

Abstract
Autism is a common neurodevelopmental disorder of complex genetic etiology. Rett syndrome, an X-linked dominant disorder caused by MECP2 mutations, and Angelman syndrome, an imprinted disorder caused by maternal 15q11–q13 or UBE3A deficiency, have phenotypic and genetic overlap with autism. MECP2 encodes methyl-CpG-binding protein 2 that acts as a transcriptional repressor for methylated gene constructs but is surprisingly not required for maintaining imprinted gene expression. Here, we test the hypothesis that MECP2 deficiency may affect the level of expression of UBE3A and neighboring autism candidate gene GABRB3 without necessarily affecting imprinted expression. Multiple quantitative methods were used including automated quantitation of immunofluorescence and in situ hybridization by laser scanning cytometry on tissue microarrays, immunoblot and TaqMan PCR. The results demonstrated significant defects in UBE3A/E6AP expression in two different Mecp2 deficient mouse strains and human Rett, Angelman and autism brains compared with controls. Although no difference was observed in the allelic expression of several imprinted transcripts in Mecp2-null brain, Ube3a sense expression was significantly reduced, consistent with the decrease in protein. A non-imprinted gene from 15q11–q13, GABRB3, encoding the β3 subunit of the GABAA receptor, also showed significantly reduced expression in multiple Rett, Angelman and autism brain samples, and Mecp2 deficient mice by quantitative immunoblot. These results suggest an overlapping pathway of gene dysregulation within 15q11–q13 in Rett, Angelman and autism and implicate MeCP2 in the regulation of UBE3A and GABRB3 expressions in the postnatal mammalian brain.
INTRODUCTION
Autism is a complex genetic neurodevelopmental disorder characterized by severe impairments in social interaction, communication and behavioral patterns that are restrictive and stereotypical (1). Rett syndrome (RTT; OMIM 312750) is an X-linked dominant neurodevelopmental disorder caused by mutations in MECP2, which encodes methyl-CpG-binding protein 2 (MeCP2) (2). Angelman syndrome (AS; OMIM 105830) is an imprinted disorder caused by maternal deficiency of chromosome 15q11–q13, methylation defects or maternal mutation of UBE3A encoding the ubiquitin ligase UBE3A/E6-AP (3). RTT and AS share overlapping clinical features with autism including developmental delay, language impairment, seizures and stereotypic behaviors (4). Furthermore, clinical assessments of social behavior have demonstrated a high frequency of autism in patients with RTT (5) and AS (6). A Mecp2 mutant mouse model of RTT also shows abnormalities in social interactions (7). In addition, MECP2 mutations have been found in a few patients diagnosed with AS (8) and autism (9,10) and 15q11–q13 duplications are present in ~1% of autism cases (11), suggesting overlap in the pathogenesis of these distinct genetic syndromes. GABRB3, encoding the GABAA receptor β subunit, is located within the 15q11–q13 (12) and shows association with autism (13,14) but is biallelically expressed (15). We have previously demonstrated MeCP2 expression defects in autism and AS brain samples by a quantitive immunofluorescence approach (16). In contrast, expression level of proteins encoded by genes within 15q11–q13 in RTT, AS and autism have not been well characterized.
MeCP2 binds to methylated CpG sites (17), represses transcription of methylated constructs (18), and associates with chromatin modifying factors histone deacetylase (19,20) and DNA methyltransferase (21,22). Mecp2-null (23,24) or mutant (25) mice recapitulate the phenotype of RTT and demonstrate that MeCP2 is essential for postnatal mammalian brain development. Although MeCP2 was predicted to be a global transcriptional repressor, a paucity of MeCP2 target genes have been identified in Mecp2-null mouse brain (26), RTT brain (27) or cell lines (28) by gene expression profiling. The gene encoding brain derived neurotropic factor has been identified as a target of MeCP2 transcriptional repression where a low basal level of transcription prior to neuronal stimulation was increased in Mecp2-null neurons (29,30). The imprinted gene H19 also showed increased expression in Mecp2-null cells (21), but it was not previously determined whether the increase was due to de-repression of the methylated paternal allele. Because of the limited number of MeCP2 target genes, the role of MeCP2 in the pathogenesis of RTT and other autism-spectrum disorders remains elusive.
Although MECP2 mutation does not affect imprinted expression of several genes within 15q11–q13 (31), we hypothesized that MeCP2 may regulate the expression level of genes in this region without necessarily affecting allele-specific expression. Here, we demonstrate that Mecp2 deficiency results in significant reduction of UBE3A/Ube3a and GABRB3/Gabrb3 expressions in mouse cerebrum without apparent alterations in allele-specific expression. Furthermore, significant reduction of UBE3A and GABRB3 expressions was observed in AS, RTT and autism human cerebral samples compared with controls. These results demonstrate overlapping epigenetic defects in these phenotypically similar but genetically distinct neurodevelopmental disorders and implicate MeCP2 in the regulation of gene expression within 15q11–q13.
RESULTS
Reduced UBE3A expression in Mecp2 deficient mice
As a non-cell-autonomous dominant effect of Mecp2 mutation in the wild-type (wt)-expressing cells of mosaic Mecp2−/+ females has been described (32), we performed single cell quantitative analyses of mutant (mt)-expressing (MeCP2-negative) and wt-expressing (MeCP2-positive) cells for UBE3A immunofluorescence on a tissue microarray by laser scanning cytometry (LSC). This approach demonstrated a reproducible 1.5–2-fold decrease in UBE3A immunoreactivity in 10 Mecp2−/+ and two Mecp2−/y cerebral samples from two different Mecp2-null strains (23,24) compared with wt controls (Fig. 1A and B). Both mt-expressing cells from Mecp2−/+ and Mecp2−/y (blue histograms) and wt-expressing cells from Mecp2−/+ cerebrum (red histograms) showed significantly lower UBE3A expression compared with wt-expressing cells from Mecp2−/+ cerebrum (orange histograms). To demonstrate that the defects observed in Mecp2−/+ and Mecp2−/y brain samples were actually due to expression differences in UBE3A, not immunofluorescence detection, immunoblot analyses were performed (Fig. 1C). Reduced UBE3A/E6-AP expression was observed by immunoblot for both Mecp2−/+ and Mecp2−/y brain samples from both Mecp2-null strains compared with controls but with lower significance than the LSC results. The precise sampling of single cells within the cerebral cortex in the LSC analysis compared with the whole brain analysis by immunoblot may explain the greater sensitivity of the LSC approach. These results demonstrate that UBE3A protein expression in the cerebral cortex is reduced by both cell-autonomous and non-cell-autonomous effects of Mecp2 mutation.
Figure 1.

UBE3A expression in Mecp2-null mouse brain. (A) A multiple tissue microarray containing wt and knock-out mouse cerebral cortex samples (16) was stained for immunofluorescence using a C-terminal reactive anti-MeCP2 and an anti-UBE3A antibody. The MeCP2-negative cells (Mecp2 mt-expressing, blue histograms, percentage shown) were separately gated from MeCP2-positive cells (Mecp2 wt-expressing, red histograms) and the total population (black histograms) and compared with age-matched wt control female or male samples (orange histograms). (B) The graph shows combined LSC results (mean ± SEM) from four replicate slides using two different anti-UBE3A antibodies. (C) Protein extracts from whole adult mouse brain were probed on an immunoblot with anti-UBE3A or anti-GAPDH. A representative image shows lower expression in brain of UBE3A in both hemizygous male (−/y) and heterozygous female (−/+) brain compared with wild-type (wt, +/y and +/+) littermate controls for both Mecp2tm1.1Bird and Mecp2tm1.1Jae strains. The graph demonstrates combined GAPDH-normalized results (mean ± SEM, quantitation as previously described (32), example ratios shown below each lane) from eight different −/+ and six different +/+ brain samples from Mecp2tm1.1Bird and Mecp2tm1.1Jae strains. *P < 0.05, ****P < 0.0001 by t-test.
Ube3a expression level is lower in Mecp2 deficient mouse brain without alterations in imprinted gene expression
As the maternal expression of Ube3a in postnatal neurons is correlated with the paternal expression of an antisense Ube3a transcript from the imprinting control region (ICR) of the Snrpn/Snurf promoter (33), we examined the role of MeCP2 on the imprinting status of this locus. Chromatin immunoprecipitation (ChIP) demonstrated that MeCP2 was bound to the promoter of Snrpn and positive control U2af1-rs1 (34) in mouse cerebrum (Fig. 2A). In contrast, neither the Ube3a nor Gabrb3 promoter was found to be associated with MeCP2 at a detectable level, consistent with a lack of methylation. Allele-specific analyses of (Mecp2−/+ × PWK)F1 progeny demonstrated the expected preferential maternal expression of Ube3a sense transcript, paternal expression of Ube3a antisense and Snrpn and biallelic expression of Gabrb3, regardless of Mecp2 genotype (Fig. 2B). These results demonstrated that reduced expression of UBE3A/Ube3a in Mecp2 deficient brain was not directly due to alterations in allele-specific expression and confirm similar results from human brain (31). Two additional imprinted genes from other loci (Rasgrf1 and H19) were also tested for allele-specific expression changes in Mecp2-null mice with similar results (Fig. 2B). These results further demonstrate that Mecp2 is not required for maintenance of imprinted gene expression.
Figure 2.
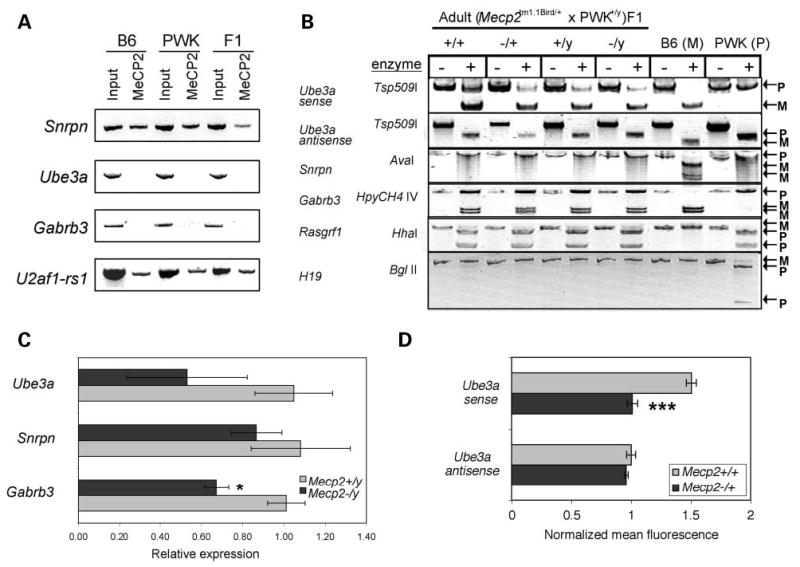
Imprinting and transcriptional analyses in mouse brain. (A) Chromatin from adult mouse cerebrum samples [C57B6, PWK or (B6 × PWK)F1] was isolated for ChIP. Anti-MeCP2 (C-terminal) was used to immunoprecipitate DNA fragments from ‘Input’ control. Ube3a and Gabrb3 promoters were not detected in the anti-MeCP2 precipitated chromatin, in contrast to the Snrpn promoter sequences that showed association with MeCP2. U2af1-rs1 was a positive control for a promoter previously demonstrated to bind MeCP2 in brain (34). (B) Mecp2tm1.1Bird/+ (B6) females were crossed with wt PWK males to obtain F1 mice heterozygous for single nucleotide polymorphisms in the coding regions of several imprinted genes. RT–PCR followed by restriction enzyme digestion (+) was performed on RNA from adult brain samples of all Mecp2 genotypes as well as parental B6 and PWK samples (P, paternal; M, maternal). As reported previously for wt (B6 × PWK)F1 brain (67), Ube3a sense exhibited preferential maternal expression, Ube3a antisense and Snrpn were exclusively paternal and Gabrb3 was biallelic, with no significant effect of Mecp2 genotype on these imprints. In addition, Rasgrf1 showed preferential paternal expression (68), whereas H19 showed exclusive maternal expression (70) in all samples. Results are representative of adult and neonatal F1 brain samples. (C) TaqMan PCR was used to quantitatively determine expression levels of Ube3a, Snrpn and Gabrb3 in 10-week-old Mecp2−/y and Mecp2+/y brain RNA. Primers spanned intron/exon boundaries and are specific to the Ube3a sense transcript. Results shown are for the average ± SEM of four experimental replicates of two mice per genotype. Although Ube3a and Gabrb3 showed consistently lower expression in Mecp2-deficient brain compared with controls, Snrpn was not significantly changed. (D) Sense and antisense transcripts of Ube3a were detected by fluorescence in situ hybridization using single stranded riboprobes and quantitated by LSC as described previously (16). Results (mean ± SEM of 3 wt and 10 Mecp2−/+ samples) were normalized to control β-actin probe. *P < 0.05, ***P < 0.001 by t-test.
To determine whether Ube3a transcript levels were also reduced, TaqMan PCR was performed on RNA samples obtained from Mecp2−/y and Mecp2+/y mice. The results showed a reduction in Ube3a (sense) and Gabrb3 transcripts, but not in Snrpn, in Mecp2 deficient brain (Fig. 2C). Fluorescence in situ hybridization with single stranded riboprobes was then performed on the tissue microarray in order to separately analyze the sense and antisense transcripts of Ube3a and confirm the reduction of the Ube3a sense transcript by another method. Figure 2D shows the results demonstrating a significantly lower expression of Ube3a sense but not Ube3a anti-sense transcripts. These combined results suggest that the decrease in UBE3A protein expression in Mecp2 deficient brain is due to decreased expression of the Ube3a sense transcript without a significant change in Snrpn or the Ube3a anti-sense transcript or the maternal-biased expression of Ube3a.
UBE3A expression is deficient in RTT, AS and autism cerebral samples
To determine whether significant reductions in UBE3A expression were also observed in human brain samples, a postmortem brain tissue microarray described previously containing human cerebral samples (16) was analyzed by immunofluorescence and LSC. The histograms in Figure 3A demonstrate significant reductions in UBE3A expression of RTT, AS and autism samples (black) compared with age-matched controls (gray). Ages of each sample are shown, but no significant differences were observed in the level of UBE3A expression with increasing age of the controls (data not shown), unlike the striking increase with age observed for MeCP2 (16,35). Mean UBE3A fluorescence was normalized to control histone H1 expression and graphed (Fig. 3B), demonstrating significant UBE3A reductions in RTT and autism samples. The reduced expression of UBE3A in the AS 15q11–q13 deletion sample was expected and consistent with a low level of paternal UBE3A expression in the cerebral cortex (36). The LSC results were confirmed by immunoblot (Fig. 3C shows a representative blot). Furthermore, reduced UBE3A expression in AUT1174 was also shown by immunoblot in a recent report (37).
Figure 3.
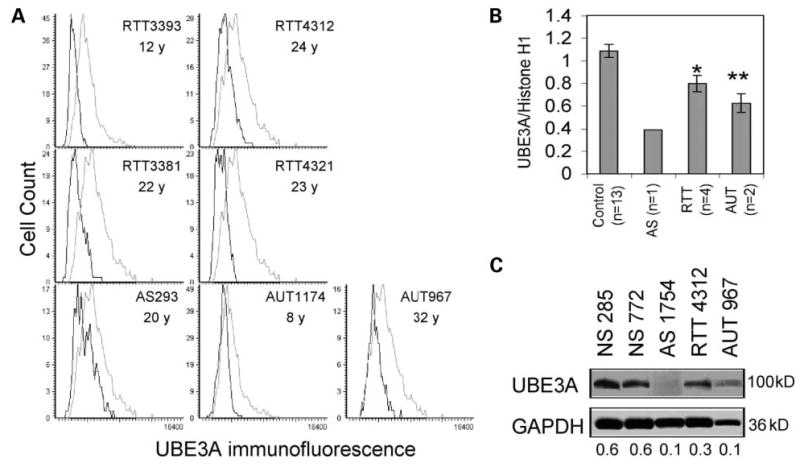
Quantitation of UBE3A expression in Rett, Angelman and autism brain samples. (A) The human tissue microarray previously described (16) containing frontal cortex layers III–V from Brodman area (BA9) was analyzed for UBE3A expression by LSC. Histograms show lower expression of patient samples (black) from Rett (RTT), Angelman (AS) and autism (AUT) compared with age-matched controls (gray). (B) UBE3A immunofluorescence values were normalized to those of control histone H1 and the results of four replicate slides using two different anti-UBE3A antibodies (Affinity Bioreagents and BD Biosciences) were averaged (mean ± SEM) for each brain sample shown in (A) (n, number of different samples). (C) Immunoblot analysis of UBE3A expression (anti-UBE3A, Affinity, recognizing all known isoforms) confirmed decreased expression in AS, RTT and AUT compared with controls (UBE3A/GAPDH ratios are shown). *P < 0.05, **P < 0.01 by t-test.
GABRB3 expression defects are observed in Mecp2 deficient mouse and RTT, AS and autism human brain
GABRB3 has been strongly implicated in autism by multiple association studies (14,38,39), but alterations in expression level have not been previously demonstrated. Because Mecp2 deficiency affected not only Ube3a expression but also the biallelically expressed gene, Gabrb3 (Fig. 2C), we further investigated its protein product, the gamma aminobutyric acid (GABA) receptor β3 subunit, for defects in Mecp2/MeCP2 deficient brain. Immunoblot analyses demonstrated reduced-GABRB3 expression in Mecp2−/+ and Mecp2−/y mouse brain compared with controls (Fig. 4A), suggesting an additional target from the 15q11–q13 locus may also be dysregulated in RTT, AS and autism.
Figure 4.

Analysis of GABRB3 expression in mouse and human brain. (A) Immunoblot analysis of GABRB3 expression (anti-GABRB3, Affinity Bioreagents, recognizing all known isoforms) shows lower expression in Mecp2−/+ and Mecp2−/y adult mouse brain compared with controls. Combined GAPDH-normalized results from eight different Mecp2−/+ and six different Mecp2+/+ adult brain samples from Mecp2tm1.1Bird and Mecp2tm1.1Jae strains were significantly lower than wild-type controls (P < 0.01). (B) Representative immunoblot analyses of GABRB3 and GAPDH in AS, RTT, autism and control (NS) cerebral samples with GABRB3/GAPDH ratios shown. Replicate immunoblot results for all samples and significance tests are shown in Table 1. Additional information about postmortem brain samples used in these studies is provided online (Supplementary Material, Table S1).
As a transmembrane protein, GABRB3 was less amenable to LSC analysis than nuclear or cytoplasmic proteins because individual cells are identified by nuclear contouring. Immunoblot analyses were, therefore, performed on frozen frontal cerebral cortex, Brodman area 9 (BA9), from a total of three RTT (with MECP2 mutations), two AS (with 15q11–q13 deletions), nine autism (no detectable MECP2 mutations or 15q11–q13 duplications) and 11 different age-matched normal controls. Two representative immunoblots are shown in Figure 4B. Table 1 shows the combined data of normalized GABRB3/GAPDH ratios for all individual samples. When analyzed as combined groups, RTT, AS and autism samples showed significantly lower GABRB3 expression compared with controls. The autism samples showed the highest significant decrease in GABRB3 as a group (P < 0.0001) despite higher variability between samples, with some showing extremely low expression (i.e., 797, 1174, 967 and 5173, with 5–10% of control expression) and others showing little change (i.e., 3924 and 5342, Fig. 4B and Table 1). The ages of samples are shown in Table 1, but reveal no significant change in GABRB3 expression with age of the control samples tested. The average GABRB3 expression for each patient category is, therefore, also represented as a group for statistical comparisons. Although GABRB3 expression deficiencies are not universal in all autism brain samples, the defects are more significant than in RTT and AS and occur at an unexpectedly high frequency (5/9 or 56%). The specificity of GABRB3 defects to AS, RTT and autism was tested by analyzing three Down syndrome (DS) brain samples with trisomy 21. Table 1 shows no significant defects in GABRB3 expression in DS compared with controls. These results, therefore, demonstrate that reduced-GABRB3 expression is a common characteristic of RTT and autism brain.
Table 1.
Significant differences in GABRB3 expression in RTT, AS and autism brain by quantitative immunoblot
| Category | Sample no. | Age (years) | No. of replicates | GABRB3/GAPDH (mean ± SEM) |
|---|---|---|---|---|
| Control | 629 | 7 | 6 | 0.75 ± 0.016 |
| 738 | 8 | 5 | 0.64 ± 0.064 | |
| 3835 | 9 | 1 | 0.78 | |
| 1297 | 15 | 1 | 0.84 | |
| 1065 | 15 | 1 | 0.87 | |
| 602 | 27 | 5 | 0.69 ± 0.047 | |
| 285 | 31 | 5 | 0.80 ± 0.081 | |
| 1104 | 35 | 1 | 0.57 | |
| 772 | 36 | 1 | 0.79 | |
| 4192 | 46 | 2 | 0.75 ± 0.025 | |
| 4503 | 56 | 2 | 0.52 ± 0.001 | |
| Control average | 0.73 ± 0.032 | |||
| Rett | 3393 | 12 | 2 | 0.61 ± 0.001a* |
| 4312 | 23 | 3 | 0.53 ± 0.017a** | |
| 5020 | 24 | 2 | 0.43 ± 0.093 | |
| Rett average | 0.52 ± 0.031b** | |||
| Angelman | 1754 | 4 | 2 | 0.51 ± 0.037a*** |
| 1494 | 43 | 2 | 0.13 ± 0.078a*** | |
| AS average | 0.32 ± 0.120b*** | |||
| Autism | 797 | 9 | 3 | 0.17 ± 0.103a*** |
| 1174 | 7 | 3 | 0.25 ± 0.099a*** | |
| 967 | 32 | 2 | 0.04 ± 0.011a* | |
| 5000 | 27 | 3 | 0.55 ± 0.117 | |
| 5342 | 11 | 2 | 0.79 ± 0.085 | |
| 5173 | 30 | 2 | 0.05 ± 0.014a* | |
| 4925 | 9 | 2 | 0.56 ± 0.097 | |
| 3924 | 16 | 2 | 0.69 ± 0.004 | |
| 1638 | 20 | 2 | 0.31 ± 0.001a* | |
| Autism average | 0.39 ± 0.060b* | |||
| Down | 1267 | 10 | 3 | 0.76 ± 0.046 |
| 707 | 23 | 3 | 0.80 ± 0.005 | |
| 1753 | 24 | 3 | 0.71 ± 0.027 | |
| Down average | 0.76 ± 0.028 |
Replicate results from each individual sample were compared with the three closest age-matched controls by t-test.
Total sample replicates were combined from each category and compared with total control sample replicates by t-test.
P < 0.01.
P < 0.05.
P < 0.001.
P < 0.0001.
DISCUSSION
Uncovering the genetic basis of autism has been challenging because of its complex etiology. Although the heritability of autism in families is high, multiple gene loci and environmental influences are expected (1). Even for the autism-spectrum disorders with known genetic causes, the molecular pathogenesis is not well understood. Because of the overlap in phenotype among RTT, AS and autism (4), we tested the hypothesis that gene expression in the 15q11–q13 region may be altered in all three syndromes. We show for the first time that the AS gene, UBE3A/Ube3a, is significantly reduced in RTT and Mecp2-null mice, demonstrating that MECP2/Mecp2 deficiency causes reduced expression of Ube3a/UBE3A. In addition, we demonstrate UBE3A deficiency in autism brain samples, consistent with a recent report (37). Furthermore, we also show for the first time deficiencies in the expression of the GABA receptor β3 subunit gene (GABRB3) in RTT and autism brain and Mecp2-null mice.
Maternal duplications in 15q11–q13 have been observed in ~1% of autism cases as the most common cytogenetic abnormality in autism (11). Genome-wide linkage studies have identified the 15q11–q13 locus in some but not all scans (40–43). Furthermore, higher resolution scans of linkage within 15q11–q13 in autism families have identified significant linkage to UBE3A and GABRB3 in some studies but not others (14,44–48). While the linkage of autism to 15q11–q13 may be somewhat disputed at the genetic level, here we show that expression level defects of two genes within 15q11–q13 are common to RTT, AS and autism. The reduction of UBE3A and GABRB3 in AS cerebrum was expected because of the maternal 15q11–q13 deletion in these samples. Interestingly, neither Ube3a deficient nor uniparental disomy mice showed evidence for decreased Gabrb3 expression (49,50), but the lack of AS brain samples with uniparental disomy or UBE3A mutations prevented our confirmation of this finding in human samples.
Our results are consistent with a recent report showing decreased UBE3A in autism brain (37), but extend the observation to demonstrate GABRB3 deficiency as a common defect in autism. The GABAA receptor genes are attractive candidate genes for autism as several studies have demonstrated linkage to one or more of these genes (13,14,48). Autoradiography has demonstrated reduced GABAA binding in autism brain (51) and elevated circulating GABA levels have been found in autistic children (52–54). Although our sample size was small owing to the requirement for postmortem human brain tissue, the frequency of UBE3A and GABRB3 expression defects within autism brain samples was much higher than for previously described genetic defects. The defects in UBE3A and GABRB3 expressions were unlikely to be due to degradation in postmortem tissue as they were not observed in multiple normal and DS controls and because the results were all normalized to housekeeping gene controls. These results suggest that reduced expression of UBE3A and GABRB3 are correlated and may be a downstream consequence of multiple different genetic or epigenetic etiologies. The phenotypic consequence of reduced expression of these genes is expected to be less severe than gene mutation (UBE3A mutations in AS) or nullisomy. Both Ube3a−/+ and Gabrb3+/−, −/− mice have learning and motor defects and inducible seizures, similar to AS and some cases of autism (55,56). While our autism brain sample population seems to be somewhat enriched for associated seizure disorders (Supplementary Material, Table S1), significantly reduced-GABRB3 expression was observed in brain samples from individuals with and without seizures.
We also identify the X-linked gene, MECP2, as one genetic cause of UBE3A and GABRB3 expression defects in the mammalian cerebrum. We have used two Mecp2 deficient RTT mouse strains (23,24) to demonstrate that UBE3A/Ube3a and GABRB3/Gabrb3 are significantly decreased in adult mouse brain compared with wt controls. Furthermore, we show significant defects in expression of UBE3A and GABRB3 in human RTT brain samples with MECP2 mutations. Our results are consistent with GABRB3 showing reduced expression in RTT brain by genome-wide expression profiling in one report (27). The relatively subtle changes in expression of UBE3A and GABRB3 observed in this study may explain why these genes were not identified in other genome-wide profiling studies of MECP2/Mecp2 mutant tissues (26,28). The observation of down-regulation as well as up-regulation of MeCP2 target genes has also been observed in expression profiling studies, arguing against a single role for MeCP2 as a transcriptional repressor.
As we have previously demonstrated MECP2 expression defects in the same autism brain samples (16), perhaps MECP2 expression defects could be responsible for the lower UBE3A and GABRB3 expressions in these autistic individuals without characterized genetic defects. Alternatively, defects in expression of all three genes may be a downstream consequence of abnormal brain development. As the Mecp2308/y RTT mouse model has been demonstrated to show some autistic behaviors, including forepaw stereopathies and social avoidance (7,25), the role of MeCP2 in the pathogenesis of at least some cases of autism appears likely (4).
In this report, we also begin to uncover a rather complex and unexpected mechanism by which MeCP2 regulates expression of UBE3A and GABRB3. Although MeCP2 binds to methylated DNA and is a predicted transcriptional repressor (17,18), it is not required for maintaining parental imprints (31). Here, we demonstrate that MeCP2 binds to the ICR in the Snrpn promoter, yet it is not required for silencing the maternal allele of Snrpn and Ube3a antisense or the paternal allele of the Ube3a sense transcript. Furthermore, expression level of Gabrb3 is affected even though this gene is biallelically expressed. We also demonstrate the maintenance of exclusive maternal expression of H19 in Mecp2-null mice despite its description as a ‘bona fide’ MeCP2 target gene (21). These results suggest that the effect of MeCP2 binding to the ICR is more complex than simply repressing imprinted genes in cis and may instead involve long-range chromatin interactions. Perhaps chromatin loop formation such has been recently demonstrated in the imprinted Igf2/H19 region (57) is adversely affected by Mecp2 deficiency, as MeCP2 has been previously described to have matrix binding activity (58). The ~1 Mb region from UBE3A to GABRB3 also includes multiple small nucleolar RNA genes (33,59,60) and ATP10A (61,62), and two other GABAA receptors (GABRA5 and GABRG3) located telomeric of GABRB3 (63) that also could be dysregulated by MeCP2 deficiency. In addition to the three CpG islands investigated here for association with MeCP2, there are 13 other CpG islands in this region that remain to be investigated as potential MeCP2 binding sites (UCSC Genome Browser, May 2004 assembly). In addition to potential long-range chromatin organization in cis, 15q11–q13 homologous trans interactions have been demonstrated in human lymphocytes (64) and brain (Thatcher et al., submitted for publication) and mouse ES cells (65) that may be important in regulating the expression level of multiple genes. Although autistic individuals with 15q11–q13 duplications have been predicted to express higher levels of UBE3A and GABRB3 compared with controls, lack of proper biparental homologous pairing could instead result in lower expression of these genes similar to the autism brain samples in our study.
In conclusion, in this report, we have made an important connection by uncovering a molecular brain alteration common to three different autism-spectrum neurodevelopmental disorders of different genetic etiologies. The expression defects in UBE3A and GABRB3 observed in RTT and autism brain are likely to be consequences of abnormal neuronal function due to deficiencies in MECP2 or other genetic or environmental causes. Further understanding of how MeCP2 regulates expression of these and other genes within 15q11–q13 may help in the identification of genetic causes of autism and aid in the design of therapies for all three disorders.
MATERIALS AND METHODS
Immunofluorescence analysis of tissue microarrays
Construction of tissue microarrays used in this study have been previously described for Mecp2-null mouse (32) and human neurodevelopmental disorders (16). Briefly, for the mouse array, triplicate 600 μm cores of cerebral cortex were removed from five Mecp2tm1.1Bird/+, five Mecp2tm1.1Jae/+, and three Mecp2+/+ controls (P20 w–P27 w); one Mecp2tm1.1Bird/y, and one Mecp2+/y littermates (P10 w); one Mecp2tm1.1Jae/y and one Mecp2+/y littermate controls (P10 w). The human tissue microarray contains triplicate frontal cerebral cortex (BA9, layers III–V) obtained frozen <30 h postmortem. 5 μm section was processed for immunofluorescence as described previously for anti-MeCP2 (C-terminal, Affinity Bioreagents) and anti-histone H1 (Upstate Biotechnology) antibodies (66). For mouse tissues, combined staining was performed with anti-UBE3A antibody (BD Biosciences and monoclonal gift from Howley, Harvard Medical School) and a cascade-blue labeled goat-anti-mouse IgG (Molecular Probes). For human tissues, an anti-UBE3A antibody (Affinity Bioreagents) was used. LSC analysis of MeCP2 negative and positive populations was performed as described previously (32).
In situ hybridization analysis of tissue microarrays
Sense and antisense transcripts of Ube3a were detected by fluorescence in situ hybridization using single stranded riboprobes from a 796 bp cloned fragment spanning exons 6–10 (primers, 5′-tcacatatgatgaagctacgaa-3′ and 5′-ttctttgcttgaatattccgg-3′) labeled with biotin (Lofstrand Laboratories). A control β-actin probe digoxigenin labeled (Roche) was simultaneously hybridized at 55°C. Hybridization conditions, fluorescent detection and quantitation by LSC were performed as described previously (16).
Immunoblot analyses
A total of 10 μg of protein extract from whole adult mouse brain was analyzed per lane by immunoblot as described previously (32) using anti-UBE3A (BD Biosciences), anti-GABRB3 (Affinity Bioreagents) or anti-GAPDH (Advanced Immunochemical) antibodies at recommended concentrations. Protein extracts were isolated from frozen human postmortem cerebral cortex (BA9) from samples previously described (16) and additional samples (Harvard Brain Bank and University of Maryland Brain and Tissue Bank). Similar to mouse samples, 10 μg of human protein was analyzed per lane with the same reagents except the substitution of a human-specific anti-UBE3A antibody (Affinity Bioreagents). Additional human cerebral (BA9) samples were obtained from autism, Down and age-matched control samples from the Autism Tissue Program for immunoblot analyses. Quantitation was performed using Nucleotech Gel Expert version 2.0.
Allelic expression of imprinted genes in interspecies hybrids
Mecp2tm1.1Bird/+ (C57B6) female mice (23) were mated to wt PWK males and the resulting offsprings were genotyped for Mecp2 as described previously (32). RNA was isolated from adult (Mecp2 tm1.1Bird/+ × PWK)F1 brain samples from all four genotypes as well as parental B6 and PWK mice. RT–PCR followed by restriction enzyme digestion was performed as described previously for Ube3a, Snrpn, Gabrb3 (67) and Rasgrf1 (68). H19 primers (5′-GAACCACTAACACTACCTGCC-3′ and 5′-GGAACTGCTTCCAGACTAGG-3′, 58°C with 30 cycles) amplified a 585 bp fragment that was digested with Bgl II.
Chromatin immunoprecipitation
Chromatin from adult mouse cerebrum samples [C57B6, PWK or (B6 × PWK)F1] was isolated for ChIP as described previously (34,69). Anti-MeCP2 (C-terminal) was used to immunoprecipitate DNA fragments from ‘Input’ control as described previously for primers spanning 5′ CpG islands for Snrpn and positive control U2af1-rs1(34), Ube3a (5′-CCCTTCTGCTTCTCTTCGGAGT-3′, 5′-CAGAAGCAGCACACGAATAAA-3′, 58°C anneal, 37 cycles) and Gabrb3 (5′-CCTCAGAGCCACCCGTAC-3′, 5′-GTCTAGGACCCCGCGACA-3, 60°C anneal, 40 cycles).
TaqMan PCR
Each PCR contained 400 nm of each primer, 80 nm of the TaqMan probe and commercially available PCR mastermix (TaqMan Universal PCR Mastermix, Applied Biosystems) containing 10 mm Tris–HCl (pH 8.3), 50 mm KCl, 5 mm MgCl2, 2.5 mm deoxynucleotide triphosphates, 0.625 U AmpliTaq Gold DNA polymerase per reaction, 0.25 U AmpErase UNG per reaction and 5 μl of the diluted cDNA sample in a final volume of 25 μl. Ube3a forward primer spanned exons 7–8 (5′-TCCAGATATTGGTATGTTCACATATG-3′), reverse primer (5′-GGGAAAATGGACATCCAGTATACAA-3′) and probe (5′-CTGATTGGCATAGTCCTGGGTCTGGC-3′) were from exon 8 (NM_173010). Snrpn forward primer spanned exons 3–4 (5′-TCAGGAAGATCAAGCCAAAGAATG-3′), reverse primer (5′-AAGTTCTCCCCACGTAGCAAGAC-3′) and probe (5′-ACAGCCAGAACGTGAAGAAAAACGGGT-3′) were from exon 4 (NM_013670). Gabrb3 forward primer spanned exons 6–7 (5′-CCGTCTGGTCTCCAGGAATGTT-3′), reverse primer (5′-CCGATATTTCTCTTCAACCGAAAA-3′) and probe (5′-TTCGCCACAGGTGCCTATCCTCGAC-3′) were from exon 7. The samples were placed in 96 well plates and amplified in an automated fluorometer (ABI PRISM 7700 Sequence Detection System, Applied Biosystems). Amplification conditions were 2 min at 50°C, 10 min at 95°C, 40 cycles of 15 s at 95°C and 60 s at 60°C. Final quantitation was done using the comparative CT method (Applied Biosystems) and is reported as relative transcription or the n-fold difference relative to a calibrator cDNA (Mecp2+/y of each pair) following normalization to HPRT1 housekeeping control gene.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
Acknowledgments
The authors thank M. Lalande and D. Yasui for critical review of the manuscript and S. Peddada for technical expertise. This work was supported in part by the UC Davis MIND Institute, the RTT Research Foundation and the NIH (1R01HD/NS41462). Mecp2tm1.1Jae mice were obtained from the Mouse Mutant Regional Resource Center at UC Davis (supported in part by NIH U42 RR14905). Human tissue samples and clinical information were generously provided by the Autism Tissue Program, the University of Maryland Brain and Tissue Bank for Developmental Disorders (supported by NIH N01-HD-1-3138), Harvard Brain Tissue Resource Center (supported in part by PHS MH/NS 31862).
References
- 1.Volkmar FR, Pauls D. Autism. Lancet. 2003;362:1133–1141. doi: 10.1016/S0140-6736(03)14471-6. [DOI] [PubMed] [Google Scholar]
- 2.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 3.Lalande M. Parental imprinting and human disease. Annu Rev Genet. 1996;30:173–195. doi: 10.1146/annurev.genet.30.1.173. [DOI] [PubMed] [Google Scholar]
- 4.Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 5.Mount RH, Charman T, Hastings RP, Reilly S, Cass H. Features of autism in Rett syndrome and severe mental retardation. J Autism Dev Disord. 2003;33:435–442. doi: 10.1023/a:1025066913283. [DOI] [PubMed] [Google Scholar]
- 6.Peters SU, Beaudet AL, Madduri N, Bacino CA. Autism in Angelman syndrome: implications for autism research. Clin Genet. 2004;66:530–536. doi: 10.1111/j.1399-0004.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 7.Moretti P, Bouwknecht JA, Teague R, Paylor R, Zoghbi HY. Abnormalities of social interactions and home cage behavior in a mouse model of Rett syndrome. Hum Mol Genet. 2005;14:205–220. doi: 10.1093/hmg/ddi016. [DOI] [PubMed] [Google Scholar]
- 8.Watson P, Black G, Ramsden S, Barrow M, Super M, Kerr B, Clayton-Smith J. Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. J Med Genet. 2001;38:224–228. doi: 10.1136/jmg.38.4.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam C, Yeung W, Ko C, Poon P, Tong S, Chan K, Lo I, Chan L, Hui J, Wong V, et al. Spectrum of mutations in the MECP2 gene in patients with infantile autism and Rett syndrome. J Med Genet. 2000;37:E41. doi: 10.1136/jmg.37.12.e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beyer KS, Blasi F, Bacchelli E, Klauck SM, Maestrini E, Poustka A, International Molecular Genetic Study of Autism. Mutation analysis of the coding sequence of the MECP2 gene in infantile autism. Hum Genet. 2002;111:305–309. doi: 10.1007/s00439-002-0786-3. [DOI] [PubMed] [Google Scholar]
- 11.Schroer RJ, Phelan MC, Michaelis RC, Crawford EC, Skinner SA, Cuccaro M, Simensen RJ, Bishop J, Skinner C, Fender D, et al. Autism and maternally derived aberrations of chromosome 15q. Am J Med Genet. 1998;76:327–336. doi: 10.1002/(sici)1096-8628(19980401)76:4<327::aid-ajmg8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 12.Wagstaff J, Chaillet JR, Lalande M. The GABAA receptor beta 3 subunit gene: characterization of a human cDNA from chromosome 15q11q13 and mapping to a region of conserved synteny on mouse chromosome 7. Genomics. 1991;11:1071–1078. doi: 10.1016/0888-7543(91)90034-c. [DOI] [PubMed] [Google Scholar]
- 13.Cook EH, Courchesne RY, Cox NJ, Lord C, Gonen D, Guter SJ, Lincoln A, Nix K, Haas R, Leventhal BL, et al. Linkage-disequilibrium mapping of autistic disorder, with 15q11–q13 markers. Am J Hum Genet. 1998;62:1077–1083. doi: 10.1086/301832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buxbaum JD, Silverman JM, Smith CJ, Greenberg DA, Kilifarski M, Reichert J, Cook EH, Fang Y, Song CY, Vitale R. Association between a GABRB3 polymorphism and autism. Mol Psychiat. 2002;7:311–316. doi: 10.1038/sj.mp.4001011. [DOI] [PubMed] [Google Scholar]
- 15.Nicholls RD, Gottlieb W, Russell LB, Davda M, Horsthemke B, Rinchik EM. Evaluation of potential models for imprinted and nonimprinted components of human chromosome 15q11–q13 syndromes by fine-structure homology mapping in the mouse. Proc Natl Acad Sci USA. 1993;90:2050–2054. doi: 10.1073/pnas.90.5.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum Mol Genet. 2004;13:629–639. doi: 10.1093/hmg/ddh063. [DOI] [PubMed] [Google Scholar]
- 17.Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 18.Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 19.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 20.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 21.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 22.Kimura H, Shiota K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J Biol Chem. 2003;278:4806–4812. doi: 10.1074/jbc.M209923200. [DOI] [PubMed] [Google Scholar]
- 23.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 24.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 25.Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- 26.Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci USA. 2002;99:15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colantuoni C, Jeon OH, Hyder K, Chenchik A, Khimani AH, Narayanan V, Hoffman EP, Kaufmann WE, Naidu S, Pevsner J. Gene expression profiling in postmortem Rett syndrome brain: differential gene expression and patient classification. Neurobiol Dis. 2001;8:847–865. doi: 10.1006/nbdi.2001.0428. [DOI] [PubMed] [Google Scholar]
- 28.Traynor J, Agarwal P, Lazzeroni L, Francke U. Gene expression patterns vary in clonal cell cultures from Rett syndrome females with eight different MECP2 mutations. BMC Med Genet. 2002;3:12. doi: 10.1186/1471-2350-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 30.Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 31.Balmer D, Arredondo J, Samaco RC, LaSalle JM. MECP2 mutations in Rett syndrome adversely affect lymphocyte growth, but do not affect imprinted gene expression in blood or brain. Hum Genet. 2002;110:545–552. doi: 10.1007/s00439-002-0724-4. [DOI] [PubMed] [Google Scholar]
- 32.Braunschweig D, Simcox T, Samaco RC, LaSalle JM. X-chromosome inactivation ratios affect wild-type MeCP2 expression within mosaic Rett syndrome and Mecp2−/+ mouse brain. Hum Mol Genet. 2004;13:1275–1286. doi: 10.1093/hmg/ddh142. [DOI] [PubMed] [Google Scholar]
- 33.Runte M, Huttenhofer A, Gross S, Kiefmann M, Horsthemke B, Buiting K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol Genet. 2001;10:2687–2700. doi: 10.1093/hmg/10.23.2687. [DOI] [PubMed] [Google Scholar]
- 34.Gregory RI, Randall TE, Johnson CA, Khosla S, Hatada I, O’Neill LP, Turner BM, Feil R. DNA methylation is linked to deacetylation of histone H3, but not H4, on the imprinted genes Snrpn and U2af1-rs1. Mol Cell Biol. 2001;21:5426–5436. doi: 10.1128/MCB.21.16.5426-5436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Balmer D, Goldstine J, Rao YM, LaSalle JM. Elevated methyl-CpG-binding protein 2 expression is acquired during postnatal human brain development and is correlated with alternative polyadenylation. J Mol Med. 2003;81:61–68. doi: 10.1007/s00109-002-0396-5. [DOI] [PubMed] [Google Scholar]
- 36.Rougeulle C, Glatt H, Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain (letter) Nat Genet. 1997;17:14–15. doi: 10.1038/ng0997-14. [DOI] [PubMed] [Google Scholar]
- 37.Jiang YH, Sahoo T, Michaelis RC, Bercovich D, Bressler J, Kashork CD, Liu Q, Shaffer LG, Schroer RJ, Stockton DW, et al. A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. Am. J Med Genet. 2004;131A:1–10. doi: 10.1002/ajmg.a.30297. [DOI] [PubMed] [Google Scholar]
- 38.Cook EH, Jr, Courchesne RY, Cox NJ, Lord C, Gonen D, Guter SJ, Lincoln A, Nix K, Haas R, Leventhal BL, et al. Linkage-disequilibrium mapping of autistic disorder, with 15q11–q13 markers. Am J Hum Genet. 1998;62:1077–1083. doi: 10.1086/301832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCauley JL, Olson LM, Delahanty R, Amin T, Nurmi EL, Organ EL, Jacobs MM, Folstein SE, Haines JL, Sutcliffe JS. A linkage disequilibrium map of the 1-Mb 15q12 GABA(A) receptor subunit cluster and association to autism. Am J Med Genet. 2004;131B:51–59. doi: 10.1002/ajmg.b.30038. [DOI] [PubMed] [Google Scholar]
- 40.IMGSoA Consortium. A full genome screen for autism with evidence for linkage to a region on chromosome 7q. Hum Mol Genet. 1998;7:571–578. doi: 10.1093/hmg/7.3.571. [DOI] [PubMed] [Google Scholar]
- 41.Philippe A, Martinez M, Guilloud-Bataille M, Gillberg C, Rastam M, Sponheim E, Coleman M, Zappella M, Aschauer H, Van Maldergem L, et al. Genome-wide scan for autism susceptibility genes. Paris Autism Research International Sibpair Study. Hum Mol Genet. 1999;8:805–812. doi: 10.1093/hmg/8.5.805. [DOI] [PubMed] [Google Scholar]
- 42.IMGSoA Consortium. A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet. 2001;69:570–581. doi: 10.1086/323264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yonan AL, Alarcon M, Cheng R, Magnusson PK, Spence SJ, Palmer AA, Grunn A, Juo SH, Terwilliger JD, Liu J, et al. A genomewide screen of 345 families for autism-susceptibility loci. Am J Hum Genet. 2003;73:886–897. doi: 10.1086/378778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Salmon B, Hallmayer J, Rogers T, Kalaydjieva L, Petersen PB, Nicholas P, Pingree C, McMahon W, Spiker D, Lotspeich L, et al. Absence of linkage and linkage disequilibrium to chromosome 15q11–q13 markers in 139 multiplex families with autism. Am J Med Genet. 1999;88:551–556. [PubMed] [Google Scholar]
- 45.Shao Y, Cuccaro ML, Hauser ER, Raiford KL, Menold MM, Wolpert CM, Ravan SA, Elston L, Decena K, Donnelly SL, et al. Fine mapping of autistic disorder to chromosome 15q11–q13 by use of phenotypic subtypes. Am J Hum Genet. 2003;72:539–548. doi: 10.1086/367846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nurmi EL, Amin T, Olson LM, Jacobs MM, McCauley JL, Lam AY, Organ EL, Folstein SE, Haines JL, Sutcliffe JS. Dense linkage disequilibrium mapping in the 15q11–q13 maternal expression domain yields evidence for association in autism. Mol. Psychiat. 2003;8:624–634. doi: 10.1038/sj.mp.4001283. 570. [DOI] [PubMed] [Google Scholar]
- 47.Veenstra-VanderWeele J, Gonen D, Leventhal BL, Cook EH., Jr Mutation screening of the UBE3A/E6-AP gene in autistic disorder. Mol Psychiat. 1999;4:64–67. doi: 10.1038/sj.mp.4000472. [DOI] [PubMed] [Google Scholar]
- 48.Nurmi EL, Bradford Y, Chen Yh, Hall J, Arnone B, Gardiner MB, Hutcheson HB, Gilbert JR, Pericak-Vance MA, Copeland-Yates SA, et al. Linkage disequilibrium at the Angelman syndrome gene UBE3A in autism families. Genomics. 2001;77:105–113. doi: 10.1006/geno.2001.6617. [DOI] [PubMed] [Google Scholar]
- 49.Sinkkonen ST, Homanics GE, Korpi ER. Mouse models of Angelman syndrome, a neurodevelopmental disorder, display different brain regional GABA(A) receptor alterations. Neurosci Lett. 2003;340:205–208. doi: 10.1016/s0304-3940(03)00123-x. [DOI] [PubMed] [Google Scholar]
- 50.Buettner VL, Longmate JA, Barish ME, Mann JR, Singer-Sam J. Analysis of imprinting in mice with uniparental duplication of proximal chromosomes 7 and 15 by use of a custom oligonucleotide microarray. Mamm Genome. 2004;15:199–209. doi: 10.1007/s00335-003-2322-8. [DOI] [PubMed] [Google Scholar]
- 51.Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML. Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord. 2001;31:537–543. doi: 10.1023/a:1013238809666. [DOI] [PubMed] [Google Scholar]
- 52.Moreno-Fuenmayor H, Borjas L, Arrieta A, Valera V, Socorro-Candanoza L. Plasma excitatory amino acids in autism. Invest Clin. 1996;37:113–128. [PubMed] [Google Scholar]
- 53.Dhossche D, Applegate H, Abraham A, Maertens P, Bland L, Bencsath A, Martinez J. Elevated plasma gamma-aminobutyric acid (GABA) levels in autistic youngsters: stimulus for a GABA hypothesis of autism. Med. Sci. Monit. 2002;8:PR1–6. [PubMed] [Google Scholar]
- 54.Aldred S, Moore KM, Fitzgerald M, Waring RH. Plasma amino acid levels in children with autism and their families. J Autism Dev Disord. 2003;33:93–97. doi: 10.1023/a:1022238706604. [DOI] [PubMed] [Google Scholar]
- 55.Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 56.DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD, Olsen RW. Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci. 1998;18:8505–8514. doi: 10.1523/JNEUROSCI.18-20-08505.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murrell A, Heeson S, Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat Genet. 2004;36:889–893. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- 58.Weitzel JM, Buhrmester H, Stratling WH. Chicken MAR-binding protein ARBP is homologous to rat methyl-CpG- binding protein MeCP2. Mol Cell Biol. 1997;17:5656–5666. doi: 10.1128/mcb.17.9.5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cavaille J, Buiting K, Kiefmann M, Lalande M, Brannan CI, Horsthemke B, Bachellerie JP, Brosius J, Huttenhofer A. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc Natl Acad Sci USA. 2000;97:14311–14316. doi: 10.1073/pnas.250426397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de los Santos T, Schweizer J, Rees CA, Francke U. Small evolutionarily conserved RNA, resembling C/D box small nucleolar RNA, is transcribed from PWCR1, a novel imprinted gene in the Prader–Willi deletion region, which is highly expressed in brain. Am J Hum Genet. 2000;67:1067–1082. doi: 10.1086/303106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meguro M, Kashiwagi A, Mitsuya K, Nakao M, Kondo I, Saitoh S, Oshimura M. A novel maternally expressed gene, ATP10C, encodes a putative aminophospholipid translocase associated with Angelman syndrome. Nat Genet. 2001;28:19–20. doi: 10.1038/ng0501-19. [DOI] [PubMed] [Google Scholar]
- 62.Herzing LBK, Kim SJ, Cook EH, Ledbetter DH. The human aminophospholipid-transporting ATPase gene ATP10C maps adjacent to UBE3A and exhibits similar imprinted expression. Am J Hum Genet. 2001;68:1501–1505. doi: 10.1086/320616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sinnett D, Wagstaff J, Glatt K, Woolf E, Kirkness EJ, Lalande M. High-resolution mapping of the gamma-aminobutyric acid receptor subunit beta 3 and alpha 5 gene cluster on chromosome 15q11–q13, and localization of breakpoints in two Angelman syndrome patients. Am J Hum Genet. 1993;52:1216–1229. [PMC free article] [PubMed] [Google Scholar]
- 64.LaSalle J, Lalande M. Homologous association of oppositely imprinted chromosomal domains. Science. 1996;272:725–728. doi: 10.1126/science.272.5262.725. [DOI] [PubMed] [Google Scholar]
- 65.Tsai TF, Bressler J, Jiang YH, Beaudet AL. Disruption of the genomic imprint in trans with homologous recombination at Snrpn in ES cells. Genesis. 2003;37:151–161. doi: 10.1002/gene.10237. [DOI] [PubMed] [Google Scholar]
- 66.LaSalle J, Goldstine J, Balmer D, Greco C. Quantitative localization of heterologous methyl-CpG-binding protein 2 (MeCP2) expression phenotypes in normal and Rett syndrome brain by laser scanning cytometry. Hum Mol Genet. 2001;10:1729–1740. doi: 10.1093/hmg/10.17.1729. [DOI] [PubMed] [Google Scholar]
- 67.Yamasaki K, Joh K, Ohta T, Masuzaki H, Ishimaru T, Mukai T, Niikawa N, Ogawa M, Wagstaff J, Kishino T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet. 2003;12:837–847. doi: 10.1093/hmg/ddg106. [DOI] [PubMed] [Google Scholar]
- 68.Yoon BJ, Herman H, Sikora A, Smith LT, Plass C, Soloway PD. Regulation of DNA methylation of Rasgrf1. Nat Genet. 2002;30:92–96. doi: 10.1038/ng795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yasui D, Miyano M, Cai S, Varga-Weisz P, Kohwi-Shigematsu T. SATB1 targets chromatin remodelling to regulate genes over long distances. Nature. 2002;419:641–645. doi: 10.1038/nature01084. [DOI] [PubMed] [Google Scholar]
- 70.Yang Y, Hu JF, Ulaner GA, Li T, Yao X, Vu TH, Hoffman AR. Epigenetic regulation of Igf2/H19 imprinting at CTCF insulator binding sites. J Cell Biochem. 2003;90:1038–1055. doi: 10.1002/jcb.10684. [DOI] [PubMed] [Google Scholar]