

Abstract
Lethal factor, the principal virulence factor of Bacillus anthracis, inhibits mitogen-activated protein kinase (MAPK) signaling by proteolytically cleaving MAPK kinases. Edema factor, another component of anthrax toxin, is an adenylate cyclase, which increases intracellular cAMP. Inhibition of MAPK signaling with either anthrax lethal toxin (LeTx) or small molecule MAPK kinase inhibitors triggers apoptosis in human melanoma cells. Normal melanocytes do not undergo apoptosis in response to MAPK inhibition but arrest in the G1 phase of the cell cycle. Importantly, in vivo treatment of human melanoma xenograft tumors in athymic nude mice with LeTx results in significant or complete tumor regression without apparent side effects, suggesting that inhibiting the MAPK signaling pathway may be a useful strategy for treating melanoma. Additionally, interrupting MAPK signaling with LeTx and elevating cAMP with anthrax edema toxin in both melanoma cells and melanocytes lead to dramatic melanin production, perhaps explaining the formation of blackened eschars in cutaneous anthrax.
Mitogen-activated protein kinase (MAPK) pathways are highly conserved among all eukaryotes and are integral cassettes in the transduction of a variety of extracellular signals (1, 2). Constitutive activation of MAPK signaling also contributes to many aspects of human cancers, hence the pathway has been identified as a potential therapeutic target for intervention (3–7). However, cancer cells typically exhibit a cytostatic response to inhibition of MAPK signaling (2, 3, 8–11). Anthrax toxin of Bacillus anthracis consists of three proteins—lethal factor (LF), edema factor (EF), and protective antigen (PA). LF, the principal virulence factor, inhibits MAPK signaling by proteolytically cleaving MAPK kinases (MEKs) (12–15). EF is an adenylate cyclase that increases intracellular cAMP, whereas PA, nontoxic by itself, binds to the cell surface and functions to translocate LF and EF into the cell (16, 17).
We observed that the human melanoma cell lines in the National Cancer Institute's Antineoplastic Drug Screen (18) were especially sensitive to the MEK-directed anthrax lethal toxin (LeTx) and the small molecule MEK inhibitor, PD98059 (9) (Fig. 1). Here we show that inhibition of the MAPK pathway triggers apoptosis in virtually all melanoma cell lines tested in vitro and in vivo. Moreover, inhibition of MEK activity induces melanin production that is significantly enhanced by edema toxin (EdTx) or a small molecule that elevates cAMP.
Figure 1.

Relative sensitivity of the National Cancer Institute's Antineoplastic Drug Screen human tumor cell lines to LeTx and PD98059. The sensitivity pattern is depicted as a Mean Graph (29), which was adapted from the data available in the Developmental Therapeutics Program, National Cancer Institute/National Institutes of Health (http://dtp.nci.nih.gov). Cancer panels (CNS, central nervous system) are indicated, and the melanoma cell lines are marked with red bars. MDA-MB-435 and MDA-N cell lines in the breast cancer panel are indicated with blue bars.
Materials and Methods
Cell Cultures and Inhibitors.
Human melanoma cell lines were obtained from the Developmental Therapeutics Program, National Cancer Institute/National Institutes of Health, and cultured as recommended (18). Neonatal human epidermal melanocytes were obtained from BioWhittaker and cultured and assayed in the recommended Melanocyte Growth Medium 3. Cells were treated with individually purified anthrax toxin components at 0.1 μg/ml each in 10 ml of growth medium for 72 h unless indicated otherwise. Cells were treated with nontoxic PA alone as control, LeTx (PA + LF), or EdTx (PA + EF) (16, 17), and for a combined treatment, the quantity of PA was doubled. PD98059 (Cell Signaling Technology, Beverly, MA) was solubilized in DMSO. Because PD98059 is not sufficiently stable in culture media to maintain inhibition of MEK activation (3), it was directly added to cultures at a final concentration of 20 μM each time at 24-hr intervals for the duration of an experiment. A single treatment of U0126 (Promega) in DMSO at a final concentration of 10 μM was used. A final concentration of 200 μM isobutylmethylxanthine (IBMX) (Calbiochem) in DMSO was used. Controls were treated with an equivalent volume of DMSO.
Cell Cycle, Apoptosis, and Immunoblot Analysis.
Cell cycle analysis by flow cytometry, apoptosis quantification assessed by the nuclear morphological changes, and immunoblot analysis were performed as described (19). Apoptosis in PA-, DMSO-, or EdTx-treated cells was approximately equal to that detected in untreated cells (background apoptosis). The primary antibodies used for immunoblot analysis were anti-phospho-MAPK (Cell Signaling Technology) to detect the activated ERK1 and -2, and anti-ERK1 (K-23) and anti-ERK2 (C-14) (Santa Cruz Biotechnology) to detect total ERK1 and -2.
In Vivo Xenograft Tumor Treatment and Histological Analysis.
Female athymic nude (NCr nu/nu) mice (8 wk old) were injected s.c. on the right upper dorsal area with SK-MEL-28 or M14-MEL cells, or on both right and left upper dorsal areas with MALME-3M cells (1 × 107 cells/mouse) (20, 21). When xenograft tumor growth was established, treatment by direct intratumoral injection was initiated. SK-MEL-28 and M14-MEL tumors were treated with 6 μg of PA + 2 μg of LF/mouse or 6 μg of PA/mouse for control animals at 2-day intervals. On only the right side, tumors in MALME-3M animals were treated with 10 μg of PA + 2 μg of LF/mouse or 10 μg of PA/mouse as control at 2-day intervals. PA and LF were prepared in Hanks' balanced salt solution. Tumor size was evaluated by caliper measurements, and tumor volume was calculated by length × width × depth. For histological examination, tumors were dissected, fixed in 10% neutral-buffered formalin overnight, embedded in paraffin blocks, and cut into thin sections. After deparafinization and rehydration, the sections were stained either with hematoxylin/eosin or for terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL) (22). TUNEL staining was performed by using the In Situ Cell Death Detection POD kit (Roche Molecular Biochemicals) following the manufacturer's recommended protocol, and the sections were counterstained with hematoxylin.
Intracellular Melanin Determination.
Intracellular melanin was solubilized by lysing cells in 0.2 M NaOH and incubating at 60°C for 1 h. A ratio of the absorbance at 405 nm for melanin, relative to that at 280 nm for protein content, was determined. Melanin content in each treatment was expressed as a ratio to the A405/A280 obtained in control cells treated with PA alone, which was set as 1.0. Compared with untreated cells, PA alone did not stimulate melanin production in any of the cell lines tested. To visualize melanin production, 4 × 106 cells/treatment were concentrated into a well of a 96-well plate and photographed.
Results
Apoptosis Triggered by Inhibition of MAPK Signaling in Melanoma Cells.
To study how inhibition of MAPK signaling preferentially targets melanoma cells (Fig. 1), MALME-3M melanoma cells were monitored for 72 h after exposure to LeTx or PD98059. Under these conditions, both LeTx and PD98059 inhibited MAPK (ERK1 and -2) activation (Fig. 2B). Within 24 h of treatment with either LeTx or PD98059, cell cycle progression was delayed in G1 (Fig. 2A) as typically observed in other cell types treated with MEK inhibitors (2, 3, 8–11). Importantly, however, we detected apoptotic cells by 48 h, and within 72 h a significant fraction of the cells were undergoing apoptosis (Fig. 2A). No apoptosis was detected in the untreated or PA- or DMSO-treated control cells (data not shown). We determined the apoptotic sensitivity of eight human melanoma cell lines treated for 72 h with LeTx, PD98059 or a third MEK inhibitor, U0126 (10, 23). Significant apoptosis was detected in most of the treated melanoma cell lines (Table 1, Experiment I). These results show that inhibition of the MAPK pathway induces apoptosis in human melanoma cells and suggest that their sensitivity to LeTx or PD98059 as revealed in National Cancer Institute's Antineoplastic Drug Screen (Fig. 1) is mediated by apoptosis.
Figure 2.

(A) Induction of G1 arrest and apoptosis in MALME-3M melanoma cells by LeTx or PD98059. Response of MALME-3M cells to LeTx (Upper) or PD98059 (Lower) was analyzed by flow cytometry. On the basis of the untreated control (0 h), 2C (G1) and 4C (G2/M) DNA content is indicated. A percentage of apoptosis quantified independently is indicated in each histogram. (B) Active phospho-MAPK (ERK1P and -2P) immunoblot of the cells treated with LeTx or PD98059 for the indicated durations as in A. The cells treated with PA alone or DMSO for 72 h were used for comparison. As loading control, the blot was reprobed with anti-MAPK (ERK1 and -2) antibodies (an 1:1 mixture of anti-ERK1 and anti-ERK2 antibodies). (C) The antagonistic effect of cAMP-elevating EdTx or IBMX on apoptosis triggered by LeTx or PD98059. (D) Active phospho-MAPK and total MAPK immunoblots of the cells treated as in C.
Table 1.
Apoptosis induced by the anthrax toxins and MEK inhibitors PD98059 and U0126 in human melanoma cell lines
| Melanoma cell lines | Experiment I
|
Experiment II
|
|||
|---|---|---|---|---|---|
| Apoptosis ± SD
(%)
|
Apoptosis (%)
|
||||
| LeTx | PD98059 | U0126 | LeTx | LeTx + EdTx | |
| LOX-IMVI* | 49.2 ± 4.5 | 3.6 ± 1.8 | 2.3 ± 1.8 | 47.2 | 20.8 |
| MALME-3M | 55.2 ± 2.6 | 47.9 ± 3.3 | 48.3 ± 10.3 | 31.5 | 1.9 |
| M14-MEL | 51.9 ± 0.9 | 53.0 ± 2.7 | 36.0 ± 8.7 | 37.1 | 42.4 |
| SK-MEL-2 | 14.9 ± 3.0 | 23.4 ± 2.3 | 35.4 ± 4.4 | 19.1 | 8.5 |
| SK-MEL-28 | 8.2 ± 1.4 | 2.5 ± 0.6 | 1.1 ± 0.2 | 8.4 | 0 |
| SK-MEL-5 | 6.7 ± 0.6 | 9.1 ± 1.2 | 1.9 ± 0.5 | 3.7 | 1.5 |
| UACC-257 | 8.6 ± 1.1 | 19.7 ± 4.0 | 4.7 ± 1.2 | 32.1 | 1.0 |
| UACC-62 | 28.1 ± 1.2 | 30.5 ± 1.8 | 8.3 ± 0.9 | 20.4 | 17.3 |
Experiment I. Percentage apoptosis shown is an average of four replicates minus background apoptosis (i.e., PA alone or DMSO). Standard deviation (SD) of the replicate samples is indicated. Experiment II. Apoptosis was quantified from a single sample treated with each toxin.
Elevation of cAMP in the melanoma cell lines appeared to interfere with apoptosis induced by the MEK inhibitors (Fig. 2C). Thus EdTx did not induce apoptosis but markedly interfered with LeTx-induced apoptosis (Fig. 2C), even though MAPK was inactivated (Fig. 2D). IBMX, a phosphodiesterase inhibitor that elevates intracellular cAMP (24), showed the same effect on PD98059-induced apoptosis as EdTx did to LeTx-induced apoptosis (Fig. 2 C and D). We further tested the LeTx and EdTx combination on all eight melanoma cell lines. With the exceptions of M14-MEL and UACC-62 melanoma cell lines, EdTx largely suppressed LeTx-induced apoptosis (Table 1, Experiment II).
Normal Melanocytes Are Resistant to MEK Inhibitor-Induced Apoptosis.
We incubated normal human melanocytes for 72 h with PD98059 (or U0126; data not shown) or 96 h with LeTx (Fig. 3). Although MAPK activation was inhibited, and the cells were growth arrested in G1, no apoptosis was observed (Fig. 3), even with 192 h of continuous LeTX treatment (data not shown). Although the level of MAPK was similar in melanoma cells and in normal melanocytes, the level of activated MAPK was significantly higher in the tumor cells (see the DMSO- and PA-treated cells in Fig. 3 B and D, respectively). These results demonstrate that the activated MEK/MAPK pathway is required for the survival of melanoma cells but not for normal melanocytes.
Figure 3.

Response of normal melanocytes to inhibition of MAPK signaling. Neonatal human epidermal melanocytes were treated either with PD98059 for 72 h (A and B) or with LeTx for 96 h (C and D), during which additional PA and LF (0.1 μg/ml each) were added after 72-h incubation. For comparison, melanoma cells were treated either with PD98059 for 48 h (M14-MEL) or with LeTx for 72 h (MALME-3M). Controls were treated with DMSO (A and B) or PA alone (C and D). (A and C) Cell cycle profiles and apoptosis. (B and D) Active phospho-MAPK and total MAPK immunoblots.
LeTx Induces Regression of Human Melanoma Xenograft Tumors in Nude Mice.
We tested the antitumor activity of LeTx on three different human melanoma xenografts grown in athymic nude mice. SK-MEL-28 melanoma cells were injected into nude mice, and after tumors were established, the animals were treated by direct intratumoral injection of LeTx at 2-day intervals. Control tumors were treated with PA alone. Dramatically, all LeTx-treated SK-MEL-28 tumor xenografts completely regressed, and the animals remained tumor-free for up to 5 wk (Fig. 4A). PA alone had no effect on the tumor growth (Fig. 4A).
Figure 4.

In vivo antitumor effects of LeTX on melanoma xenograft tumors. (A) SK-MEL-28 melanoma xenograft tumors at 310 mm3 (day 27) were intratumorally injected with PA alone (PA, ○) as control or with LeTx (LeTx, ■) (n = 7). Five doses were given at 2-day intervals (days 27–35, ↑) and then an additional dose 3 days later (day 38, ↓). Tumor volume (mm3) is shown with standard deviations. (B) MALME-3M melanoma xenograft tumors were established on both the right and left dorsal upper flank areas of nude mice. When the left-side tumor reached 70 mm3 in size, treatment was initiated by direct intratumoral (IT) injection into the right-side tumors only, with either LeTx (■) or PA alone as control (○) at 2-day intervals (n = 5). When the right-side tumors completely regressed in the LeTx-treated group (day 8), the treatment route was changed to s.c. (SC) injection on the right side of animals (day 10). Individual tumors (small squares) are shown for the left side from day 28 (gray area).
To determine whether the antitumor effect of LeTx is systemic, we used a contralateral xenograft tumor model (21). MALME-3M melanoma xenograft tumors were established on both the right and left upper dorsal areas of nude mice. At 2-day intervals, we injected LeTx intratumorally only into tumors on the right side. While these tumors completely regressed after six doses and remained undetectable throughout the experiment, the tumors on the left side also regressed (Fig. 4B). Moreover, when the experiment was terminated on day 36, two tumors on the left side had completely regressed, whereas three animals relapsed after initial regression (Fig. 4B). PA alone had no effect on tumor growth (Fig. 4B). A total of 19 doses of LeTx were administered over a 5-wk period without obvious adverse effects on the general health or behavior of the animals. As previously demonstrated (21), these results show that antitumor effects of LeTx are systemic. Moreover, the tumor size of the melanoma xenografts directly influenced tumor regression (Table 2), suggesting that the antitumor effects of LeTx are dose-dependent.
Table 2.
Relationship between tumor size and response to LeTx treatment
| Group | Treatment | n | Tumor
volume, mm3
|
Tumor response† | |
|---|---|---|---|---|---|
| Beginning of protocol | End of protocol* | ||||
| I | PA | 4 | 76 ± 11 | 419 ± 110 | Progression |
| LeTx | 4 | 76 ± 10 | 0 | CR | |
| II | PA | 4 | 186 ± 19 | 726 ± 109 | Progression |
| LeTx | 4 | 187 ± 18 | 0 | CR | |
| III | PA | 4 | 278 ± 31 | 853 ± 216 | Progression |
| LeTx | 4 | 278 ± 37 | 0, 0, 40, 66‡ | 90% reduction | |
| IV | PA | 6 | 448 ± 52 | 1,585 ± 263 | Progression |
| LeTx | 6 | 437 ± 131 | 266 ± 74 | 39% reduction | |
M14-MEL melanoma xenograft tumors were allowed to grow until the average tumor size became approximately 76 mm3 (Group I), 187 mm3 (II), 278 mm3 (III), or 443 mm3 (IV). The tumors were treated by direct intratumoral injection with LeTx or PA alone as control at 2-day intervals for 14 days.
Tumor volume 2 days after last treatment.
Tumor response is indicated as percentage of the final tumor volume/initial tumor volume. CR, complete regression.
All tumors in Group III completely regressed with five doses. However, two of the tumors relapsed as soon as treatment was halted. Individual tumor size is indicated.
Histological characterization revealed qualitative differences between the LeTx- and PA-treated tumors. The control tumors treated with PA alone showed hemorrhage and necrosis (Fig. 5 A and B), and tumor cells were sporadically TUNEL positive (Fig. 5C). By contrast, the LeTx-treated SK-MEL-28 tumors remaining after 37 days (Fig. 4A) were devoid of organized cellular structures, contained fragmented nuclei, and were TUNEL positive (Fig. 5 D–F), indicating apoptotic cell death. The left-side MALME-3M tumors and M14-MEL tumors treated with LeTx were all strongly TUNEL positive (data not shown). Therefore, blocking MAPK signaling in melanoma xenograft tumors with LeTx triggers apoptosis in vivo and results in tumor regression. Interestingly, SK-MEL-28 cells that were weakly apoptotic to LeTx in vitro (Table 1) displayed significant sensitivity in vivo (Fig. 4A).
Figure 5.
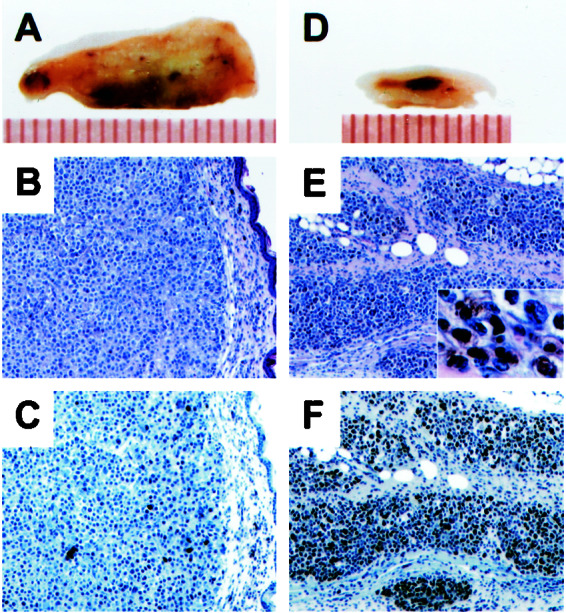
Histological examination of the SK-MEL-28 xenograft tumors treated with PA or LeTx. (A and D) Cross sections (mouse skin to the top) of tumors (a scale in millimeters shown below). (B and E) Hematoxylin/eosin staining of the tumor sections. (E Inset) Melanin deposits in higher magnification. (C and F) TUNEL staining of the adjacent sections (counterstained with hematoxylin).
Cooperation Between LeTx and EdTx Stimulates Melanin Production.
We observed, after treatment with LeTx, that several melanoma cell lines turned black because of melanin production (data not shown). Thus, UACC-257 cells treated with LeTx or PD98059 showed a dramatic increase in melanin production (Fig. 6A). In addition, we found in vitro (Fig. 6A) that EdTx or IBMX significantly enhanced melanin production, whereas EdTx or IBMX did not stimulate melanogenesis de novo (Fig. 6A). Consistent with LeTx inducing melanin production in melanoma cells in vitro, a melanoma tumor xenograft treated with LeTx in vivo and had subsequently regressed (Group III, Table 2) generated a patch of melanin (Fig. 6B) that faded away over time. Moreover, melanin deposits were evident throughout the LeTx-treated melanoma xenografts (Fig. 5E Inset). We conclude that MAPK signaling is a negative regulator of melanin production and that, in the absence of active MAPK, increased cAMP by EdTx or IBMX enhances melanin production. Similarly, PD98059 has been shown to induce melanogenesis and differentiation of B16 mouse melanoma cells (25).
Figure 6.

Melanogenesis induced by anthrax toxins. (A) UACC-257 melanoma cells were treated with PA alone as control, LeTx, EdTx, or LeTX + EdTx, as well as DMSO as control, PD98059, IBMX, or PD98059 + IBMX for 72 h, and intracellular melanin was quantified. Melanin content is expressed as arbitrary ratio units compared with PA control, which is set as 1.0. (Upper) Photographs depict the corresponding cells in the wells of a 96-well plate. (B) A dark patch on a M14-MEL animal (from Table 2).
Discussion
LeTx has been shown to prevent growth in vivo of ras transformed NIH 3T3 cells (ref. 21 and reviewed in ref. 26), but cancer cells in general display a cytostatic response to disruption of MAPK signaling (2, 3, 8–11). We show in this report that inhibition of MAPK signaling with anthrax LeTx or small molecule MEK inhibitors evokes an apoptotic response in human melanoma cells (Fig. 2 and Table 1). Significantly, even cell lines (MDA-MB-435 and MDA-N) thought to be of breast cancer origin but highly sensitive to LeTx or PD98059 (Fig. 1) turn out to be melanoma cells on the basis of cDNA microarray expression analysis (27). Normal melanocytes do not undergo apoptosis but arrest in the G1 phase of the cell cycle in response to MAPK inhibition (Fig. 3). These findings indicate that the MAPK pathway provides an essential survival signal for melanomas. Importantly, inhibition of MAPK signaling in vivo with LeTx also induced apoptosis (Fig. 5), resulting in either complete or significant regression of human melanoma xenograft tumors established in athymic nude mice (Fig. 4 and Table 2). Surprisingly, SK-MEL-28 melanoma cells were more sensitive to LeTx in vivo (Fig. 4A) than in vitro (Table 1), suggesting that different in vitro and in vivo factors significantly influence melanoma cell survival.
Increase in cAMP level interferes with the MEK inhibitor-induced apoptosis, despite MAPK inactivation (Fig. 2 and Table 1). These results suggest that MAPK-mediated survival signaling may be substituted by a cAMP-dependent pathway, perhaps by protein kinase A activation. We also find that along with apoptosis, melanin production is triggered by inactivation of the MAPK pathway in melanoma cells (Fig. 6). However, whereas cAMP increased by EdTx or IBMX blocks apoptosis induced by MAPK inhibition, melanogenesis is enhanced by both pathways (see Figs. 2C and 6A), indicating that at least two different signaling pathways operate downstream of MAPK for these responses. Cutaneous anthrax is characterized by the formation of a blackened eschar in the center of skin lesions, from which anthrax derives its name (from the Greek Anthrakos, coal) (16, 17). Our data suggest that the blackened eschar may result from melanin production in melanocytes stimulated by LeTx inhibiting MAPK activity that is further enhanced by EdTx elevating cAMP.
Our in vivo results demonstrate that LeTx confers a systemic antitumor effect in a dose-dependent manner without obvious side effects on the host animals (Fig. 4B and Table 2). Furthermore, as previously reported (21), it is possible that LeTx targets tumor neovasculature as well, which may be synergistic with the MAPK inhibition-induced apoptosis to cause tumor regression. Collectively, these findings present the interesting prospect that anthrax LeTx may be developed as a therapeutic but for this purpose better referred to as “tumor lethal toxin.” In addition, it is important to point out that LF inactivates most known members of the MEK family (12–15), and we cannot exclude that targeting other family members may also contribute to the antitumor effects of LeTx.
The incidence of melanoma is increasing dramatically in all parts of the world (28). Despite improved early diagnosis, long-term survival from the advanced disease remains clinically challenging. Recent advances in our understanding of the molecular pathophysiology of human cancers present opportunities to develop molecular targets and mechanism-based approaches for the intervention of cancers. Here, our in vitro and in vivo findings that inhibition of MAPK signaling effectively triggers cell death by apoptosis in human melanoma cells, but not in normal melanocytes, offer a potential molecular target-based therapeutic strategy for treating melanomas.
Acknowledgments
We thank J. Bosman, L. Ruff, D. Dylewski, J. Martin, and B. Buckner for technical assistance, C. Miranti and K. Eisenmann for critical comments on the manuscript, and L. Ritsema for preparing the manuscript. A portion of this research was supported in part by the National Cancer Institute, Department of Health and Human Services, under contract with the former Advanced Bioscience Laboratories.
Abbreviations
- MAPK
mitogen-activated protein kinase
- MEK
MAPK kinase
- LF
lethal factor
- EF
edema factor
- PA
protective antigen
- LeTx
lethal toxin
- EdTx
edema toxin
- DMSO
dimethyl sulfoxide
- IBMX
isobutylmethylxanthine
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
References
- 1.Lewis T S, Shapiro P S, Ahn N G. In: Advances in Cancer Research. Vande Woude G F, Klein G, editors. Vol. 74. San Diego: Academic; 1998. pp. 49–139. [DOI] [PubMed] [Google Scholar]
- 2.English J, Pearson G, Wilsbacher J, Swantek J, Karandikar M, Xu S, Cobb M H. Exp Cell Res. 1999;253:255–270. doi: 10.1006/excr.1999.4687. [DOI] [PubMed] [Google Scholar]
- 3.Cohen P. Curr Opin Chem Biol. 1999;3:459–465. doi: 10.1016/S1367-5931(99)80067-2. [DOI] [PubMed] [Google Scholar]
- 4.Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J, Kohno M. Oncogene. 1999;18:813–822. doi: 10.1038/sj.onc.1202367. [DOI] [PubMed] [Google Scholar]
- 5.Mandell J W, Hussaini I M, Zecevic M, Weber M J, VandenBerg S R. Am J Pathol. 1998;153:1411–1423. doi: 10.1016/S0002-9440(10)65728-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sivaraman V S, Wang H-y, Nuovo G J, Malbon C C. J Clin Invest. 1997;99:1478–1483. doi: 10.1172/JCI119309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Licato L L, Brenner D A. Digest Dis Sci. 1998;43:1454–1464. doi: 10.1023/a:1018894227169. [DOI] [PubMed] [Google Scholar]
- 8.Roussel M F. In: Advances in Cancer Research. Vande Woude G F, Klein G, editors. Vol. 74. San Diego: Academic; 1998. pp. 1–24. [DOI] [PubMed] [Google Scholar]
- 9.Dudley D T, Pang L, Decker S J, Bridges A J, Saltiel A R. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukazawa H, Uehara Y. Cancer Res. 2000;60:2104–2107. [PubMed] [Google Scholar]
- 11.Sebolt-Lepold J S, Dudley D T, Herrera R, Van Becelaere K, Wiland A, Gowan R C, Tecle H, Barrett S D, Bridges A, Przybranowski S, et al. Nat Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 12.Duesbery N S, Webb C P, Leppla S H, Gordon V M, Klimpel K R, Copeland T D, Ahn N G, Oskarsson M K, Fukasawa K, Paull K D, et al. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 13.Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, Montecucco C. Biochem Biophys Res Commun. 1998;248:706–711. doi: 10.1006/bbrc.1998.9040. [DOI] [PubMed] [Google Scholar]
- 14.Pellizzari R, Guidi-Rontani C, Vitale G, Mock M, Montecucco C. FEBS Lett. 1999;462:199–204. doi: 10.1016/s0014-5793(99)01502-1. [DOI] [PubMed] [Google Scholar]
- 15.Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Biochem J. 2000;352:739–745. [PMC free article] [PubMed] [Google Scholar]
- 16.Leppla S H. In: Bacterial Toxins and Virulence Factors in Disease. Moss J, Iglewski B, Vaughan M, Tu A T, editors. Vol. 8. New York: Dekker; 1995. pp. 543–572. [Google Scholar]
- 17.Duesbery N S, Vande Woude G F. Cell Mol Life Sci. 1999;55:1599–1609. doi: 10.1007/s000180050399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigro-Wolff A, et al. J Natl Cancer Inst. 1991;83:757–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 19.Koo H-M, Gray-Goodrich M, Kohlhagen G, McWilliams M J, Jeffers M, Vaigro-Wolff A, Alvord W G, Monks A, Paull K D, Pommier Y, et al. J Natl Cancer Inst. 1999;91:236–244. doi: 10.1093/jnci/91.3.236. [DOI] [PubMed] [Google Scholar]
- 20.Plowman J, Dykes D J, Hollingshead M, Simpson-Herren L, Alley M C. In: Anticancer Drug Development Guide. Teicher B, editor. Totowa, NJ: Humana; 1997. pp. 101–125. [Google Scholar]
- 21.Duesbery N S, Resau J, Webb C P, Koochekpour S, Koo H-M, Leppla S H, Vande Woude G F. Proc Natl Acad Sci USA. 2001;98:4089–4094. doi: 10.1073/pnas.061031898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gavrieli Y, Sherman Y, Ben-Sasson S. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Favata M, Koriuchi K Y, Manos E J, Daulerio A J, Stradley D A, Feeser W S, Van Dyk D E, Pitts W J, Earl R A, Hobbs F, et al. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 24.Beavo J, Reifsnyder D. Trends Pharmacol Sci. 1990;11:150–155. doi: 10.1016/0165-6147(90)90066-H. [DOI] [PubMed] [Google Scholar]
- 25.Englaro W, Bertolotto C, Busca R, Brunet A, Pages G, Ortonne J-P, Ballotti R. J Biol Chem. 1998;273:9966–9970. doi: 10.1074/jbc.273.16.9966. [DOI] [PubMed] [Google Scholar]
- 26.Bodart, J. F., Chopra, A., Liang, X. & Duesbery, N. (2002) Cell Cycle, in press. [PubMed]
- 27.Ross D T, Scherf U, Eisen M B, Perou C M, Rees C, Spellman P, Iyer V, Jeffrey S S, Van de Rijn M, Waltham M, et al. Nat Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 28.Parker S, Tong T, Bolden S, Wingo P. CA Cancer J Clin. 1997;47:5–27. doi: 10.3322/canjclin.47.1.5. [DOI] [PubMed] [Google Scholar]
- 29.Paull K D, Hamel E, Malspeis L. In: Cancer Chemotherapeutic Agents. Foye W O, editor. Washington, DC: Am. Chem. Soc.; 1995. pp. 9–45. [Google Scholar]
- 30.Stinson S F, Alley M C, Kopp W C, Fiebig H-H, Mullendore L A, Pittman A F, Kenney S, Keller J, Boyd M R. Anticancer Res. 1992;12:1035–1054. [PubMed] [Google Scholar]