

Summary
MAPK cascades play the critical role in regulating Ras oncogene activity by phosphorylation-dependent mechanisms. Whereas the ERK MAPK pathway is required for Ras transformation, our previous works established that the p38 activity is inhibitory to Ras signaling in both experimental and ras-mutated cancer cells [Chen, G., Hitomi, M., Han, J., and Stacey, D. W. (2000) J. Biol. Chem. 275, 38973–38980; Qi, X., Tang, J., Pramanik, R., Schultz, R. M., Shirasawa, S., Sasazuki, T., Han, J., and Chen, G. (2004) J. Biol. Chem., 279, 22138–22144]. Here we report that K-Ras activates p38γ, a p38 MAPK family member, by inducing its expression without increasing its phosphorylation and depletion of induced p38γ suppresses Ras transformation in rat intestinal epithelial cells. This p38γ activity contrasts with that of its family member p38α, which is activated by Ras through phosphorylation, leading to an inhibition of Ras transformation. Mechanistic analyses show that unphosphorylated p38γ may promote Ras transformation through an increased complex formation with ERK proteins. Significantly, functional p38γ protein is expressed only in K-ras mutated human colon cancer cells, and p38γ transcripts are ubiquitously increased in a set of primary human colon cancer tissues. These studies thus demonstrate the essential role of p38γ in K-Ras transformation independent of phosphorylation and elevated p38γ may serve as a novel diagnostic marker and therapeutic target for human colon cancer.
Introduction
p38 MAPK (mitogen-activated protein kinase) was first identified in studies of endotoxin-induced cytokine expression (1,2). So far, four p38 isoforms have been cloned and characterized, including p38α, p38β, p38γ, and p38δ (3). The p38 upstream activators include MKK3 (MAPK kinase 3) and MKK6 (4). Its downstream effectors consist of kinases such as MK2 (MAPK-activating protein kinase 2) and PRAK (p38-related/activated protein kinase) and transcription factors including ATF-2 (activating transcription factor-2) and MEF2 (myocyte enhancement factor 2) (3). In addition to these effectors, p38 can also signal through cross-talk with JNK (c-Jun NH2-terminal kinase) (5,6) and ERK (extracellular signal-regulated kinase) pathways (7,8). p38 MAPK is mostly responsive to cytokines and inflammatory stress and plays an important role in regulating inflammation and immuno-response (3,9). p38 activation, however, also triggers other biological effects, such as cell death, differentiation, and proliferation by a cell-type specific mechanism (3,10). To date, biological functions of p38 MAPK have mostly been demonstrated by analyzing p38α, and studies of other p38 isoform proteins will contribute to understanding pleiotropic activities of p38 activation.
All MAPKs are activated by dual phosphorylation on threonine and tyrosine residues within a Thr-Xaa-Tyr motif without significant alterations of their expression (11–13). p38γMAPK (also called SAPK3 or ERK6), which shares about 60% identity with p38α and p38β(14,15), has several unique properties. First, in contrast to the ubiquitously expressed p38α, p38γ mRNA is only detectable in normal skeletal muscle (14–16). Recent studies, however, demonstrate that p38γ protein is highly expressed in several human malignant cell lines (17–22), indicating its possible role in tumorigenesis. Secondly, expression levels of p38γ mRNA and/or protein are increased by a differentiation-associated process, an effect distinct from all other MAPKs (14,16,23). Furthermore, elevation of p38γ concentration by transfection, in the absence of upstream activators, induces C2C12 cell differentiation (14). These results together suggest that in addition to phosphorylation-dependent kinase activity (17,20), an increased expression of p38γ may regulate life-important biological processes such as malignant transformation and cell differentiation.
The Ras family of proteins consists of three isoforms (H-, K-, and N-Ras) that play a critical role in controlling normal and malignant cell growth (24). K-ras mutation is one of the most common abnormal genetic events in human cancer, with the highest incidence in pancreatic carcinomas (90%) and colorectal tumors (50%) (25,26). MAPKs (ERK, JNK, and p38) are the best-characterized signal pathways in transduction and regulation of Ras activity (12,27). ERK/MAPK activation has been shown to be both necessary and sufficient in transforming experimental NIH 3T3 cells (28,29), whereas the JNK pathway is also required for Ras transformation (30,31). On the contrary, activation of p38 MAPK is antagonistic to Ras activity, including inhibition of Ras-induced proliferation in NIH 3T3 cells (6), suppression of Ras transformation in RIE cells (30), and induction of K-Ras-dependent cell death in human colon cancer cells (32). In this study, we sought to test the hypothesis that endogenous p38 family members may regulate Ras activity by an isoform-specific mechanism. Our results show that in contrast to p38α, K-Ras activates p38γ by inducing its expression without increasing its phosphorylation, and induced p38γ is required for K-Ras transformation by a mechanism possibly involving a complex formation with ERK proteins.
Materials and Methods
Reagents, Cell lines and cDNA constructs
Cell culture materials were supplied by Gibco and chemicals purchased from Sigma. Fetal bovine serum (FBS) was obtained from BioWhittaker. Protein sepharose G and protein A-sepharose 4B beads were purchased from ZYMED. p38 specific antibodies were described previously (17,18). Their specificity were further confirmed by a rabbit polyclonal antibody from BD Clontech. ERK1 and ERK2 antibodies were from Santa Cruz. Phosphor-p38 and phosphor-ERK antibodies were from Cell Signaling. Rat intestinal epithelial IEC-6, mouse fibroblast NIH 3T3, human colon cancer HCT116, and HT-29 cells were purchased from ATCC and maintained in modified Eagle medium (MEM) containing 10% FBS and antibiotics at 37°C, 5% CO2. Flag-tagged p38 isoform cDNAs in pcDNA3 vector and their dominant negative AGF counterparts were previously described (17,18). The adenovirus vector containing HA-tagged constitutively active MKK6 was used as previously described (17,32). HA-tagged K-Ras and H-Ras cDNAs (both with G12V mutation) were provided by Guthrie cDNA Resource Center and sub-cloned into a retroviral vector LZRS (33). Retroviral vector pLHCX was used to express the wild-type and non-phosphorylated p38γ cDNA as previously described (34). After transfection into Phoenix-Ampo retrovirus packaging cells (ATCC), supernatants were used to infect IEC-6 and NIH 3T3 cells. To establish stable Ras-transformed cell lines, transduced cells were selected with puromycin for about two weeks.
Assays for Ras transformation and cell proliferation
Morphological transformation following retroviral infection was examined under the phase-contrast microscope. For anchorage-independent growth, IEC6/K-Ras cells were either treated with different inhibitors for 24 h or infected with adenovirus (vector and ad-MKK6) for 5 h, or pSUPER siRNA overnight. 2 × 104 cells were plated in growth medium containing 0.33% Sea-plaque-agarose. Formation of multi-cellular colonies was visualized and quantitated about two weeks later (30). The colonies formed on an entire 60-mm plate were photographed and manually counted, and the number of colonies per field shown came from 2 to 3 plates of one experiment and analyzed for statistical significance using student’s t test. Similar results were obtained from at least two additional experiments. To analyze effects of p38γ overexpression on cell proliferation, normal IEC-6 cells were infected with the retrovirus pLHCX, pLHCX-p38γ or pLHCX-p38γ/AGF. The protein expression was assessed by Western blot 48 h and [3H]-thymidine incorporation for DNA synthesis was determined 72 h later, as previously described (35). To assess effects of depleting endogenous p38γ protein on human colon cancer cell proliferation, HCT116 cells were infected with pSR or pSR-siRNA for 72 h. Cells were then pulse-incubated with [3H]-thymidine and DNA synthesis was measured (35).
Tansfection, immunoprecipitation, immunostaining, and Western blot
Cells were transfected with flag-tagged p38 isoforms, with and without K-Ras, and collected in modified RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP40, 0.25% sodium deoxycholate, 1 mM EGTA, 10 mM NaF, 1 mM Na3VO4, 1 mM PMSF and 1 μg/ml aprotinin, Leupeptin, and Pepstain). The flag precipitates were analyzed for p38 isoform protein expression using a flag antibody, for their phosphorylation using a specific p-p38 antibody, and for bound endogenous ERK/p-ERK proteins by Western blot. The endogenous phosphorylated p38 proteins were isolated with a mouse monoclonal p-p38 specific antibody, and precipitates were analyzed for the presence of p38α and p38γ using specific antibodies (17,18). To isolate endogenous p38γ and p38α proteins, lysates were incubated with respective specific antibodies and precipitates were examined for the presence of ERK/p-ERK protein by Western blot. To detect cellular distributions of ERK and p38γ, normal IEC-6 cells were transfected with HA-ERK1 in the presence and absence of flag-p38γ/AGF. Thereafter, cells were fixed and co-stained with mouse anti-flag antibody plus anti-mouse IgG Cy3 to detect transfected flag-p38γ/AGF and with anti-HA-IgG-FITC conjugate to detect transfected HA-ERK1, as previously described (36). For Western Blot analyses, cells were directly lysed in 1X loading buffer and separated on SDS-PAGE. All following procedures were the same as described previously (18,35).
Experiments with siRNA to inhibit p38γ expression
To deplete endogenous p38γ protein, a retroviral vector pSUPER (pSR) (OligoEngine, Cat# VEC-pRT-0002) was used as previously described (37). The target sequence (5′-AAG GAG ATC ATG AAG GTG ACG-3′) was cloned into the pSR vector, which was transfected into the packaging cells to produce the virus-containing supernatants for infection, as we previously described (32). Among four sequences analyzed, this siRNA yields the most substantial effect on p38γ depletion in several cell types and consequently used for all analyses. Typically, cells were analyzed for p38γ protein depletion 72 h after infection.
Northern blot and Gene expression array
For Northern blot, total RNA was prepared by using the Trizol kit. Human p38γ cDNA was used to generate a 520-bp fragment by PCR (primers: FW: 5′-GGC TTT TAC CGC CAG GAG-3′; RE: 5′-GTC ATC TCA CTG TCT GCC TGC CT-3′;). The probe was labeled with [32P] dCTP using the high Prime Kit and purified with Quick Spin Columns (35). A matched tumor/normal expression array kit was purchased from BD Clontech (catalog number 7840-1). The membrane containing the matched cDNA samples was incubated with the [32P] dCTP-labeled p38γ probe according to Manufacturer instructions. The specific radioactivity was measured with a Phosphor-Imager (Amersham Bioscience). Results were measured with Scion Image software and normalized by ubiquitin.
Results
K-Ras induces p38γ protein expression without increasing its phosphorylation in IEC-6 epithelial cells
To analyze signaling interactions between Ras and the p38 family, HA-tagged activated K-Ras and H-Ras cDNAs were sub-cloned into a retroviral vector LZRS (33). After transfection into Phoenix packaging cells, supernatants were used to infect rat intestinal epithelial IEC-6 and mouse fibroblast NIH 3T3 cells, followed by selection with puromycin. We used Western blot to examine these transfected cells for p38 family protein expression and phosphorylation. The specificity of p38 isoform-specific antibodies (17) has been established in our previous publication (18). Results in Fig. 1A show that the predominant form of p38 in vector transfected IEC-6 cells is p38α, with p38γ barely detectable, whereas p38α, p38β, and p38γ (but not p38δ data not shown) proteins are expressed in 3T3 cells. Consistent with our previous finding (6), increased p38 phosphorylation was observed in both cell lines transfected with either K-Ras or H-Ras oncogene, together with increased phosphorylated ERK proteins. Levels of total p38α and ERK1/2 proteins, however, remain relatively consistent with and without Ras transfections (Fig.1A and data not shown). Surprisingly, p38γ protein expression was specifically induced by K-Ras in IEC-6 but not 3T3 cells, suggesting its potential role in K-Ras tumorigenesis in epithelial cells. Furthermore, levels of both p38γ protein (Fig.1A) and p38γ RNA (Fig.1B) were increased in K-Ras- as well as H-Ras-transfected IEC-6 cells, indicating a Ras-isoform independent p38γ induction.
Fig.1. K-Ras selectively induces unphosphorylated p38γ protein expression in epithelial cells.
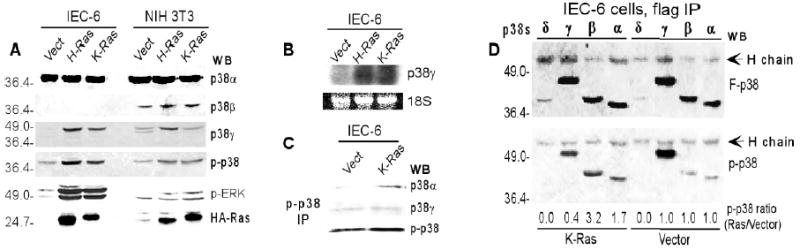
A. K-Ras selectively induces p38γ protein expression in IEC-6 cells. The vector and Ras stably transfected IEC-6 and 3T3 cells were analyzed by Western blot for protein expression and phosphorylation. Similar results were obtained from additional two experiments. B. Ras oncogene induces p38γ RNA expression in IEC-6 cells. C. The phosphorylated p38 in IEC6/K-Ras cells is p38α and not p38γ. Cell lysates were immunoprecipitated with a mouse phospho-p38 antibody, and the precipitates were examined for the presence of p38αand p38γ proteins by Western blot. D. Transient K-Ras expression phosphorylates p38α/β but dephosphorylates p38γ. IEC-6 cells were transiently transfected with flag-tagged p38 isoforms with and without K-Ras, and the flag precipitates were examined for p38 expression and phosphorylation. The p-p38 ratio was calculated by dividing each p-p38 band of the K-Ras transfected group by that of the corresponding vector group after normalization to the respective flag-p38 band.
The phospho-p38 antibody used in the previous analysis reacts with all p38 family members dual-phosphorylated at the Thr and Tyr residues. A single phosphorylated p38 band around 39 kd in IEC-6 cells (Fig.1A) suggests that this phosphor-protein may be p38α since only p38α and p38γ proteins are expressed in these cells, and p38γ (about 45 kd) migrates more slowly than phosphor-p38. To further confirm this speculation, an equal amount of lysates from the vector- and K-Ras transfected-IEC-6 cells was incubated with a mouse phosphor-p38 antibody, and the precipitates were examined for their reactivity with a rabbit antibody against p38α, p38γ, and p-p38 by Western blot. Results in Fig.1C show that the recovery of p38α was greater in K-Ras transfected cells than that in mock-transfection, where no change in p38γ was observed. These results thus demonstrate that K-Ras selectively induces p38α, not p38γ, phosphorylation in IEC-6 cells.
So far, almost all studies of MAPK signaling have focused on regulation of MAPK activity by phosphorylation (12,38). To understand mechanisms by which Ras-induced p38γ protein becomes unphosphorylated, a transient transfection experiment was performed. Normal IEC-6 cells in this case were transfected with flag-tagged p38 isoforms, with and without the K-Ras expressing plasmid. The anti-flag precipitates were analyzed by Western blot for phosphorylation of transfected p38 proteins using a phosphor-specific p38 antibody (Fig.1D). Comparison of the flag-p38 (top) to the phospho-p38 (p-p38) band (bottom) shows that phosphorylated p38α and p38β signals were increased 1.7- and 3.2-fold by K-Ras respectively. Surprisingly, K-Ras did not increase p38γ protein phosphorylation, but, instead, decreased its level by 60% (bottom, Fig.1D). These experiments clearly demonstrate that K-Ras selectively phosphorylates p38α and p38β but dephosphorylates p38γ in these epithelial cells. Inhibition of p38γ phosphorylation by Ras may be due to limited amounts of endogenous upstream kinases to phosphorylate p38γ upon Ras transfection and/or Ras activation of p38γ specific phosphotases, leading to its dephosphorylation. Stimulation of p38α phosphorylation and inhibition of p38γ phosphorylation by transient K-Ras expression provide an explanation for increased p38α phosphorylation and elevated unphosphorylated p38γ protein expression in stably transfected IEC6/K-Ras cells.
p38γ is selectively induced by Ras oncogene and its over-expression does not lead to cell proliferation nor malignant transformation
That Ras induces p38γ protein expression without increasing its phosphorylation is a novel observation. To explore whether this induction is specific to Ras, normal IEC-6 cells were treated with mitogens (serum, TPA) and stresses {arsenite (ARS), tumor necrosis factor-α (TNFα)} and examined for p38γ protein expression by Western blotting. As shown in Fig.2A, p38γ protein level was not increased by these stimuli, although ERK and/or p38/JNK phosphorylation was strongly increased under the same conditions (also no substantial p38γ protein increase 24 h later, data not shown). Interestingly, transient Ras infection led to a substantial p38γ protein elevation (Fig.2B). Thus, p38γ protein expression is selectively induced by Ras oncogene and not by other mitogens, pointing to its potential role in Ras malignant transformation.
Fig.2. p38γ protein is not mitogenic in IEC-6 cells.
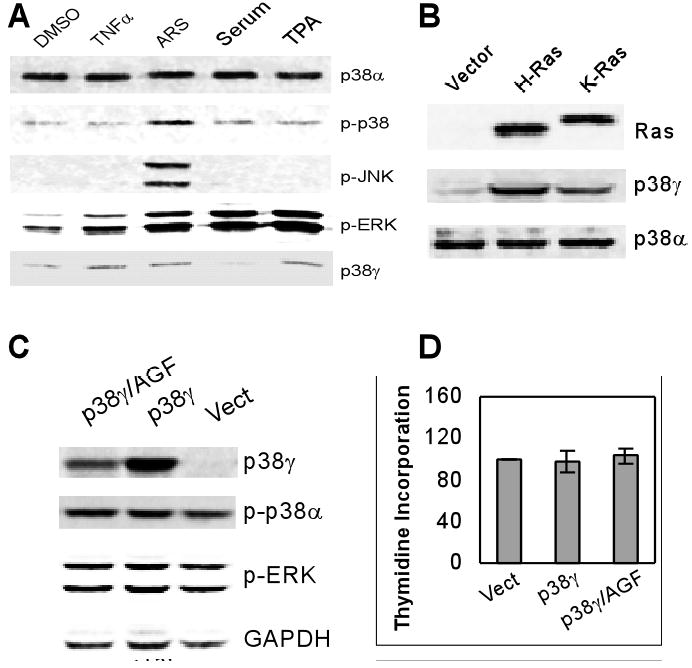
A. Serum and TPA do not induce p38γ protein expression. IEC-6 cells were treated with 100 ng/ml of TNFα, 100 μM of ARS, 20% of serum, or 100 ng/ml of TPA and examined for p38γ protein expression as well as MAPK phosphorylation by Western blot. B. Transient Ras infection induces p38γ protein expression. IEC-6 cells were infected with the retroviral vector LZRS or the Ras containing virus for 48 h and analyzed for p38γ protein expression by Western. C. p38γ over-expression does not affect ERK or p38α phosphorylation. Cells were infected with the pLHCX vector or the vector containing p38γ or p38γ/AGF expressing cDNA and prepared for Western blot 48 h later. D. p38γ over-expression does not increase DNA synthesis in IEC-6 cells. Following the retroviral infection, cells were pulse-labeled with [3H]-thymidine and DNA synthesized was estimated by a Scintillation Counter (35). Results are shown as % of the vector control (means of three separate experiments).
Activated Ras induces cell transformation through its constitutively proliferative signaling. To explore whether p38γ alone is mitogenic or oncogenic by a phosphorylation-dependent mechanism, p38γ and its non-phosphorylated mutant p38γ/AGF were over-expressed in normal IEC-6 cells by a retroviral vector pLHCX (34) and their effects on cell proliferation were assessed by thymidine incorporation. Results in Fig.2C and 2D show that higher levels of p38γ proteins have no significant effects on DNA synthesis (thymidine incorporation). Furthermore, p38γ over-expression did not lead to the soft-agar growth (data not shown). Consistent with this observation, both p38γ and p38γ/AGF did not increase ERK phosphorylation, which is strongly induced by TPA and serum (Fig.2A and 2C). These studies thus reveal that p38γ per se is not mitogenic or oncogenic.
Regulations of K-Ras transformation by PD and SB suggest the required role of p38γ in Ras transformation
Studies from others (30,39) and ours (6,40) have established the required role of ERK phosphorylation and the inhibitory role of p38α (also called p38) phosphorylation in Ras proliferative and transforming activity. Since Ras stimulates ERK/p38α phosphorylation and induces p38γ expression, we sought to explore whether p38γ is involved in their Ras regulatory activities.
To explore potential roles of p38γ in these regulations, stable K-Ras transformed IEC-6 cells (IEC6/K-Ras) were incubated with a specific ERK inhibitor PD98059 (PD) or a p38α/β inhibitor SB203580 (SB), their effects on Ras transformation and p38γ protein expression were examined. PD treatment almost completely reversed the transformed morphology as compared to normal IEC-6 cells, whereas SB increased the transformation (cells became more refractile) (Fig.3A). The morphological alterations were further confirmed by an increased soft-agar formation by SB and a decreased anchorage-independent growth of IEC6/K-Ras cells by PD (Fig.3B). Of great interest, inhibition of ERK phosphorylation by PD couples with a substantial decrease in p38γ protein expression (Fig.3C). These results suggest that the ERK kinase activity, as least as demonstrated with PD, may promote Ras transformation through increasing p38γ expression. In the case of SB, on the other hand, it increases ERK phosphorylation and stimulates p38γ expression (Fig.3C). In addition to suppressing p38α/β activity, SB can also activate c-Raf in cellular systems (41), which may contribute to the increased ERK phosphorylation. Because PD decreases, while SB increases, p38γ protein expression as well as Ras transformation, results from these correlative analyses support the notion that p38γ may be required for Ras malignant transformation.
Fig.3. Regulations of Ras transformation by PD and SB suggest a role of p38γ in Ras transforming activity.
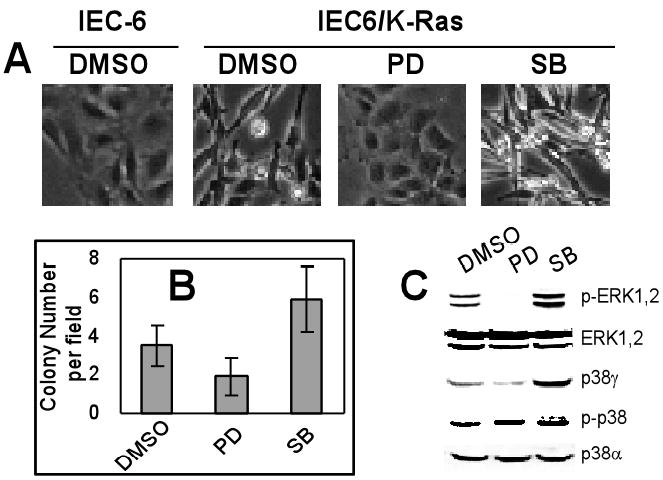
A. Increased and decreased morphological transformation by SB203580 (SB) and PD98058 (PD). Cells were grown in normal medium containing 10 μM of inhibitors or DMSO for 24 h, and their morphology was photographed. B. The opposing roles of SB and PD in regulating anchorage-independent growth of IEC/K-Ras cells. Cells were treated with inhibitors for 24 h and plated for soft-agar growth. Results are the colony number per field (mean ± SD, n=10 from 2 separate plates, with p<0.05 between DMSO and PD, and between DMSO and SB, analyzed with Student t test). Similar results were obtained from two additional experiments. C. PD decreases while SB increases p38γ protein expression. IEC6/K-Ras cells were treated with DMSO or inhibitors for 24 h and analyzed for protein expression and phosphorylation. Similar results were obtained from additional two separate experiments.
Depletion of p38γ protein demonstrates its essential role in K-Ras transformation in IEC-6 cells and in K-Ras-dependent proliferation in human colon cancer cells
To directly prove requirements of p38γ protein in Ras activity, endogenous p38γ protein was depleted from IEC6/K-Ras cells and its effects on Ras transformation were next determined. To silence p38γ protein expression, a pSUPER (pSR) small interference RNA (siRNA) retroviral vector was used (37). As shown in Fig.4A, the p38γ protein decreased by about 80% in comparison to the vector control 72 h after the viral infection. The siRNA-mediated p38γ depletion is specific, as it has no effects on p38α or ERK protein expression. More importantly, p38γ depletion led to a reversion of the morphological transformation, which was reflected by a reduction in cell density and a loss of spindly appearance (Fig.4B). To further confirm the morphological reversion, cells were infected and plated on soft-agar for their anchorage-independent growth. Results in Fig.4C reveal that the colony-forming activity of IEC6/K-Ras cells was substantially inhibited by p38γ protein depletion, which correlates with a decreased DNA synthesis (data not shown). These results therefore directly demonstrate the essential role of p38γ in K-Ras transformation.
Fig.4. Depletion of p38γ protein suppresses K-Ras-induced transformation in IEC-6 cells and inhibits K-Ras dependent proliferation in human colon cancer cells.
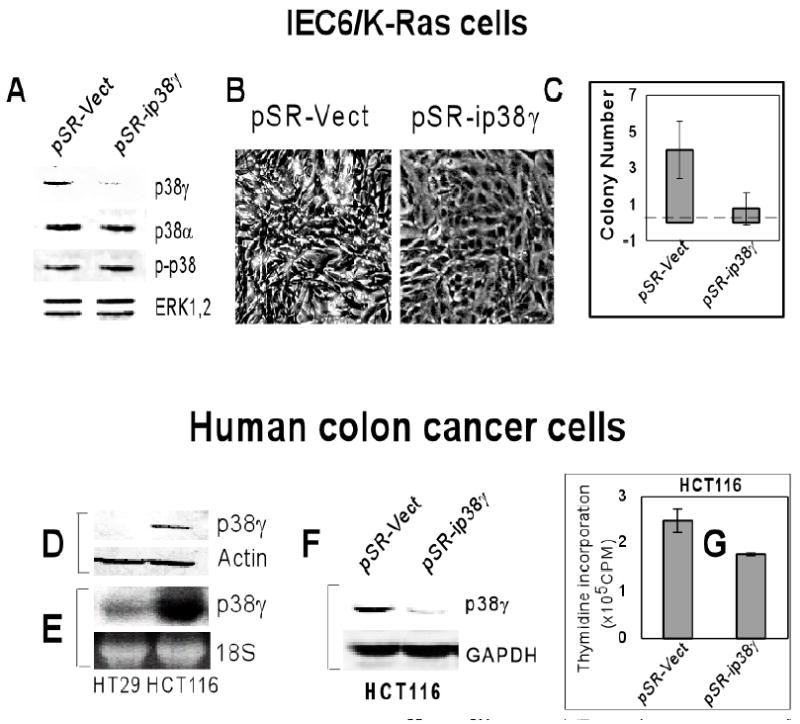
A. Depletion of p38γ protein expression by the siRNA retroviral infection in IEC6/K-Ras cells. Cells were infected with the retroviral vector pSR or the pSR-ip38γ (siRNA) and analyzed for protein expression/phosphorylation by Western blot. Results shown are representative of three separate experiments. B & C. p38γ protein depletion reverses the transformed morphology (B) and inhibits IEC6/K-Ras cell growth on soft agar (C). Cells were infected overnight and plated for soft-agar colony formation (C). For morphological observation, the picture was taken 72 h after infection (B). Results shown in C are means of colony number per field (± SD, n=30 from six plates in three separate experiments, p<0.01). D & E. Levels of p38γ protein (D) and p38γ mRNA (E) are higher in K-ras mutated HCT116 cells than normal K-ras containing HT29 human colon cancer cells. F & G. p38γ depletion inhibits DNA synthesis in HCT116 cells. HCT116 cells were infected with pSR or pSR-ip38γ, and the protein expression was examined 72 h later by Western blot (F). DNA synthesis was measured by thymidine incorporation (G). Results are means of triplicate infections (±SD, p<0.01).
Human colon cancer cells were further utilized to explore the roles of p38γ in natural K-ras mutation- induced tumorigenesis. Consistent with our results in rat IEC-6 cells, there are higher levels of p38γ protein (Fig.4D) and RNA (Fig.4E) in K-ras mutated HCT116 than in wild-type K-ras containing HT-29 human colon cancer cells. Furthermore, depletion of p38γ protein in HCT116 cells significantly inhibited DNA synthesis (Fig.4F and 4G), indicating that p38γ protein is not only expressed in K-ras-mutated human colon cancer cells but also required for K-Ras-dependent malignant proliferation. Because the K-ras gene disruption inhibits HCT116 tumor growth in vitro and in vivo (42), these results suggest that p38γ may be required for K-ras-dependent malignant proliferation in human colon cancers.
p38γ may promote Ras transformation through its complex formation with ERK proteins
Previous studies have shown a physical interaction between p38δ and ERK proteins, which may play a role in regulating keratinocyte differentiation (43,44). Since the ERK activity is essential for Ras transformation (Fig.3), p38 may interact with ERK proteins and thereby regulate Ras transforming activity. To this end, endogenous p38γ and p38α from IEC6/K-Ras cells were isolated with respective antibodies, and the precipitates were examined for the presence of ERK proteins. Results in Fig.5A show that both p38γ and p38α have the ability to bind ERK proteins. To demonstrate whether these bindings are involved in regulating Ras transformation, IEC-6/K-Ras cells were transiently over-expressed with MKK6, a p38 activator, using an adenovirus-mediated gene delivery (35,36). Consistent with previous reports (6,30), MKK6 over-expression completely abolishes the soft-agar growth of the Ras transformed cells (Fig.5D and E). Of great interest, the ERK and/or p-ERK protein in the p38γ complex was significantly diminished, while it remained unchanged (p-ERK undetected, data not shown) in the p38α precipitates, in response to the MKK6 infection (Fig.5B and C). Since MKK6 induces p38α but not p38γ phosphorylation as well as inhibits Ras transformation (Fig.5D-F, and data not shown), these results further established an inhibitory role of phosphorylated p38α. Moreover, MKK6 over-expression does not alter levels of ERK/p-ERK and p38γ proteins but decreases the p38γ-bound ERK/p-ERK protein (Fig.5B and 5F), pointing to a critical role of ERK-p38γ complex formation in Ras transformation. Because the positive correlation has been established between p38γ protein levels and Ras transforming activity (as demonstrated with PD, SB and the siRNA), cellular p38γ protein may promote Ras transformation through its ERK/p-ERK binding activity. Thus, either depleting p38γ protein or disrupting p38γ-ERK binding can inhibit Ras transforming activity.
Fig.5. Roles of p38γ-ERK complex formation in K-Ras transformation.
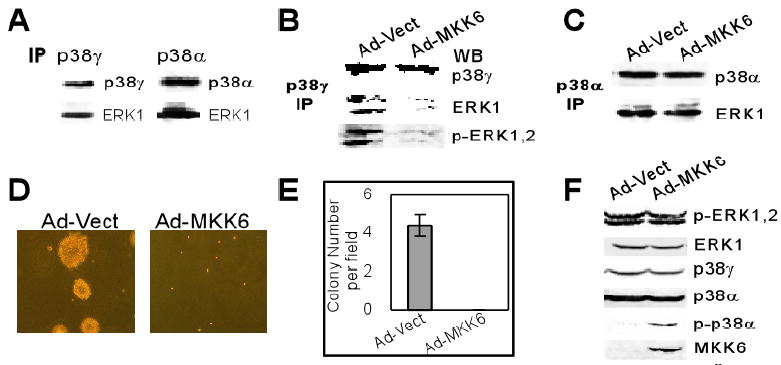
A. Both p38γ and p38α bind to ERK proteins. Lysates of IEC6/K-Ras cells were incubated with p38γ and p38α antibody, and ERK protein in the precipitates was examined by Western blot. B & C. The ERK and/or p-ERK protein in p38γ but not p38α complex is diminished by MKK6. Cells were infected with Ad-Vect or Ad-MKK6 for 48 h and the equal amount of lysates was immunoprecipitated with a p38γ or p38α antibody, followed by Western blot for the presence of ERK/p-ERK protein in the precipitates. Similar results were obtained from one additional experiment. D & E. Inhibition of soft-agar growth of IEC6/K-Ras cells by MKK6. Cells were infected and plated in growth medium containing Sea-plaque-agarose for anchorage-independent growth. Pictures shown (D) were taken about two weeks later. The numbers in (E) are mean of colony numbers from 15 different fields of two separate experiments and there was no single colony formation in MKK6 group. F. Infection with ad-MKK6 phosphorylates p38α without affecting p38γ protein expression. Cells were infected with ad-Vect or ad-MKK6 and analyzed for protein expression/phosphorylation by Western blot. Results are representative from two separate experiments.
Cellular co-localization is a strong indication for protein-protein interaction. ERK proteins are known to be predominantly cytoplasmic in unstimulated cells, which are translocated into the nucleus following activation (45,46). p38γ, on the other hand, has been shown to be both cytoplasmic and nuclear in PC12 cells (47). To explore whether p38γ is localized similarly as ERK protein in IEC-6 cells, HA-tagged ERK1 and flag-tagged p38γ/AGF were co-transfected and their expression detected by fluorescence microscopy as previously described (36). p38γ/AGF over-expression was used to mimic higher levels of induced non-phosphorylated p38γ proteins in IEC6/K-Ras cells. Results in Fig.6 showed that transfected ERK proteins are mostly in the cytoplasm (top panel, left). Although the signal is still strong in cytoplasm after the p38γ co-expression, its substantial portion became nuclear and around the nuclear membrane (Fig.6, top panel, right). Moreover, transfected p38γ/AGF and HA-ERK1 exhibited a similar distribution pattern. These results thus further confirm a physical interaction between p38γ and ERK proteins and indicate that p38γ may act as a Ras effector through increasing nuclear ERK accumulation.
Fig.6. Localization of ERK and p38γ proteins in IEC-6 cells.
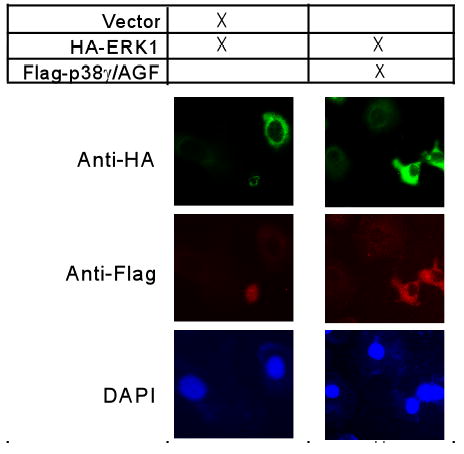
Normal IEC-6 cells were transfected with HA-ERK1 in the presence and absence of flag-p38γ/AGF. Cells were fixed and co-immunostained for transfected ERK1 using anti-HA antibody and for transfected p38γ using anti-flag antibody as described in Materials and Methods.
Our results in Fig.1 show that in IEC6/K-Ras cells endogenous p38α is phosphorylated, whereas p38γ remains in an unphosphorylated form. If p38γ-ERK complex plays a role in Ras transformation, the ERK binding activity of p38γ should be increased when it becomes unphosphorylated. To explore this possibility, normal IEC-6 cells were transiently transfected with flag-tagged wild-type p38γ and its non-phosphorylated AGF mutant in the absence and presence of K-Ras by including p38α for comparison. Anti-flag precipitates were examined for the presence of endogenous ERK proteins. Results in Fig.7 show that transfected p38γ and p38α bind to endogenous ERK proteins independent of their phosphorylation status and independent of K-Ras transfection. Strikingly, the p38γ/AGF binds much more ERK/p-ERK proteins following Ras transfection (lane 4, left panel), an event not observed with the wild-type p38γ transfection. This effect is opposite to p38α, as ERK proteins are only phosphorylated in the wild-type p38α-complex and in the absence of K-Ras, indicating a possibility that p-ERK protein may be relocated from the p38α to the unphosphorylated p38γ complex in response to Ras activation. These results together suggest a scenario where p38γ expression is induced while its phosphorylation is concomitantly inhibited by Ras oncogene, and induced unphosphorylated p38γ may promote Ras transformation through ERK binding, leading to increased/sustained ERK phosphorylation.
Fig.7. Increased ERK binding by non-phosphorylated p38γ in response to transient K-Ras expression.
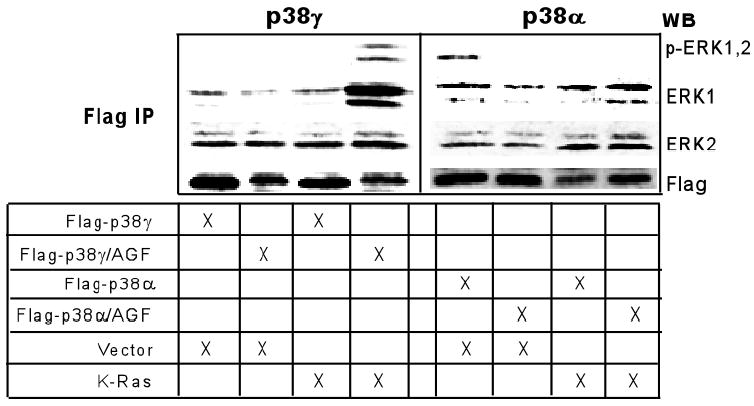
Normal IEC-6 cells were transiently transfected with flag-tagged wild-type or the mutant p38s (AGF) in the absence or presence of K-Ras as indicated. Flag precipitates were analyzed for the presence of endogenous ERK/p-ERK proteins. Similar results were obtained from a separate experiment.
p38γ transcripts are elevated in primary human colon cancer tissues
To further explore the role of p38γ in human cancer development, we used a matched tumor/normal expression array to examine p38γ gene expression. In this array, cDNA samples representing 11 different tissues of 68 cancer patients were immobilized to a nylon membrane. From each patient, a pair of cDNA samples was derived from the tumor and corresponding normal tissue. After hybridization with a radiolabeled p38γ probe, the membrane was scanned with a phosphor-Imager for specific binding. As shown in Fig.8A, p38γ transcript was detected in every tissue examined. Most strikingly, 100% of colon cancer patients (total 11) showed higher levels of p38γ signal in tumor tissues than in matched normal tissues (underlined). Furthermore, levels of p38γ mRNA were also higher in most breast cancer tissues (8/9), a result similar to that previously reported about the hyper-expressed ERK MAPK in human breast cancer tissues (48). Expression levels of a housekeeping gene, ubiquitin, however, were similar between normal and tumorous tissues (Fig.8B). The p38γ fold-increase in human colon cancers varied from 1.3 to 4.0 after normalization to ubiquitin, with an average 2.21 ± 0.79 fold (SD, p<0.01) (Fig.8C). Since K-ras mutation is frequently observed in human colon cancers (25) and p38γ expression is increased in K-ras mutated HCT116 cells (Fig.4), these results strongly suggest that elevated p38γ is likely to play a role in K-Ras-induced human colon tumorigenesis.
Fig.8. Roles of p38γ in primary human colon cancer.
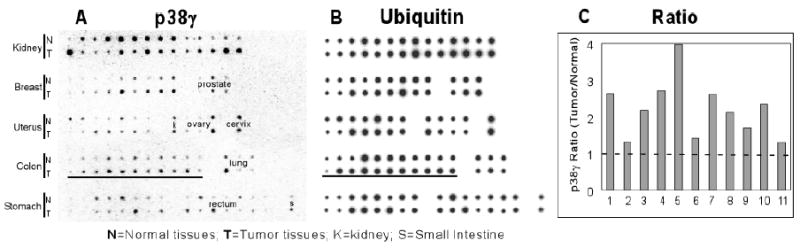
A. p38γ transcripts are increased in primary human colon cancer tissues as demonstrated by the Matched Tumor/Normal expression array. A nylon membrane containing cDNA samples was incubated with a human p38γ cDNA probe, and the radioactivity was measured by a phosphor-image. B. Expression of the housekeeping gene ubiquitin as a loading control. C. The p38γ mRNA ratio (tumor/normal) in colon tissues after normalization with ubiquitin (calculated from A and B).
Discussion
Demonstrating the essential role of p38γ in K-Ras transformation independent of phosphorylation will greatly impact our understanding of MAPK signaling. These results suggest that in addition to phosphorylation-dependent effects, stress p38γ MAPK can execute phosphorylation-independent functions as a Ras effector. A phosphorylation-dependent activity of p38γ may be primarily regulating stress response through transient activation and inactivation (17,20). In response to Ras oncogene, however, p38γ expression is induced, while its phosphorylation is inhibited, and elevated p38γ proteins consequently play an essential role in maintaining Ras transformed phenotype (Fig.9). Although increased p38γ RNA in K-Ras-transformed and -mutated cells suggests a trans-activation mechanism, we cannot rule out the involvement of Ras-induced p38γ dephosphorylation in elevating its protein concentration. The phosphorylation-independent activity of p38γ is consistent with our previous observation that over-expression of both wild-type and non-phosphorable p38γ showed a similar regulatory effect on gene expression (18). Thus, p38γ MAPK has dual activity: it serves as a kinase to regulate stress response by a phosphorylation-dependent mechanism and acts as a Ras effector to promote Ras transformation through increased expression without phosphorylation.
Fig.9. An experimental model shows a requirement of p38γfor K-Ras transformation as opposite to the inhibitory activity of phosphorylated p38α.
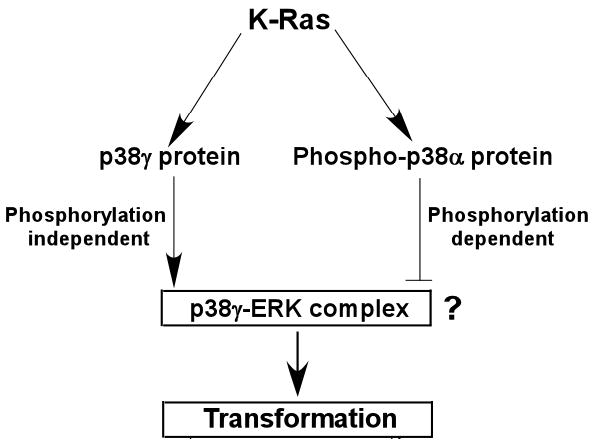
K-Ras activates p38γ by increasing its expression without phosphorylation but stimulates p38α by phosphorylation. Increased non-phosphorylated p38γ protein promotes Ras transformation, whereas induced phosphorylated p38α inhibits Ras transforming activity. Experiments with several agents suggest the critical role of p38γ-ERK complex formation in p38γ promoting Ras transformation, albeit this mechanism remains to be further established (?). This model suggests that Ras transforming activity in a given system will be determined by the signaling integration of p38 family members.
Previous studies have shown that p38 family members can either collaborate or oppose each other in regulating gene expression in response to various signaling. Hypoxia, for example, induces p38α/γ phosphorylation in PC12 cells, both of which inhibit cyclin D1 expression (20). Our previous work further showed that MKK6 and arsenite stimulate phosphorylations of all four p38 family members in human breast cancer cells, in which p38β increases but p38γ/δ inhibits or has no effects on AP-1 dependent gene expression (18). A similar opposing effect of p38 isoforms was also demonstrated in stress regulating hemo-oxygenase-1 expression (49). All of these analyses, however, were carried out by p38 over-expressions and physiological relevance of these observations has not consequently been established. Our present studies, on the other hand, demonstrate that K-Ras stimulates endogenous p38α phosphorylation while inducing endogenous p38γ expression. Furthermore, experiments with SB to inhibit, and with MKK6 to stimulate, p38α phosphorylation, reveal its Ras inhibitory role dependent of phosphorylation, whereas the p38γ depletion analyses show its requirement for Ras transformation independent of phosphorylation (Fig.9). These results indicate that the transforming activity of Ras oncogene in a given system will be determined by the signaling integration among endogenous p38 family members. A higher ratio of unphosphorylated p38γ proteins over phosphorylated p38α proteins would favor Ras transforming activity and vice versa.
It is unlikely that p38γ promotes Ras transformation through its intrinsic mitogenic activity. This is because p38γ expression is not induced by other mitogens and p38γ over-expression does not increase DNA synthesis nor lead to transformation. Our results do suggest, on the other hand, that a complex formation between p38γ and ERK proteins may play a role in p38γ maintaining Ras transformed phenotype (Fig.9). This is indicated by reduced ERK/p-ERK proteins in the p38γ but not p38α complex following MKK6-induced inhibition of Ras-dependent growth and by increased ERK/pERK binding through the p38γ/AGF over-expression in response to Ras signaling. This conclusion, however, remains to be further proven by directly demonstrating the Ras-transformation inhibitory activity of ERK-binding deficient p38γ mutants. Since both p38γ and p38α bind to ERK proteins, and only p38γ/AGF has an increased affinity to ERKs in the presence of Ras, future analyses should focus on the structural differences between p38α and p38γ proteins as well as their relationships with phosphorylation on the kinase subdomain.
p38γ contains a PDZ-domain binding motif (ETPL) in its C-terminus, which is absent in p38α protein (50). This motif has been shown to be important for its sub-cellular localizations and/or its interactions with other PDZ-domain containing proteins to maintain certain structures of cytoskeleton, a process that is important for Ras transformation (51,52). Moreover, these interactions are also regulated by protein phosphorylations and dephosphorylations (51,53). Since ERK protein requires a complex formation with other proteins for its activities such as nuclear localizations and induced epithelial morphogenesis (54,55), p38γ may promote Ras transformation through its C-terminal mediated ERK binding by a scaffold-like mechanism.
Human colorectal cancer is the second leading cause of cancer death in the United States, to which K-ras mutation is the most established contributing factor (56). While various approaches have been tested to inhibit Ras oncogene activity (57,58), effective therapeutics to selectively inhibit activated ras oncogene in human cancer remains to be established (59). This slow progress is mainly due to lack of specific Ras oncogene effectors, since many of those are shared by normal cellular Ras proteins in response to mitogenic signaling (26). p38γ, however, appears to be a specific Ras oncogene effector because it is not expressed in normal cells/tissues (3) and is induced selectively by Ras oncogene and not by other mitogens. Most significantly, our results further show that p38γ gene expression is elevated ubiquitously in a set of primary human colon cancer tissues over matched normal tissues. Thus, p38γ may serve as a novel diagnostic marker and therapeutic target for human colon cancer.
Acknowledgments
We would like to thank Drs T. Patel, Z. Luo, A. Lin, and K-L. Guan for useful discussions, Susie Chen, Linda. Qi for critically reading the manuscript, and Hines VA Medical Center for facility support.
Footnotes
This work was supported by a grant from NIH (CA91576 to G.C.).
References
- 1.Han J, Lee JD, Bibbs L, Ulevitch RJ. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 2.Lee JC, Laydon JT, McDonnel PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 3.Ono K, Han J. Cell Sign. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 4.Wang L, Ma R, Flavell R, Choi ME. J Biol Chem. 2002;277:42257–47262. doi: 10.1074/jbc.M208573200. [DOI] [PubMed] [Google Scholar]
- 5.Nemoto S, Sheng Z, Lin A. Mol Cell Biol. 1998;18:3518–3526. doi: 10.1128/mcb.18.6.3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen G, Hitomi M, Han J, Stacey DW. J Biol Chem. 2000;275:38973–38980. doi: 10.1074/jbc.M002856200. [DOI] [PubMed] [Google Scholar]
- 7.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 8.Oh C, Chang S, Yoon Y, Lee S, Lee Y, Kang S, Chun J. J Biol Chem. 2000;275:5613–5619. doi: 10.1074/jbc.275.8.5613. [DOI] [PubMed] [Google Scholar]
- 9.Nebreda AR, Porras A. TIBS. 2000;25:257–260. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- 10.Whitmarsh AJ, Davis RJ. Nature. 2000;403:255–256. doi: 10.1038/35002220. [DOI] [PubMed] [Google Scholar]
- 11.Robinson MJ, Cobb MH. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 12.Chang L, Karin M. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 13.Weston CR, Lambright DG, Davis RJ. Science. 2002;296:2346–2347. doi: 10.1126/science.1073344. [DOI] [PubMed] [Google Scholar]
- 14.Lechner C, Zahalka MA, Giot J, Moller NP, Ullrich A. Pro Natl Acad Sci. 1996;93:4355–4359. doi: 10.1073/pnas.93.9.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Z, Jiang Y, Ulevitch RJ, Han J. Biochem Biophys Res Commun. 1996;228:334–340. doi: 10.1006/bbrc.1996.1662. [DOI] [PubMed] [Google Scholar]
- 16.Tortorella LL, Lin CB, Pilch PF. Biochem Biophy Res Comm. 2003;306:163–168. doi: 10.1016/s0006-291x(03)00936-7. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, McGowan CH, Zhao M, He L, Downey JS, Fearns C, Wang Y, Huang S, Han J. Mol Cell Biol. 2000;20:4543–4552. doi: 10.1128/mcb.20.13.4543-4552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pramanik R, Qi X, Borowicz S, Choubey D, Schultz RM, Han J, Chen G. J Biol Chem. 2003;278:4831–4839. doi: 10.1074/jbc.M207732200. [DOI] [PubMed] [Google Scholar]
- 19.Abdollahi T, Robertson NM, Abdollahi A, Litwack G. Cancer Res. 2003;63:4521–4526. [PubMed] [Google Scholar]
- 20.Conrad PW, Rust RT, Han J, Millhorn DE, Beitner-Johnson D. J Biol Chem. 1999;274:23570–23576. doi: 10.1074/jbc.274.33.23570. [DOI] [PubMed] [Google Scholar]
- 21.Simon C, Simon M, Vucelic G, Hicks MJ, Plinkert PK, Koitchev A, Zenner HP. Exp Cell Res. 2001;271:344–355. doi: 10.1006/excr.2001.5374. [DOI] [PubMed] [Google Scholar]
- 22.Pillaire M, Nebreda AR, Darbon J. Biochem Biophy Res Comm. 2000;278:724–728. doi: 10.1006/bbrc.2000.3877. [DOI] [PubMed] [Google Scholar]
- 23.Cuenda A, Cohen P. J Biol Chem. 1999;274:4341–4346. doi: 10.1074/jbc.274.7.4341. [DOI] [PubMed] [Google Scholar]
- 24.Boguski MS, McCormick F. Science. 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- 25.Bos JL, Fearon ER, Hamilton SR, Verlaan–de Vries M, van Boom JH, van der Eb AJ, Vogelstein B. Nature. 1987;327:293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- 26.Downward J. Nature Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 27.Vojtek AB, Der CJ. J Biol Chem. 1998;273:19925–19928. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- 28.Mansour S, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 29.Cowley S, Paterson H, Kemp P, Marshall CJ. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 30.Pruitt K, Pruitt WM, Bilter GK, Westwick JK, Der CJ. J Biol Chem. 2002;277:31808–31817. doi: 10.1074/jbc.M203964200. [DOI] [PubMed] [Google Scholar]
- 31.Johnson R, Spiegelman B, Hanahan D, Wisdom R. Mol Cell Biol. 1996;16:4504–4511. doi: 10.1128/mcb.16.8.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qi X, Tang J, Pramanik R, Schultz RM, Shirasawa S, Sasazuki T, Han J, Chen G. J Biol Chem. 2004;279:22138–22144. doi: 10.1074/jbc.M313964200. [DOI] [PubMed] [Google Scholar]
- 33.Tang J, Gordon GM, Muller MG, Dahiya M, Foreman KE. J Virol. 2003;77:5975–5984. doi: 10.1128/JVI.77.10.5975-5984.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayakawa J, Depatie C, Ohmichi M, Mercola D. J Biol Chem. 2003;278:20582–20592. doi: 10.1074/jbc.M210992200. [DOI] [PubMed] [Google Scholar]
- 35.Qi X, Pramank R, Wang J, Schultz RM, Maitra RK, Han J, DeLuca HF, Chen G. J Biol Chem. 2002;277:25884–25892. doi: 10.1074/jbc.M203039200. [DOI] [PubMed] [Google Scholar]
- 36.Qi X, Borowicz S, Pramanik R, Schultz RM, Han J, Chen G. J Biol Chem. 2004;279:6769–6777. doi: 10.1074/jbc.M311492200. [DOI] [PubMed] [Google Scholar]
- 37.Brummelkamp TR, Bernards R, Agami R. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 38.Hazzalin CA, Mahadevan LC. Nature Reviews. 2002;3:30–40. doi: 10.1038/nrm715. [DOI] [PubMed] [Google Scholar]
- 39.Sheng H, Shao J, DuBois R. J Biol Chem. 2001;276:14498–14504. doi: 10.1074/jbc.M010093200. [DOI] [PubMed] [Google Scholar]
- 40.Chen G, Templeton D, Suttle DP, Stacey D. Oncogene. 1999;18:7149–7160. doi: 10.1038/sj.onc.1203149. [DOI] [PubMed] [Google Scholar]
- 41.Kalmes A, Deou J, Clowes AW, Daum G. FEBS Let. 1999;444:71–74. doi: 10.1016/s0014-5793(99)00034-4. [DOI] [PubMed] [Google Scholar]
- 42.Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Science. 1993;260:85–88. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- 43.Efimova T, Broome A, Eckert RL. J Biol Chem. 2003;278:34277–34285. doi: 10.1074/jbc.M302759200. [DOI] [PubMed] [Google Scholar]
- 44.Efimova T, Broome A, Eckert RL. Mol Cell Biol. 2004;24:8167–8183. doi: 10.1128/MCB.24.18.8167-8183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson M, Stippec SA, Goldsmith E, White MA, Cobb MH. Curr Biol. 1998;8:1141–1150. doi: 10.1016/s0960-9822(07)00485-x. [DOI] [PubMed] [Google Scholar]
- 46.Wolf IW, Rubinfeld H, Yoon S, Marmor G, Hanoch T, Seger R. J Biol Chem. 2001;276:24490–24497. doi: 10.1074/jbc.M103352200. [DOI] [PubMed] [Google Scholar]
- 47.Sabio G, Reuver S, Feijoo C, Hasegawa M, Thomas GM, Centeno F, Kuhlendahl S, Leal–Ortiz S, Goedert M, Garner C, Cuenda A. Biochem J. 2004;380:19–30. doi: 10.1042/BJ20031628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sivaraman VS, Wang H, Nuovo GJ, Malbon CC. J Clin Invest. 1997;99(7):1478–1483. doi: 10.1172/JCI119309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kietzmann T, Samoylenko A, Immenschuh S. J Biol Chem. 2003;278:17927–17936. doi: 10.1074/jbc.M203929200. [DOI] [PubMed] [Google Scholar]
- 50.Hasegawa M, Cuenda A, Spillantini MG, Thomas GM, Buee-Scherrer V, Cohen P, Goedert M. J Biol Chem. 1999;274:12626–12631. doi: 10.1074/jbc.274.18.12626. [DOI] [PubMed] [Google Scholar]
- 51.Radziwill G, Erdmann RA, Margelisch U, Moelling K. Mol Cell Biol. 2003;23:4663–4672. doi: 10.1128/MCB.23.13.4663-4672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowititz M, Hisaminato A, Fujiwara T, to Y, Cantely LC, Yaffe MB. EMBO J. 2000;19:6778–6791. doi: 10.1093/emboj/19.24.6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Das S, Dixon JE, Cho W. Pro Natl Acad Sci USA. 2003;100:7491–7496. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chou F, Hill JM, Hsieh J, Pouyssegur J, Brunet A, Glading A, Uberall F, Ramos JW, Werner MH, Ginsberg MH. J Biol Chem. 2003;278:52587–52597. doi: 10.1074/jbc.M309322200. [DOI] [PubMed] [Google Scholar]
- 55.Ishibe S, Joly D, Liu Z, Cantley LG. Mol Cell. 2004;16:257–267. doi: 10.1016/j.molcel.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 56.Lamprecht SA, Lipkin M. Nature Rev Cancer. 2003;3:601–614. doi: 10.1038/nrc1144. [DOI] [PubMed] [Google Scholar]
- 57.Koller E, Gaarde WA, Monia BP. Trends in Pharmacological Sciences (TIPS) 2000;21:142–148. doi: 10.1016/s0165-6147(00)01448-6. [DOI] [PubMed] [Google Scholar]
- 58.Sebolt-Leopold J, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S, Leopold WR, Sattiel AR. Nat Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 59.Sebti SM, Der CJ. Nature Rev Cancer. 2003;3:945–951. doi: 10.1038/nrc1234. [DOI] [PubMed] [Google Scholar]