspected paradigm for the pathogenesis of sickle cell vasoocclusion in which adherent leukocytes play a direct role and suggest that drugs targeting SS RBC–leukocyte or leukocyte–endothelial interactions may prevent or treat the vascular complications of this debilitating disease.
Sickle cell disease, the first molecular disease identified in humans, is among the most common inherited hematological disorder in the United States. It results from a single amino acid substitution in the β-chain of hemoglobin (HbβS). HbβS polymerizes on deoxygenation, producing less deformable sickle red blood cells (SS RBCs) that can obstruct blood vessels (1). More than 20 years ago, it was demonstrated that SS RBCs displayed increased adherence to endothelial cells (2, 3). Subsequent studies recognized the importance of several adhesion pathways in these interactions (4–10), and revealed that young (low-density) SS RBC were more adherent to the endothelium than dense (often irreversibly sickled) SS RBCs (11–13). Collectively, these observations led to the current multistep model for sickle cell vasoocclusion in which light-density cells first adhere in postcapillary venules and secondary trapping of dense cells produces vascular obstruction and ischemia.
To test this model in vivo, we applied the technique of intravital microscopy, which allows direct visualization of the microvasculature, to mice genetically engineered to exclusively express human (h) HbβS. Such mice have circulating irreversibly sickled cells and manifest several of the cardinal features of sickle cell disease, including anemia, reticulocytosis, and organ damage (14, 15). To overcome the limitations imposed by the high perinatal mortality and low breeding efficiency, we generated sickle cell mice by transplantation of sickle cell bone marrow precursors into lethally irradiated normal adult recipients. Unexpectedly, we observed that SS RBCs interacted primarily with adherent leukocytes in postcapillary and collecting venules rather than the endothelial surface, and that these interactions led to vascular occlusion. These data indicate a direct role for leukocytes in the pathogenesis of sickle cell vasoocclusion and provide a potential explanation for the clinical association between leukocytosis and poor prognosis in sickle cell disease.
Methods
Mice.
Wild-type (WT) C57BL/6 mice were obtained from Charles River Breeding Laboratories. SS [Tg(Hu-miniLCRα1GγAγδβS) mα −/− mβ −/−] and SA [Tg(Hu-miniLCRα1GγAγδβS) mα −/− mβ −/+] mice (14, 15) and SAD transgenic mice (16) were bred in the animal facilities at Mount Sinai School of Medicine and Berkeley laboratories. Both SA and SS mice are from a similar mixed background (H2b with contributions from C57BL/6, 129Sv, FVB/N, DBA/2, Black Swiss) and SAD animals were backcrossed (>20 generations) into the C57BL/6 strain. All experimental procedures were approved by the Animal Care and Use Committee of Mount Sinai.
Bone Marrow Transplantation.
Femoral bone marrow nucleated cells from WT C57BL/6, SA, and SS mice were harvested and injected (1.5 × 106 cells/recipient) via the lateral tail vein of lethally irradiated (1,200 cGy, two split doses) C57BL/6 recipient male animals (5–6 weeks of age). Following the procedure, transplanted animals were transferred to sterile cages containing sterile food and water, and allowed to recover for 2 months, the minimum time for full replacement of circulating RBCs with those of donor origin based on a normal mouse RBC survival of approximately 55 days (17). Using this approach, one donor can generate 10–15 gender- and age-matched recipients.
Evaluation of Hemoglobin Chimerism.
Between 2 and 3 months after transplantation, blood (≈20 μl) was obtained by tail vein sampling into 1 ml of PBS. After two washings in PBS, RBCs were lysed by resuspending the cell pellet in water. The amount of Hb in lysates was quantitated using a spectrophotometer (540 nM). Hb was electrophoresed (20 μg per lane) on an acidic polyacrylamide gel containing urea and Triton X-100 (18). Under these conditions, mouse (m) Hbβ and h HbβS comigrate, but m Hbα and h Hbα can be distinguished in Coomassie-stained gels, and thus the degree of chimerism can be determined by comparing the density of m Hbα band of tested samples with control mixtures of h and m Hb.
Intravital Microscopy.
Transplanted mice (>97% donor hemoglobin) were anesthetized with a mixture of 2% α-chloralose and 10% urethane in PBS (6 ml/kg), and a polyethylene tube was inserted into the trachea to facilitate spontaneous respiration. Following scrotal incision, the testicle and the surrounding cremaster muscle were exteriorized and the testis and epididymis were removed after ligation of their feeding vessels. The cremaster muscle was longitudinally opened by cautery, carefully stretched, and pinned across a Plexiglas intravital microscopy stage. The cremaster tissue was continuously superfused throughout the experiment with bicarbonate-buffered saline (37°C) equilibrated with a mixture of 95% N2 and 5% CO2. Microvessels were visualized using a custom-designed intravital microscope (MM-40, Nikon), using 40× Nikon water immersion objective. Images were captured using a CCD camera (Hamamatsu, Bridgewater, NJ) and recorded on a Sony SVHS video recorder (SVO-9500). Fifteen minutes following surgery, several postcapillary and collecting venules were videotaped for 75 min [first intravital microscopy monitoring period (IVM 1)], after which murine rTNF-α (0.5 μg i.p.; R & D Systems) was injected. After exposure to TNF-α for 90 min, venules were recorded for an additional 90 min (IVM 2). Whenever possible, the same cremasteric venules were recorded during IVM 1 and IVM 2.
Wall shear rates (γ) were calculated based on Poiseuille's Law for a Newtonian fluid [γ = 8 (Vmean/Dv), where Dv is the diameter of the venule and Vmean is estimated from the centerline red blood cell velocities (VRBC), using the empirical correlation Vmean = VRBC/1.6 (19)]. VRBC was measured for each venule in real time using an optical Doppler velocimeter (Texas A&M, College Station, TX).
RBCs were identified based on their size and shape (discoid and sickle-shaped cells) by playback analyses of videotapes. An interaction between RBC and WBC was defined as the arrest of an RBC on an adherent leukocyte for >0.07 s (>2 video frames). This time interval was selected because it correlates with a readily discernable adhesion event when the videotape is played in real time. RBC–WBC interactions were counted over a 100-μm venular segment for three consecutive 1-min intervals per venule.
Blood counts were determined at the end of the intravital experiment using an automatic cell counter (System 9018CP, Serono–Baker Diagnostics, Allentown, PA).
Statistical Analysis.
Data are presented as mean ± SE. Parametric comparisons were performed using ANOVA. The Kruskal–Wallis test was used for nonparametric comparisons among more than two groups, and subsequent Mann–Whitney test with the Bonferroni correction for paired comparisons.
Results
Intravital Microscopy Model of Sickle Cell Disease.
We generated cohorts of age- and gender-matched SS and control mice, using a bone marrow transplantation strategy. Bone marrow-derived hematopoietic precursors from WT animals, SS mice, and mice expressing both h αβS hemoglobin and a hybrid hemoglobin containing h α and wild-type m β-globin (hereinafter termed SA mice) were injected into lethally irradiated WT adult mice. Two months after transplantation, the majority of recipient mice displayed >97% donor hemoglobin (Fig. 1), and phenotypes resembling those of the respective bone marrow donors. In some cases, however, we have seen further increments in the expression of HbβS between 60 and 80 days after transplantation, perhaps reflecting incomplete ablation of endogenous committed erythroid precursors. These results thus indicate that sickle cell hematopoietic precursors can reconstitute the entire erythroid compartment of normal recipients.
Figure 1.

Analysis of donor chimerism by Hb electrophoresis. Hb lysates were electrophoresed (20 μg per lane) on an acidic polyacrylamide gel containing urea and Triton X-100. The m Hbβ (lane 1) and h HbβS (lane 2) comigrate, but m Hbα and h Hbα can be distinguished. Various m:h Hb mixtures (lanes 3–7) were used to define the sensitivity of detecting residual m Hb (≈1.5%). Three representative (*) wild-type mice that received SS bone marrow demonstrated >97% donor h Hb. Only mice with >97% h Hb were used in the studies.
To observe dynamic interactions between the flowing blood cells and the vessel wall, the cremasteric microcirculation was evaluated by intravital microscopy. The surgical preparation of the cremaster muscle itself initiates an inflammatory response, leading to progressive recruitment of adherent leukocytes. Because inflammation is associated with human sickle cell crises (1), we reasoned that vasoocclusion in SS mice might be accentuated by administering a pro-inflammatory agent. We chose TNF-α, a cytokine that induces P- and E-selectin-mediated leukocyte rolling (20, 21). In preliminary experiments, mice were pretreated with the cytokine for 2–3 h before the surgical preparation. In contrast to control animals and SS mice that did not receive TNF-α, SS mice pretreated with the cytokine died during or soon after the cremasteric preparation. These results suggest that SS mice are highly susceptible to systemic inflammation. We thus designed an experimental protocol that allowed us to monitor cell adhesion and hemodynamics in the microcirculation of SS and control animals during inflammation induced by surgical manipulation and then augmented by the administration of TNF-α.
Sickle Erythrocytes Interact with Adherent Leukocytes.
Leukocyte rolling is a required initial step for leukocyte adhesion to the vessel wall and subsequent diapedesis (22, 23). During IVM 1, the absolute number of leukocyte rolling per minute and adherent leukocytes per 100 μm in postcapillary and collecting venules were significantly higher in SS mice compared with WT animals (Fig. 2A). The increased number of rolling leukocytes in SS mice likely originates from higher peripheral blood WBC counts (Table 1) because the percentage of rolling leukocytes (e.g., leukocyte flux fractions), which take into account volumetric flow rates and differences in systemic WBC counts (24), were similar between these two groups (rolling leukocyte flux fractions: 6.5 ± 1.1% and 5.3 ± 2.0% for WT and SS mice, respectively; P = 0.61). The absolute increase in leukocyte recruitment to the vessel wall of SS mice is consistent with the increased inflammatory response observed in another sickle cell transgenic strain after ischemia-reperfusion injury (25).
Figure 2.

Analyses of leukocyte and SS RBC adhesion events in postcapillary and collecting venules of SS, SA, and WT mice. (A) The numbers of rolling and adherent leukocytes were determined from video sequences recorded between 15 and 90 min after surgery. The numbers of rolling and adherent leukocytes were significantly higher in the venules of SS mice compared with WT controls; *, P < 0.05; #, P < 0.001. (B) SS RBC interactions with adherent leukocytes were determined in venules recorded between 30 and 90 min after the cremasteric surgery. A mean of 1.7 RBCs interacted per adherent leukocyte per minute in SS mice, whereas these interactions were virtually absent in SA or WT mice; ¥, P < 0.0001. (C) Number of RBC–WBC interactions during IVM 1 in SS mice. Each dot represents an individual venule. The number of SS RBC interactions per adherent leukocyte per minute, evaluated over 100 μm venular lengths during IVM 1, increased with time after cremasteric surgery (R = 0.46; n = 44 venules; P = 0.002). (D) Wall shear rates (γ) in venules of SS, SA, and WT mice during IVM 1 and IVM 2; ¶, P < 0.0001. Data are mean ± SE; n = 25–36 venules from four to five mice.
Table 1.
Hematologic parameters, spleen/body weight ratios, and venular hemodynamics
| Genotype | Leukocytes, ×103/μl | Hematocrit, % | Platelets, ×103/μl | Spleen/body weight ratios, g/g × 10−3 | VRBC,
mm/s
|
Venular diameters, μm
|
||
|---|---|---|---|---|---|---|---|---|
| IVM 1 | IVM 2 | IVM 1 | IVM 2 | |||||
| WT | 4.3 ± 0.6 | 41.1 ± 1.5 | 722 ± 56 | 2.5 ± 0.1 | 2.3 ± 0.1 | 2.4 ± 0.2 | 21.9 ± 0.8 | 21.8 ± 1.0 |
| SA | 7.4 ± 1.8 | 33.4 ± 2.1 | 1364 ± 126† | 2.9 ± 0.1* | 2.2 ± 0.1 | 2.0 ± 0.1 | 20.2 ± 1.2 | 21.7 ± 1.2 |
| SS | 22.3 ± 2.4* | 13.1 ± 0.8† | 287 ± 21† | 27.3 ± 2.7‡ | 2.2 ± 0.1 | 0.6 ± 0.2‡ | 20.9 ± 1.3 | 24.9 ± 1.8 |
| SSP/E −/− | 72.1 ± 11.4† | 18.2 ± 4.7† | 331 ± 72* | 36.0 ± 3.3‡ | 2.6 ± 0.1 | 1.9 ± 0.1 | 20.4 ± 1.5 | 20.3 ± 1.2 |
Blood counts were determined using an automatic cell counter. Centerline velocities (VRBC) were measured in real time by using an optical doppler velocimeter. Venular diameters were assessed using a video caliper. n = 4–9 mice for blood counts; n = 23–41 venules from 4–5 mice for hemodynamic characteristics.
, P < 0.05 compared to WT animals.
, P < 0.001 compared to WT animals.
, P < 0.0001 compared to WT animals.
Although we observed occasional SS RBCs interacting with the endothelium in venules of SS mice, the most striking finding was that circulating SS RBCs bound to adherent leukocytes (WBCs) in venules. SS RBC–WBC interactions were quantitated over 100-μm venular lengths. In the 30–90-min time interval after surgery, an average of 1.7 SS RBCs interacted per adherent leukocyte per minute, whereas RBC–WBC interactions were virtually absent in SA or WT mice (Fig. 2B). SS RBC–WBC interactions started approximately 30 min after the cremaster surgery and increased progressively with time (Fig. 2C). The increasing rate of SS RBC interactions per adherent WBC over time suggests that there are qualitative changes in adherent leukocytes, or perhaps SS RBCs (26), during the inflammatory response. After TNF-α administration, the interactions between SS RBC and WBC were either sustained or further increased and commonly produced complete venular occlusions (Fig. 3). As a result, the average shear rates in SS mouse venules were dramatically decreased during IVM 2 compared with those of IVM 1, whereas shear rates in venules of SA or WT mice were not significantly changed (Fig. 2D). Because the venular diameters were similar in IVM 1 and IVM 2, the reductions in shear rates in SS mice reflected reductions in central line RBC velocities. Interestingly, platelet counts were significantly lower in SS mice at the end of IVM 2 (Table 1), suggesting that platelets are consumed or perhaps sequestered in the spleen after treatment of SS mice with TNF-α.
Figure 3.
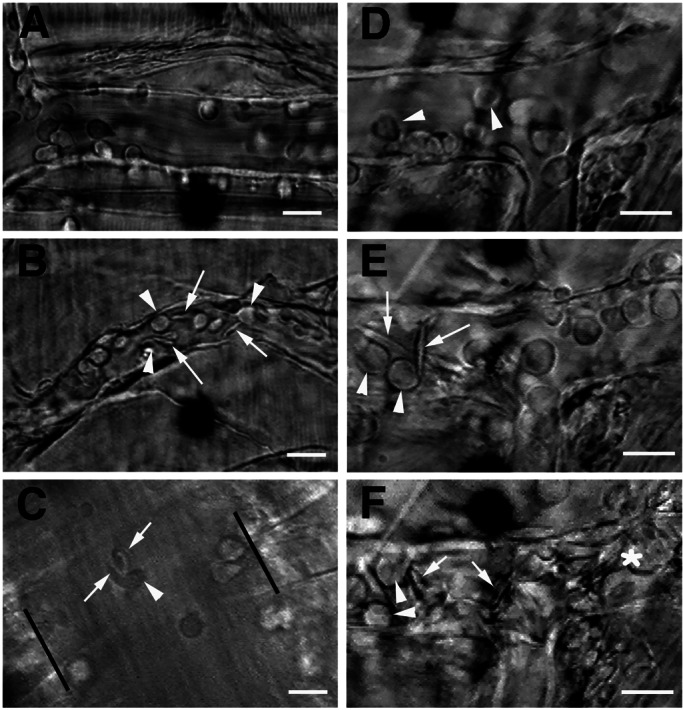
Intravital microscopy of the cremasteric microcirculation in SA- and SS-transplanted mice. (A) Still frame of a venule from an SA-transplanted mouse after TNF-α injection, during IVM 2. Numerous rolling leukocytes (round cells) and some adherent leukocytes can be observed, but RBCs flow freely. Owing to the rapid blood flow, RBCs cannot be distinguished in the microcirculation of live animals unless they are interacting. (B) Venule of a SS transplanted mouse during IVM 2. Numerous SS RBCs (arrows) bind to adherent leukocytes (arrowheads) and can resist the shear of flowing blood. (C) Large venule of an SS mouse after TNF-α stimulation. The parallel bars mark the venular walls. Both sickle- and discoid-shaped RBCs (arrow) bind to an adherent leukocyte (arrowhead). See Movie 1, which is published as supporting information on the PNAS web site (www.pnas.org), for the original video segments. (D–F) Sequential digital frames of a venular segment during the intravital microscopy protocol. (D) Fifteen minutes after surgery, only rolling and adherent leukocytes are observed and no RBC adhesion is seen. (E) At the end of IVM-1, many interactions between sickle cells (arrows) and adherent leukocytes are apparent. (F) Venular occlusion during IVM 2 (100 min after TNF-α). Individual blood cells cannot be easily distinguished in the multicellular aggregates of occluded venules (*), except for a few leukocytes (arrowheads) and sickle cells (arrows). (Scale bars, 15 μm.)
Although both discoid and sickle-shaped RBCs adhered to leukocytes, most interacting RBCs were sickle shaped (Fig. 3; ‘also see Movie 1), suggesting that sickling-induced RBC membrane changes or the ability of the sickled cells to align in the flowing stream might influence their interactions with leukocytes. Approximately 18.0% ± 3.1% of adherent leukocytes interacted with SS RBCs, raising the possibility that only subsets of leukocytes or only leukocytes in strategic locations can bind SS RBCs. To exclude the possibility that the observed SS RBC–WBC interactions were due to the transplantation procedure itself, we also studied the parental SS group of mice that had not undergone transplantation and found similar RBC–WBC interactions (see Movie 2, which is published as supporting information on the PNAS web site).
To ascertain whether SS RBC–WBC interactions were limited to our SS mouse model (15), we analyzed nontransplanted transgenic mice expressing another sickling hemoglobin (h βSAD) by using the same protocol. Owing to the relatively low concentration of HbβS (≈19%) and the inhibitory effects of murine globin on sickle hemoglobin polymerization, βSAD transgenic (SAD) mice have a milder phenotype than SS mice; thus, these mice are not anemic and do not exhibit circulating sickle cells under normoxic conditions (16). RBC–WBC interactions were also observed in SAD mice, even though none of the interacting RBCs were sickle-shaped, both before and after TNF-α administration. The rate of interactions, however, was lower (0.14 ± 0.05 RBC/WBC/min between 30 and 90 min; n = 22 venules) than that of SS mice (Fig. 2B), and RBC binding to adherent WBC did not produce vasoocclusion.
Inhibition of Leukocyte Adhesion Protects Against Vasoocclusion.
P- and E-selectins mediate leukocyte rolling on venular endothelium and play a critical role in the recruitment of leukocytes into inflammatory sites. As a result, mice deficient in both P- and E-selectins (P/E −/−) have severe defects in leukocyte rolling on, and adhesion to, endothelium (20, 21). To further investigate the role of adherent leukocytes in vasoocclusion, we generated by bone marrow transplantation SS mice lacking both endothelial selectins (SS P/E −/−). Two months after transplantation, SS P/E −/− mice expressed >97% human hemoglobins (Fig. 4A). Like nontransplanted P/E −/− mice (20, 21), SS P/E −/− animals displayed very high circulating leukocyte counts (Table 1) and markedly reduced numbers of rolling (Fig. 4B) and adherent (Fig. 4C) leukocytes. The mean number of SS RBC interactions per adherent leukocyte was 95% less in SS P/E −/− mice than in SS animals (0.06 ± 0.02 interaction/WBC/min; n = 32 venules, P = 0.002). In addition, the percentage of adherent WBCs that interacted with SS RBCs was also lower in SS P/E −/− mice compared with SS animals (4.0% ± 1.6% vs. 18.0% ± 3.1%, respectively; P = 0.002), suggesting that adhesion to (and likely signaling induced by) endothelial selectins may enhance the ability of adherent leukocytes to bind circulating SS RBCs. Most strikingly, SS mice deficient in both endothelial selectins were protected from developing vasoocclusion in response to TNF-α. In sharp contrast to the results in SS mice, the shear rates in the cremaster microcirculation of SS PE −/− animals were only mildly reduced (Fig. 4D), and blood flow was preserved after TNF-α treatment, even in the smallest venules (Fig. 4E; see also Movie 3, which is published as supporting information on the PNAS web site). Moreover, all five SS P/E −/− animals survived the entire intravital experiment, whereas four of five SS mice died during IVM 2. Thus, SS PE −/− mice which have reduced leukocyte recruitment to the venular wall, are protected from sickle cell-mediated vascular occlusion induced by TNF-α. Because SS PE −/− mice, like SS mice, displayed significantly lower platelets counts compared with WT mice at the end of the experiment, it suggests that platelets may not play a dominant role in vascular occlusion induced by TNF-α.
Figure 4.

Sickle cell mice deficient in both P- and E-selectins are protected from vasoocclusion. (A) Hb gel electrophoresis of SS P/E −/− chimeras. Blood lysates from m and h controls and three SS P/E −/− chimeric mice (*) were electrophoresed on an acid/urea/Triton 100-X gel and the gel was stained with Coomassie blue. RBCs from all studied SS P/E −/− chimeric mice contained >97% h Hb. (B) The number of rolling leukocytes in SS and SS P/E −/− mice during IVM 1 and IVM 2. The number of rolling leukocytes was drastically reduced in SS P/E −/− animals compared with SS mice during IVM 1 (P = 0.0001). The reduction in the rolling leukocytes in SS mice during IVM 2 (a), compared with IVM 1, results in part from the lower wall shear rates due to vascular occlusions in the venules of SS mice (see Fig. 2D). (C) Numbers of adherent leukocytes in SS and SS P/E −/− animals during IVM 1 and IVM 2. The number of adherent leukocytes was significantly reduced in SS P/E −/− compared with SS animals both during IVM 1 and IVM 2 (P < 0.0001). (D) Wall shear rates in venules of SS P/E −/− mice during IVM 1 and IVM 2. In contrast to SS mice (Fig. 2D), shear rates were mildly decreased during IVM 2 (P = 0.01). (E) Still photograph of a venule from a SS P/E −/− animal 90 min after TNF-α. No rolling and one adherent leukocyte (arrowhead) can be observed. Blood flow (right to left) is preserved even in small postcapillary venules (arrows). See Movie 3.
Discussion
Using two different murine models of sickle cell disease, we demonstrate that SS RBCs interact with adherent leukocytes in cremasteric postcapillary and collecting venules. These interactions occurred early (≈30 min) after the surgical manipulation and were maintained or enhanced after administration of TNF-α. Although most interactions between SS RBC and WBC were transient, some lasted several seconds, resisted the shear of flowing blood, and produced vasoocclusion (see Movies 1–3). These observations indicate that adherent leukocytes play a direct role in the vascular occlusions caused by sickle cell disease.
A number of clinical observations have linked leukocytosis with symptomatic sickle cell disease but it has been unclear whether the leukocytosis was simply a surrogate marker of inflammation rather than the leukocytes being directly engaged in the pathogenesis of vasoocclusion. Thus (i) leukocytosis in SS patients is associated with increased mortality (27), (ii) hydroxyurea-induced reductions in leukocyte counts in sickle cell anemia correlate with clinical benefit (28), (iii) severe leukocytosis is commonly present in patients with acute chest syndrome (29), (iv) acute infection is frequently suspected as triggering sickle cell pain crisis, and (v) four separate reports have linked the administration of a myeloid colony-stimulating factor (G-CSF or GM-CSF) to the induction of a severe, or even fatal, sickle cell crisis (30–33). In vitro experiments in static model systems have, in fact, demonstrated sickle cell adhesion to cultured monocytes (34) and adherent neutrophils (35). The latter was mediated at least in part by SS RBC IgG and a receptor mechanism inhibitable by Arg–Glu–Asp (RGD)-containing peptides. However, because these studies were conducted under static conditions, their in vivo significance is currently unclear. Our in vivo data indicate that leukocytes that are adherent to the vessel wall can directly contribute to vascular occlusion in the cremaster muscle of sickle cell mice by means of their interactions with SS RBCs, and that inhibition of leukocyte adhesion by targeted disruption of P- and E-selectin expression can prevent lethal vasoocclusion. Thus, these results provide a plausible mechanism to reconcile the association between leukocytosis and sickle cell disease pathogenesis and suggest that both RBC–WBC and WBC–endothelial interactions are potential targets to prevent and/or treat sickle cell vasoocclusion.
Supplementary Material
Acknowledgments
We thank Drs. Dhananjaya K. Kaul for advice on the cremasteric preparation and Frank Costantini for providing breeding pairs of SAD transgenic mice. We thank Dr. George Atweh for helpful discussions. These studies were supported in part by the Manhattan Sickle Cell Center (P60-HL28381) and National Institutes of Health RO1 Grants DK 56638 and HL 69438 (to P.S.F.), HL 19278 (to B.S.C.), and HL 31579 (to N.M.).
Abbreviations
- SS
human sickle hemoglobin
- WT
wild type
- h
human
- m
mouse
- IVM
intravital microscopy monitoring period
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Embury S H, Hebbel R P, Mohandas N, Steinberg M H. Sickle Cell Disease: Basic Principles and Clinical Practice. New York: Raven; 1994. [Google Scholar]
- 2.Hoover R, Rubin R, Wise G, Warren R. Blood. 1979;54:872–876. [PubMed] [Google Scholar]
- 3.Hebbel R P, Yamada O, Moldow C F, Jacob H S, White J G, Eaton J W. J Clin Invest. 1980;65:154–160. doi: 10.1172/JCI109646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wautier J L, Pintigny D, Wautier M P, Paton R C, Galacteros F, Passa P, Caen J P. J Lab Clin Med. 1983;101:911–920. [PubMed] [Google Scholar]
- 5.Mohandas N, Evans E. Blood. 1984;64:282–287. [PubMed] [Google Scholar]
- 6.Wick T M, Moake J L, Udden M M, Eskin S G, Sears D A, McIntire L V. J Clin Invest. 1987;80:905–910. doi: 10.1172/JCI113151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hillery C A, Du M C, Montgomery R R, Scott J P. Blood. 1996;87:4879–4886. [PubMed] [Google Scholar]
- 8.Tait J F, Smith C. J Biol Chem. 1999;274:3048–3054. doi: 10.1074/jbc.274.5.3048. [DOI] [PubMed] [Google Scholar]
- 9.Kaul D K, Tsai H M, Liu X D, Nakada M T, Nagel R L, Coller B S. Blood. 2000;95:368–374. [PubMed] [Google Scholar]
- 10.Matsui N M, Borsig L, Rosen S D, Yaghmai M, Varki A, Embury S H. Blood. 2001;98:1955–1962. doi: 10.1182/blood.v98.6.1955. [DOI] [PubMed] [Google Scholar]
- 11.Mohandas N, Evans E. J Clin Invest. 1985;76:1605–1612. doi: 10.1172/JCI112144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barabino G A, McIntire L V, Eskin S G, Sears D A, Udden M. Prog Clin Biol Res. 1987;240:113–127. [PubMed] [Google Scholar]
- 13.Kaul D K, Fabry M E, Nagel R L. Proc Natl Acad Sci USA. 1989;86:3356–3360. doi: 10.1073/pnas.86.9.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryan T M, Ciavatta D J, Townes T M. Science. 1997;278:873–876. doi: 10.1126/science.278.5339.873. [DOI] [PubMed] [Google Scholar]
- 15.Paszty C, Brion C M, Manci E, Witkowska H E, Stevens M E, Mohandas N, Rubin E M. Science. 1997;278:876–878. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- 16.Trudel M, Saadane N, Garel M C, Bardakdjian-Michau J, Blouquit Y, Guerquin-Kern J L, Rouyer-Fessard P, Vidaud D, Pachnis A, Romeo P H, et al. EMBO J. 1991;10:3157–3165. doi: 10.1002/j.1460-2075.1991.tb04877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffmann-Fezer G, Mysliwietz J, Mortlbauer W, Zeitler H J, Eberle E, Honle U, Thierfelder S. Ann Hematol. 1993;67:81–87. doi: 10.1007/BF01788131. [DOI] [PubMed] [Google Scholar]
- 18.Alter B P, Goff S C. Blood. 1980;56:1100–1105. [PubMed] [Google Scholar]
- 19.Baker M, Wayland H. Microvasc Res. 1974;7:131–143. doi: 10.1016/0026-2862(74)90043-0. [DOI] [PubMed] [Google Scholar]
- 20.Frenette P S, Mayadas T N, Rayburn H, Hynes R O, Wagner D D. Cell. 1996;84:563–574. doi: 10.1016/s0092-8674(00)81032-6. .H. R. [DOI] [PubMed] [Google Scholar]
- 21.Bullard D C, Kunkel E J, Kubo H, Hicks M J, Lorenzo I, Doyle N A, Doerschuk C M, Ley K, Beaudet A L. J Exp Med. 1996;183:2329–2336. doi: 10.1084/jem.183.5.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Butcher E C. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 23.Springer T A. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 24.Lipowsky H H, Zweifach B W. Microvasc Res. 1978;15:93–101. doi: 10.1016/0026-2862(78)90009-2. [DOI] [PubMed] [Google Scholar]
- 25.Kaul D K, Hebbel R P. J Clin Invest. 2000;106:411–420. doi: 10.1172/JCI9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar A, Eckmam J R, Swerlick R A, Wick T M. Blood. 1996;88:4348–4358. [PubMed] [Google Scholar]
- 27.Platt O S, Brambilla D J, Rosse W F, Milner P F, Castro O, Steinberg M H, Klug P P. N Engl J Med. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 28.Charache S, Barton F B, Moore R D, Terrin M L, Steinberg M H, Dover G J, Ballas S K, McMahon R P, Castro O, Orringer E P. Medicine (Baltimore) 1996;75:300–326. doi: 10.1097/00005792-199611000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Vichinsky E P, Neumayr L D, Earles A N, Williams R, Lennette E T, Dean D, Nickerson B, Orringer E, McKie V, Bellevue R, et al. N Engl J Med. 2000;342:1855–1865. doi: 10.1056/NEJM200006223422502. [DOI] [PubMed] [Google Scholar]
- 30.Pieters R C, Rojer R A, Saleh A W, Saleh A E, Duits A J. Lancet. 1995;345:528. [PubMed] [Google Scholar]
- 31.Abboud M, Laver J, Blau C A. Lancet. 1998;351:959. doi: 10.1016/S0140-6736(05)60614-9. [DOI] [PubMed] [Google Scholar]
- 32.Adler B K, Salzman D E, Carabasi M H, Vaughan W P, Reddy V V B, Prchal J T. Blood. 2001;97:3313–3314. doi: 10.1182/blood.v97.10.3313. [DOI] [PubMed] [Google Scholar]
- 33.Grigg A P. Blood. 2001;97:3998–3999. doi: 10.1182/blood.v97.12.3998. [DOI] [PubMed] [Google Scholar]
- 34.Schwartz R S, Tanaka Y, Fidler I J, Chiu D T, Lubin B, Schroit A J. J Clin Invest. 1985;75:1965–1972. doi: 10.1172/JCI111913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hofstra T C, Kalra V K, Meiselman H J, Coates T D. Blood. 1996;87:4440–4447. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.