

Abstract
Compelling data has been amassed indicating that soluble factors, or cytokines, emanating from the immune system can have profound effects on the neuroendocrine system, in particular the hypothalamic-pituitary-adrenal (HPA) axis. HPA activation by cytokines (via the release of glucocorticoids), in turn, has been found to play a critical role in restraining and shaping immune responses. Thus, cytokine–HPA interactions represent a fundamental consideration regarding the maintenance of homeostasis and the development of disease during viral infection. Although reviews exist that focus on the bi-directional communication between the immune system and the HPA axis during viral infection (188,235), others have focused on the immunomodulatory effects of glucocorticoids during viral infection (14,225). This review, however, concentrates on the other side of the bi-directional loop of neuroendocrine-immune interactions, namely, the characterization of HPA axis activity during viral infection and the mechanisms employed by cytokines to stimulate glucocorticoid release.
INTRODUCTION
Maintenance of homeostasis during immune challenge involves activation of the immune system, resolution of the challenge, and protection of the host against potentially toxic inflammatory processes. Examples of immune challenges include infection with viruses, bacteria, fungi, or parasites; tissue damage and destruction; and inappropriate responses to auto-antigens that may result in the development of autoimmune disease. Upon immune challenge, the immune system is activated to release numerous protein hormones called “cytokines.” One functional group of cytokines is that which mediates the innate immune response, including the proinflammatory cytokines tumor necrosis factor–alpha (TNF-α), interleukin-1 (IL-1), and interleukin-6 (IL-6), and the type I interferons (IFN-α/β). These cytokines are released in the early stages of an immune response from a variety of cell types, including activated immune cells, such as macrophages (and their CNS counterparts microglia), vascular endothelial cells, fibroblasts, and neurons. Another group of cytokines is that which mediates later, adaptive immunity, such as the T cell cytokines IL-2 and IFN-γ (type II interferons), which are especially important in mediating anti-viral defenses. In addition to contributing to the progression of the immune response against viral infection, cytokines released as part of the innate or adaptive immune response can activate the HPA axis, resulting in the release of adrenal glucocorticoids (37,57,120,178,258,290) (Fig. 1). In turn, glucocorticoids negatively feedback onto immune cells to suppress the further synthesis and release of cytokines, thereby protecting the host from the detrimental consequences of an overactive immune response (e.g., tissue damage, autoimmunity, septic shock). In addition, glucocorticoids play an important role in shaping immunity by influencing immune cell trafficking to sites of inflammation and altering downstream, adaptive immune responses by causing a shift from cellular (Th1/inflammatory) to humoral (Th2/anti-inflammatory) type immune responses (72). Therefore, in contrast to the traditional view of glucocorticoids as immunosuppressant hormones, they are more accurately conceptualized as immunomodulatory hormones, that can stimulate as well as suppress immune function, depending on the type of immune response, the immune compartment, and the cell type involved.
FIG. 1.
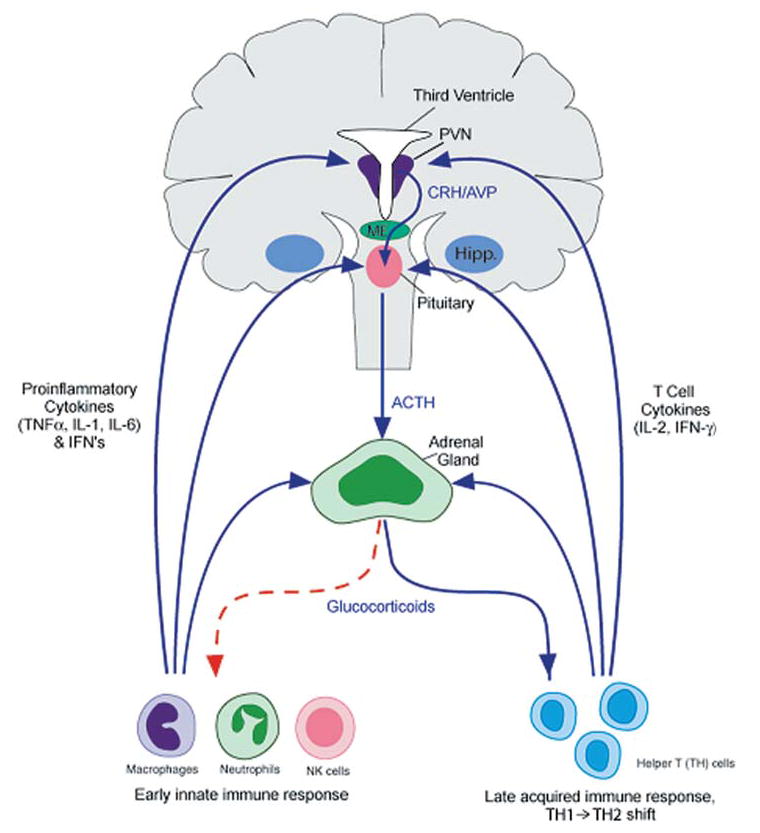
Bidirectional communication between the immune system and the hypothalamic-pituitary-adrenal (HPA) axis (human brain). The immune system, via early innate proinflammatory cytokines (TNFα, IL-1, and IL-6) and interferons, and late acquired T cell cytokines (IL-2 and INF-γ), stimulates glucocorticoid release by acting at all three levels of the HPA axis. In turn, glucocorticoids negatively feedback on the immune system to suppress the further synthesis and release of proinflammatory cytokines (red dotted line). In addition, glucocorticoids play an important role in shaping downtream acquired immune responses, by causing a shift from cellular (Th1/inflammatory) to humoral (Th2/anti-inflammatory) type immune responses. By doing so, glucocorticoids protect an organism from the detrimental consequences of overactive inflammatory immune responses. ACTH, adrenocorticotropic hormone; AVP, arginine vasopressin; CRH, corticotropin-releasing factor; hipp, hippocampus; IFN, interferon: IL, interleukin; ME, median eminence; PVN, paraventricular nucleus of the hypothalamus; TNF, tumor necrosis factor. Reprinted with modifications by permission from Silverman et al. (274).
Although hypothalamic corticotropin-releasing hormone (CRH) is considered a primary mechanism by which cytokines stimulate glucocorticoid release, increasing evidence supports a direct action of cytokines at the level of the pituitary and adrenal glands, as well. Cytokine receptors have been detected at all HPA axis levels, and therefore, each level can serve as an integration point for immune and neuroendocrine signals. In addition, cytokines are synthesized in the brain, the anterior pituitary, and the adrenal gland. The production of local cytokines in these tissues may function in a paracrine manner to amplify and maintain elevated HPA activity during chronic inflammation. Once glucocorticoids are released, maintenance of appropriate glucocorticoid activity is accomplished by local tissue regulation of glucocorticoid availability and action by factors such as corticosterone binding globulin (CBG), 11β-hydroxysteroid dehydrogenase (11β-HSD), the multidrug resistance transporter (MDR), and ultimately, the glucocorticoid receptor (GR) (274).
The kinetics and magnitude of HPA axis stimulation, and hence glucocorticoid release, induced by viral infection may be specific to the virus, the kinetics of the immune response to the virus, and the extent of virus/immune-induced pathology. Viruses that induce early, innate NK cell-mediated anti-viral defenses, characterized by early proinflammatory cytokine production, cellular infiltration, and inflammation, tend to produce early glucocorticoid responses, thereby protecting the host from proinflammatory cytokine-mediated pathology. On the other hand, viruses that induce later, adaptive T cell–mediated anti-viral defenses, characterized by late Th1/CTL cytokine production, cellular infiltration, and inflammation, tend to produce late glucocorticoid responses, thereby protecting the host from T cell–mediated pathology. Viruses that elicit both strong early proinflammatory and later T cell responses may exhibit a biphasic glucocorticoid response, whereas those that induce little or no inflammation, may not stimulate significant glucocorticoid release. Examples of these different responses are presented in this review, as we describe what is currently known about HPA axis activation during infections with Newcastle disease virus (NDV), murine cytomegalovirus (MCMV), lymphocytic choriomeningitis virus (LCMV), influenza, herpes simples virus type-1, (HSV-1), and human immunodeficiency virus (HIV).
Given the critical role of the HPA axis and glucocorticoid responses in maintaining a balance between the beneficial and detrimental effects of proinflammatory cytokines, as well as shaping downstream immune responses, it has become increasingly apparent that cytokine–HPA axis interactions are fundamental to immune regulation during viral infection. Moreover, redundant pathways of glucocorticoid induction, incorporating all three levels of the HPA axis, exist to ensure the survival of the host during immune challenge. Indeed, the essential role of glucocorticoids in protecting the host against a lethal overactivation of inflammatory responses has been demonstrated in a number of animal model systems, including viral infection (148,243,254,268).
THE HYPOTHALAMIC-PITUITARY-ADRENAL (HPA) AXIS
In considering HPA axis activity during viral infection, it is important to review the functional anatomy and physiology of the HPA axis. Activation of the HPA axis is well known to subserve the body’s response to a stressor, which can be defined as any physical or psychological stimulus that disrupts an organism’s homeostatic balance. Viral infections, in general, are physiologically stressful, as indicated by the concomitant activation of the HPA axis (89). Along with catecholamines (the end-product of sympathetic nervous system activation), glucocorticoids orchestrate the “fight or flight” response, which consists of the rapid mobilization of energy (glucose, amino acids, and fatty acids) from storage sites to critical muscles and the brain, concomitant with increased heart rate, blood pressure, and breathing rate to facilitate the rapid transport of nutrients and oxygen to relevant tissues. During such emergency situations, activation of the HPA axis also assists the body in shunting metabolic resources from growth, digestion, reproduction, and certain aspects of immunity to the more immediate challenge at hand. Particularly pertinent to infection as a stressor, according to Munck (196), “the physiological function of stress–induced increases in glucocorticoid levels is to protect not against the source of stress itself, but against the normal defense reactions (e.g., immune response/inflammation) that are activated by stress; glucocorticoids accomplish this function by turning off those defense reactions, thus preventing them from overshooting and themselves threatening homeostasis.” Therefore, some glucocorticoid actions may help mediate the recovery from the stress response, rather than mediate the stress response itself.
Activation of the HPA axis begins with the release of corticotropin-releasing hormone (CRH). CRH neurons originate in the parvocellular division of the paraventricular nucleus (PVN) of the hypothalamus and terminate in the external layer of the median eminence (ME), releasing CRH into the hypophysial-portal circulation (Fig. 2). CRH, in turn, acts on CRH-R1 receptors on anterior pituitary corticotrophs to stimulate the rapid release of adrenocorticotropic hormone (ACTH) from cellular stores, and, after a delay, the synthesis of the ACTH precursor peptide proopiomelanocortin (POMC) to replenish ACTH stores. ACTH then is released into the peripheral circulation and stimulates the release of glucocorticoids (cortisol in humans and non-human primates; corticosterone in rodents) from the adrenal cortex by acting on the MC2-R (type 2 melanocortin receptor) (for review, see (127)). Whereas both CRH-R1 and MC2-R are membrane bound, G-protein coupled receptors (linked to the adenylate cyclase–cAMP–PKA pathway), glucocorticoid receptors (GR’s) are cytosolic steroid receptors, that when activated by binding of its ligand (glucocorticoids), translocate into the nucleus to interact with other relevant transcription factors (e.g., NFκB or AP-1) or to directly alter the transcription of glucocorticoid-sensitive genes (by binding to a glucocorticoid response element [GRE] in their promoter region). Every nucleated cell in the body expresses glucocorticoid receptors; hence the widespread effects of glucocorticoids on practically every system of the body (e.g., metabolic, endocrine, nervous, cardiovascular, immune).
FIG. 2.
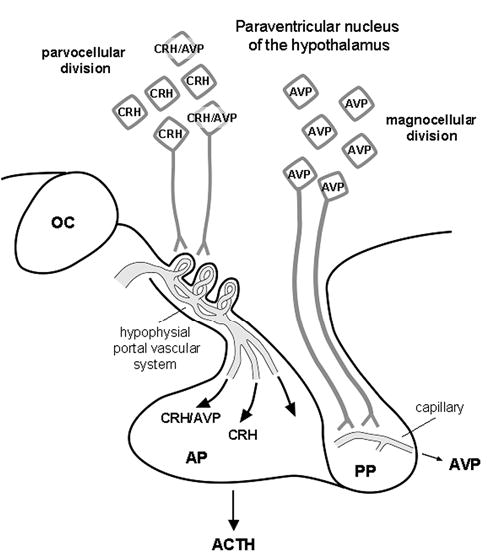
The hypothalamo-pituitary unit. CRH and CRH/AVP neurons originating from the parvocellular division of the PVN of the hypothalamus terminate in the external layer of the median eminence (ME), releasing CRH and or CRH/AVP into the hypophysial-portal circulation, which then act on corticotrophs in the AP to release ACTH into the general circulation. In addition, AVP neurons originating from the magnocellular division of the PVN pass through the internal layer of the ME and terminate on capillaries in the PP to release AVP into the general circulation. ACTH, adrenocorticotropic hormone; AP, anterior pituitary; AVP, arginine vasopressin; CRH, corticotropin-releasing factor; OC, optic chiasm; PP, posterior pituitary; PVN, paraventricular nucleus of the hypothalamus;
CRH neurons of the PVN serve as a final common stress pathway by receiving converging inputs from multiple areas of the brain, allowing CRH to coordinate the behavioral, neuroendocrine, and autonomic responses to stress (264). These afferent pathways include projections from (a) ascending brainstem noradrenergic pathways (from the ventrolateral medulla [VLM] and the nucleus of the solitary tract [NTS]) that relay visceral sensory information, (b) descending cortical and limbic pathways (from the septum, amygdala, and hippocampus) via the bed nucleus of the stria terminalis [BNST] that relay cognitive and emotional information, (c) intrahypothalmic connections receiving innervation from brainstem and limbic structures, and (d) circumventricular organs [CVO’s] (e.g., organum vascularis of the lamina terminalis [OVLT], subfornical organ [SFO]) that relay blood-borne chemosensory signals. Therefore, hypothalamic CRH neurons are strategically situated to intercept and disperse signals from and to the body about the internal and external environment.
In response to prolonged stress, the expression of hypothalamic CRH mRNA and pituitary POMC mRNA increase, along with hypersecretion of CRH and ACTH. However, upon continued exposure to CRH, pituitary CRH-R1 receptors downregulate, desensitizing the pituitary corticotroph to CRH signals and hence reducing ACTH release to the primary stressor (127). Although parvocellular PVN expression of arginine vasopressin (AVP) (co-expressed in CRH neurons) is low at baseline, it increases substantially in response to chronic stress. In the presence of CRH (although not alone), AVP acts synergistically to potentiate ACTH release via the vasopressin V1b receptor (linked to the PLC–Ca2+/DAG–PKC pathway) (without affecting POMC transcription) (127,163). Therefore, AVP action on pituitary corticotrophs is able to maintain corticotroph responsiveness, and hence ACTH release, to novel stressors following repeated activation of the HPA axis, despite concomitant CRH-R1 desensitization.
HPA axis activity itself also needs to be kept in check, therefore, glucocorticoids exert negative feedback at the hypothalamus and pituitary to inhibit the synthesis and secretion of CRH and POMC/ACTH, respectively (154). Moreover, glucocorticoid negative feedback causes a reduction in corticotroph CRH-R1 mRNA expression and an increase in CRH-R1 downregulation, leading to a decrease in CRH-R1 number, and hence, the desensitization of the corticotroph to the stimulatory effects of CRH on ACTH release. In addition, the hippocampus, which displays a high density of GR, exerts a negative influence on the PVN, and hence HPA axis activity, as well (129). According to Paul Plotsky (240), “One may speculate that attenuation of the ACTH secretory-response by glucocorticoid feedback conserves the response capacity of the HPA axis to subsequent stressors, and acts to limit the duration of total tissue glucocorticoid exposure, thus minimizing catabolic, anti-reproductive, and immunosuppressive effects, and counter-balancing the tendency of central circuitry to over-respond to a repeated stressor.”
BIDIRECTIONAL COMMUNICATION BETWEEN NEUROENDOCRINE AND IMMUNE SYSTEMS: HISTORICAL PERSPECTIVE
That substances released from the adrenal gland could affect immune function was one of the initial indications that there are meaningful interactions between the neuroendocrine and immune systems. Indeed, as early as the 1850s, Thomas Addison described a patient with adrenal insufficiency who exhibited an excess of circulating lymphocytes, thus providing some of the first evidence of a reciprocal relationship between adrenal hormones and immunologic parameters. In the 1920s, H. Jaffe demonstrated that adrenalectomized rats exhibited hypertrophy of the thymus, and conversely, in the 1930s, Hans Selye reported that animals exposed to a variety of stressors exhibited enlarged adrenal glands coupled with thymic involution. In the 1940s, Philip Hench discovered that patients with autoimmune disorders, such as rheumatoid arthritis, produced an endogenous substance under “stressful” conditions that had anti-inflammatory/immunosuppressive properties and hence, ameliorated the symptoms of the autoimmune disease. Isolation and characterization of this endogenous compound by Kendall led to the discovery of the adrenal steroid, cortisone, which along with other glucocorticoids, have become a mainstay in the treatment of autoimmune and inflammatory diseases. Of note, Hench and Kendall shared the Nobel prize in Medicine for their discovery in 1950.
Although the immunomodulatory effects of glucocorticoids initially were believed to be mediated by pharmacological rather than physiological concentrations of steroid, seminal work by Besedovsky and colleagues in the 1970s and 1980s substantiated a physiological role for glucocorticoids in regulating immune responses. For example, physiologic concentrations of glucocorticoids were found to facilitate antigenic specificity (40) and reduce splenic weight and cellularity (83). Besedovsky was also one of the first to demonstrate that immune system activity could influence the release of glucocorticoids. Indeed, his work demonstrated that circulating glucocorticoids increase in the rat during the immune response to foreign antigens (i.e., sheep red blood cells [SRBCs]) (37). Furthermore, rats injected with culture supernatants from peripheral blood or spleen cells stimulated with mitogens in vitro produce increases in plasma glucocorticoid levels similar in magnitude to those reached at the peak of the immune response after antigen exposure (41). Besedovsky and colleagues concluded that lymphoid cells produce a glucocorticoid increasing factors (GIF) (41,43), that completes an immunoregulatory feedback circuit, in which immune cells secrete “hormones” that stimulate the release of adrenal glucocorticoids, that in turn negatively feedback on immune cells to prevent overactivity and preserve specificity of immune responses.
The concept of bi-directional communication between immune and neuroendocrine systems became firmly established with the work of Blalock and colleagues in the 1980s. These investigators demonstrated the synthesis/expression of common ligands and receptors in immune and neuroendocrine systems, for example, neuropeptides in immune cells and cytokines in endocrine glands (48,327). In addition, they proposed the concept of a “lymphoid-adrenal axis,” in which ACTH produced by virus (Newcastle disease virus [NDV])–stimulated lymphocytes was able to directly stimulate corticosterone release in the absence of pituitary-derived ACTH (in hypophysectomized mice) (275,277). Of note, subsequent studies failed to replicate Blalock’s findings and reported that extra-pituitary sources of ACTH were not sufficient to stimulate NDV-induced adrenal steroidogenesis (38,88,91,220). Complementary to these latter studies, Besedovsky et al. (43) showed that rats injected with culture supernatants from mitogen-stimulated peripheral blood or spleen cells produced an increase in plasma ACTH concentrations (along with increased glucocorticoid levels—like NDV-infected animals); however, when the rats were hypophysectomized, the glucocorticoid response was abolished. These studies suggested that immune-induced glucocorticoid release is dependent on a functioning pituitary gland, and that “GIFs” do not directly stimulate the adrenals in the absence of pituitary-derived ACTH. Furthermore, Besedovsky et al. observed changes in hypothalamic electrical activity (36) and norepinephrine turnover (42) in SRBC-injected rats, concomitant with peak immune and corticosterone responses, suggesting that the brain is involved in immune-neuroendocrine regulation.
In the mid-1980s, a series of papers in the journal Science demonstrated that the monocyte-derived, proinflammatory cytokine IL-1 fulfilled the requirement of a GIF. These studies showed that administration of IL-1 to rats and mice increased plasma ACTH and corticosterone levels (32,35,262) and did so by acting at multiple levels of the HPA axis. Although none of these early studies provided evidence for a direct action of IL-1 on glucocorticoid release from the adrenal gland (262,333), some demonstrated a direct IL-1 effect on ACTH release from pituitary cells (34,333) and some did not (32,262). These latter studies confirmed a role for hypothalamic CRH in the IL-1–induced ACTH and glucococorticoid responses (32,262). In addition, IL-1 was shown to be a critical mediator of the HPA response during viral (NDV) infection (35). Subsequent to this early landmark work, a plethora of studies have examined the direct impact of various cytokines (e.g., IL-1, IL-6, TNFα, IFN’s, IL-2) alone or in combination, on HPA axis function. Interestingly, more recent data from both in vivo and in vitro studies indicate that cytokines may have the capacity to stimulate the adrenal gland directly, and therefore may activate glucocorticoid release independent of central nervous system (CNS) neuroendocrine pathways (see section on cytokine effects on the adrenal gland).
THE IMMUNE RESPONSE TO VIRUSES
Prior to considering the pathways by which cytokines activate the HPA axis, it is important to review some general features of the immune response to viral infection, including the role of relevant cytokines.
Immune responses to viral infections share certain characteristics that are distinct from bacterial and parasitic infections (1,136) (Fig. 3). The presence of a virus in infected cells is recognized by the detection of double-stranded RNA, generated during viral replication. This signal leads to the transcription and secretion of type I interferons (IFN-α and IFN-β), which protect neighboring cells not yet infected by making them resistant to viral replication, and hence, inducing an anti-viral state. In addition to inhibiting viral replication, type I IFN’s activate natural killer (NK) cell cytotoxicity and increase MHC class I expression, allowing for more effective antigen presentation to CD8+ cytolytic T cells (CTLs). NK cells are one of the principal mechanisms of innate immunity against viruses early in the course of infection, before late, specific immune responses have developed. In addition to their lytic functions, NK cells can be stimulated by IL-12 (released by macrophages and dendritic cells; acts in synergy with TNFα) to release IFN-γ (type II interferon), which is crucial in controlling infections before T cells have been activated to produce more IFN-γ. In the presence of IL-12 and IFN-γ, CD4+ T cells are encouraged to develop into inflammatory T cells (Th1), which promote cellular immunity through the activation of macrophages (via IFN-γ release) and CD8+ T cells (via IL-2 release). IL-2 is a crucial cytokine for T cell proliferation and differentiation. The principle mechanism of specific immunity against established viral infections, especially with non-cytopathic viruses, is the CD8+ CTL. During the early, innate immune response to viral infections (as well as other types of infections), the proinlfammatory cytokines, TNFα, IL-1, and IL-6, also are induced to aid in the progression of anti-viral immunity.
FIG. 3.
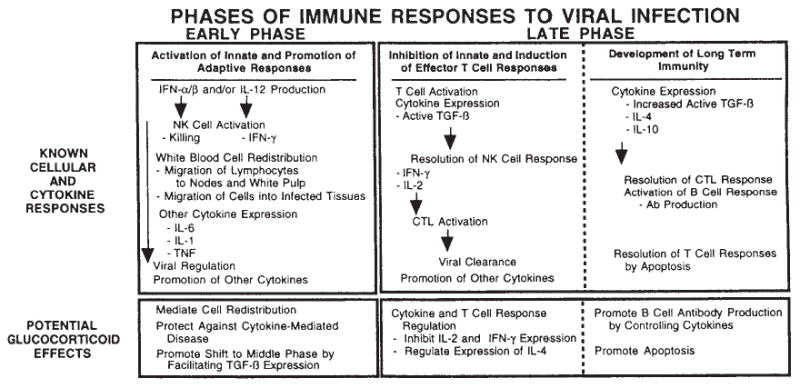
Cellular and cytokine responses to viral infection and potential immunomodulatory effects of glucocorticoids. Ab, antibody; CTL, cytotoxic T lymphocyte; IFN, interferon; IL, interleukin; NK, natural killer; TNF, tumor necrosis factor; TGF, transforming growth factor. Reprinted by permission from Miller et al. (188).
When considering the HPA axis effects of cytokines, it is important to keep in mind that (a) cytokines are pleiotropic (one cytokine can exert many actions), (b) they are redundant (different cytokines can exert the same action), (c) they often influence the synthesis of other cytokines (e.g., TNF → IL-1 → IL-6, while IL-6 inhibits TNF and IL-1 synthesis), and (d) they often influence the action of other cytokines (e.g., TNF, IL-1, and IL-6 can act synergistically). The importance of synergistic actions of cytokines in HPA axis stimulation has been demonstrated by several studies. For example, in an in vitro preparation of isolated rat hypothalami, Buckingham et al. (59) showed that the release of CRH by conditioned media from lipopolysaccharide (LPS)–stimulated peritoneal macrophages (containing multiple cytokines) was much greater than that observed in response to TNFα, IL-1, or IL-6 alone. In addition, there is in vivo evidence that supports the role of synergistic actions of these proinflammatory cytokines in stimulating a complete, LPS-induced ACTH response (236) and in stimulating greater HPA axis activity together compared to their individual effect (310).
CYTOKINE–HPA AXIS INTERACTIONS: POTENTIAL MECHANISMS
Given the importance of the glucocorticoid response in restraining potentially detrimental immune responses, redundant or complementary pathways of glucocorticoid induction are activated by cytokines at all three levels of the HPA axis, including the hypothalamus, pituitary, and adrenal gland. Although hypothalamic CRH is considered a primary mechanism by which cytokines stimulate glucocorticoid release, increasing evidence supports a direct action of cytokines at the level of the pituitary and adrenal glands as well. Cytokine receptors have been detected at all HPA axis levels and therefore, each level can serve as an integration point for immune and neuroendocrine signals. In addition to circulating cytokines being able to act upon all three levels of the HPA axis, cytokines are synthesized in the brain, the anterior pituitary, and the adrenal gland. The production of local cytokines may function in a paracrine manner to amplify and maintain elevated HPA activity during chronic inflammation. Therefore, each level of the HPA axis contains a local cytokine network, which can be stimulated by a variety of circulating cytokines. In examining the effects of cytokines on HPA axis function, the innate proinflammatory cytokines—IL-1, IL-6, and TNFα—have been the most studied. However, other cytokines (e.g., IFN’s and IL-2) also have been shown to influence HPA axis activity.
Brain/hypothalamus (PVN)
The majority of evidence indicates that activation of hypothalamic CRH is the primary means by which cytokines stimulate the release of ACTH and glucocorticoids. Since cytokines are large, soluble peptides, significant consideration has been given as to how cytokines cross the blood–brain barrier (BBB) and activate CRH-producing neurons in the PVN. Several mechanisms, which are not mutually exclusive, have been identified. Cytokines may (a) stimulate visceral (vagal) afferents that project to the nucleus tractus solitarius (NTS) in the brainstem, activating the release of norepinephrine (NE) from catecholaminergic terminals (of the ventral noradrenergic bundle [VNAB]) in the PVN, (b) passively cross the BBB at “leaky” regions where the BBB is not intact, such as the circumventricular organs (CVO’s) [e.g., organum vascularis of lateral terminalis (OVLT), subfornical organ (SFO), and area postrema (AP)], and directly activate neurons that project to the hypothalamus, (c) exert a direct effect on CRH nerve terminals in the median eminence (ME), (d) act on endothelial cells of brain vasculature or glial cells in the CVO’s, inducing the synthesis/release of secondary messengers such as central cytokines, prostaglandins (PG’s), or nitric oxide (NO), which in turn activate hypothalamic neurons, and (e) cross the BBB themselves via active transport. Evidence exists to support all of these pathways, depending on the cytokine dose, time interval, and route of administration (247,300,302,322) (Fig. 4).
FIG. 4.
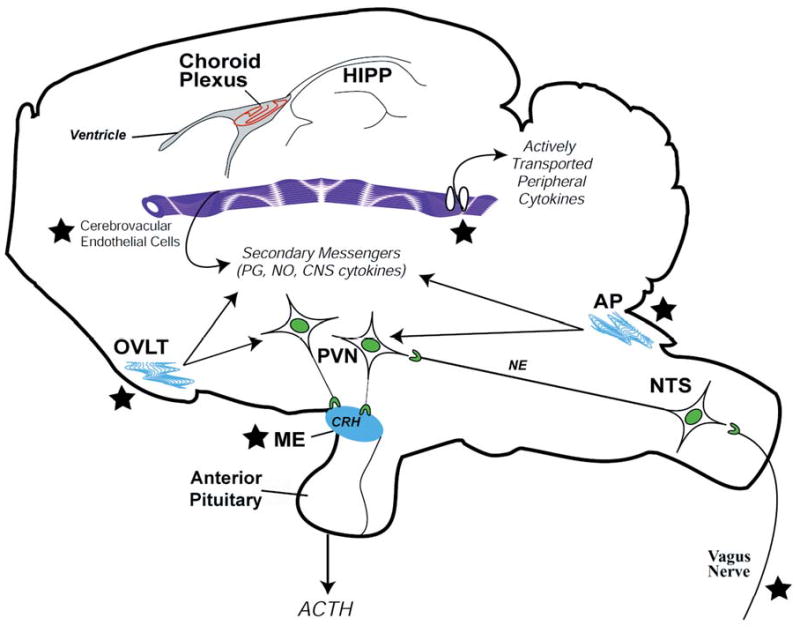
Proposed mechanisms by which peripheral cytokines activate CRH-producing neurons of the PVN in the hypothalamus (rat brain). ACTH, adrenocorticotropic hormone; AP, area postrema; CRH, corticotropin-releasing hormone; HIPP, hippocampus; ME, median eminence; NE, norepenephrine; NO, nitric oxide; NTS, nucleus tractus solitarius; OVLT, organum vascularis of lateral terminalis; PG, prostaglandins; PVN, paraventricular nucleus of the hypothalamus. ★ = site of entry of peripheral cytokine signals. Reprinted by permission from Silverman et al. (274).
Cytokine network in the brain
Cytokine receptors
Once the peripheral immune signal reaches the brain, cytokines can interact with their receptors in multiple brain regions involved in HPA axis regulation, including the hypothalamus (PVN) and the hippocampus, which provides negative feedback regulation on PVN activity (129). In addition, many of the aforementioned brain regions involved in relaying the circulating cytokine message to the PVN have been shown to express cytokine receptors.
Of the two types of IL-1 receptors, both IL-1RI (activating) and IL-IRII (decoy) mRNA and protein have been detected in the mouse brain (102,230). In the mouse, high levels of IL-1RI mRNA expression are found in the hippocampus (dentate gyrus), the midline raphe system, the choroid plexus, meninges, and endothelial cells of post-capillary venules throughout the brain (78,173). However, only a scattered signal for IL-1RI mRNA has been detected in the ME, and no signal in the hypothalamus has been found. In contrast, IL-1RI mRNA in the rat brain is predominantly expressed over blood–brain barrier–related cells and not strongly expressed in certain neuronal populations as seen in the mouse brain (173). More specifically, constitutive expression of IL-1RI mRNA has been detected at the interface of the vascular wall and perivascular glia (335) and on endothelial cells (308) of the rat brain. In addition, IL-1 binding sites are detected in vagus nerve paraganglia (115). Peripheral administration of LPS has been shown to either downregulate (178,291) or upregulate (106,200) IL-1 receptors in the mouse brain.
Binding of IL-6 to IL-6R is followed by the rapid association of the gp130 molecule, the signal transducing component of the IL-6R complex, which increases the affinity of the IL-6R for IL-6. In the untreated rat, IL-6R mRNA has been detected in numerous brain regions that have been implicated in playing a role in HPA axis regulation, including the hippocampus, hypothalamus, and circumventricular organs such as the median eminence (ME) (266,305). However, the PVN displays no specific hybridization signal for IL-6R mRNA under basal conditions (266,305). Valliere et al. (305) have demonstrated that intraperitoneal (ip) injection of LPS in rats is able to upregulate the expression of IL-6R mRNA in selective regions of the brain that constitutively express IL-6R mRNA. Interestingly, LPS also induces IL-6R mRNA expression in some areas in which no IL-6R mRNA is detected under basal conditions, such as the PVN and endothelial cells of cerebral blood vessels (305). IL-6R mRNA has been localized to glial cells, ependymal cells, and neurons (263,266). Gp130 immunoreactivity also has been observed in both glial and neural cells, and its distribution in the rat brain overlaps with that of IL-6R (320). Gp130 mRNA expression is detected throughout the rat brain (including the PVN) and is upregulated in the OVLT and in endothelial cells of cerebral microvasculature in response to peripheral administration of LPS (305).
TNF exerts its functions through two structurally related, but functionally distinct receptors, TNF-R1 (p55) and TNF-R2 (p75). Whereas TNF-R1 mRNA is constitutively expressed in the CVO’s (including the ME), choroid plexus, meninges, and microvasculature, and to a lesser extent in the PVN, TNF-RII mRNA is barely detectable in the rat brain under basal conditions (198). Systemic treatment with LPS induces an increase TNF-RI mRNA expression in cells of blood–brain barrier–associated structures (198). Specific binding sites for TNF in mouse brain homogenates have been shown to be widespread, but maximal in the brainstem (159). Both types of TNF receptors have been localized to cerebrovascular endothelial cells (22), glial cells (86), and neurons (49).
Even though IFN-α and IFN-β show little structural similarity to each other, they both bind to the same receptor, the type I IFN receptor (type I IFN-R). In humans, type I IFN-R protein is constitutively expressed in microglia and its expression is increased in central inflammatory conditions (339). In addition, binding sites for IFN-α have been identified in the rat brain, the hypothalamus in particular (142). The receptor for IFN-γ is distinct from that of IFN-α/β. IFN-γR mRNA is constitutively expressed in the mouse brain, and its expression is increased by LPS (303). mRNA for IFN-α/β and -γ receptors also have been detected in astrocytes of the human brain (290).
IL-2 can bind to two distinct receptors: the IL-2Rα and the IL-2 Rβγc. Binding of IL-2 to IL-2Rα alone does not lead to any detectable biological response, but requires binding to the IL-2 Rβγc as well. IL-2 first binds rapidly to IL-2Rα, which facilitates its association with IL-2 Rβγc, thereby increasing the affinity of the signaling receptor for IL-2. IL-2 binding sites and/or IL-2 receptors are observed in the rat hypothalamus and hippocampus (9,168). IL-2R mRNA expression in the brain has been shown to increase upon exposure to LPS in mice (303).
Cytokine synthesis
During systemic inflammation or infection, the central production of cytokines may function in a paracrine manner to amplify the signal of circulating cytokines and maintain elevated HPA axis activity. Evidence for a role in centrally synthesized cytokines comes from the observation that centrally administered IL-1, IL-6, and TNFα stimulate ACTH release at doses much lower than the effective doses administered peripherally (300). In addition, central administration of cytokine antibodies or antagonists attenuate HPA axis activity induced by the peripheral administration of LPS and particular cytokines (194). In healthy brains, cytokines are expressed at low levels or not at all (265). Constitutive expression of cytokines tends to be localized to neurons, whereas induced central cytokines are mainly produced by glial cells, especially microglia (170).
Constitutive expression of IL-1 protein has been detected in the rat hypothalamus and hippocampus (171), although some studies have failed to detect basal IL-1 expression in the rat brain (15,98,307). The discrepancy in results may be due to a difference in sensitivity of the assays used. In response to systemic LPS administration, an increase in IL-1β immunoreactivity or mRNA expression has been detected in perivascular microglia and macrophages in the meninges, choroid plexus, CVO’s, and hypothalamus in the rat brain (98,307) and in the mouse hypothalamus and hippocampus (170,239). Ip injection of LPS has been shown to induce a biphasic response in IL-1β mRNA expression in the rat brain, where an early phase of induced transcription (2 h post-injection) can be detected in areas without an effective blood–brain barrier, such as the choroid plexus, meninges, and cerebrovasculature, followed by a later phase of expression (6 h post-injection) in the parenchyma, including areas such as the hypothalamus (336).
Whereas Schobitz et al. (266) have shown that IL-6 mRNA in the rat brain is constitutively expressed in regions such as the hippocampus and hypothalamus (colocalized with IL-6R mRNA), Vallieres et al. (305) have reported that IL-6 mRNA expression is undetectable under basal conditions. Upon ip LPS administration, however, IL-6 mRNA is increased in the CVO’s (including the ME) and the choroid plexus of the rat (305) and in the brainstem, hippocampus, and hypothalamus of the mouse (170,239). Whereas LPS induces a strong and transient increase in IL-1 and TNFα mRNAs in the hypothalamus and hippocampus, the induction of IL-6 mRNA tends to be progressive over time (170). In vitro, IL-6 mRNA and release is detected in rat medial basal hypothalamic fragments under basal conditions and is further stimulated by LPS, presumably from glial cells (284).
While no expression of TNFα mRNA has been detected in the rat brain under basal conditions (199), the mouse brain has been shown to exhibit constitutive levels of TNFα mRNA (239) and TNFα immunoreactivity in neurons of the hypothalamus, caudal raphe system, and the ventral surface of the brainstem (56). Increased TNFα mRNA expression is observed in the rat (109) and the mouse (170) hypothalamus in response to systemic LPS administration. Similar to the pattern of central induction of IL-1 mRNA, ip injection of LPS tends to stimulate a biphasic response in TNFα mRNA expression in both the rat (199) and the mouse (55) brain, where an early phase of induced transcription can be detected in blood–brain barrier–related structures such as the choroid plexus, CVOs, and meninges, followed by a later phase of expression in the parenchyma, including areas such as the hypothalamus and the NTS. Naduau et al. (199) have shown that the induced TNFα is mainly a product of microglia and brain macrophages.
IFN-α protein has been localized to microglia and neurons of the human brain and its microglial expression is increased (along with the expression of IFN-R) in central inflammatory conditions (338). IFN-α mRNA also is constitutively expressed in the human brain (54). Although IFN-γ mRNA is not constitutively expressed in the mouse brain, its expression is induced by LPS (239). A neuronal IFN-γ, which has a molecular weight distinct from that of lymphocyte-derived IFN-γ but crossreacts immunologically and shares certain bioactivities with it, has been detected in the hypothalamus and hippocampus of the rat brain (31,172,221). IFN-γ–like immunoreactivity is abundantly present in the rat peripheral nervous system, while weaker staining has been detected in the brain, specifically in the hypothalamus and midbrain (155).
IL-2 immunoreactivity has been detected in the rat brain, in regions such as the hypothalamus, ME, and hippocampus (9,168). In addition, Araujo et al. (8) have shown that IL-2 is released from rat astroglial and microglial cells in culture and its release is enhanced by IL-1. IL-2 mRNA is detected in the hypothalamus, including the PVN, following iv injection of tetanus toxin in rats (162).
Cytokine induction of CRH
The proinflammatory cytokines—IL-1, IL-6, and TNFα—have all been shown to stimulate HPA axis activity in vivo in various species (39,300). Release of CRH from PVN neurons into the ME is established as a primary pathway by which cytokines stimulate the HPA axis. Early groundbreaking studies by Sapolsky et al. (262) and Berkenbosch et al. (32) concluded that ip administration of IL-1 (in rats) induces a CRH-dependent ACTH response, based on effective neutralization of the response by prior administration of CRH-antisera and detection of increased CRH release, as assessed by an increase of CRH in the hyposhysial portal blood (Sapolsky et al.) and a decrease in CRH content in the median eminence (Berkenbosch et al.). Furthermore, participation of CRH-producing neurons of the PVN is implicated by the findings of IL-1–induced expression of c-fos mRNA (a cellular marker of neuronal activation) and an increase in CRH mRNA in the PVN (97). Similarly, it has been demonstrated that PVN lesions completely prevent the increase in ACTH induced by intravenous (iv) administration of IL-6 and TNFα (164), and that immunoneutralization of CRH blocks the stimulatory effects of IL-6 and TNFα on ACTH and/or corticosterone release (33,146,205,311). IL-1, IL-6, and TNFα also have been shown to stimulate the release of CRH from rat hypothalamic explants (33,206,287). Moreover, both IL-1 and IL-6 are capable of acting directly on CRH nerve terminals in the ME (181,183,270,287), thereby increasing CRH release without necessarily increasing CRH synthesis in the PVN. However, a direct action of TNF at the level of the ME has not been demonstrated. Although iv administration of TNFα results in an increase in CRH release from the ME (321), Sharp et al. (270) failed to show an effect of TNFα on ACTH secretion when injected directly above the ME.
Some studies have reported differential effects of proinflammatory cytokines on PVN activity. While IL-1 and IL-6 were able to stimulate ACTH secretion after intra-ME delivery, only IL-1 was able to stimulate ACTH secretion after injection into the PVN (181). Callahan et al. (62) also reported an increase in Fos expression in CRH-producing neurons of the PVN in response to ip administration of IL-1β, but not to IL-6. Moreover, an ip injection of IL-1β, in addition to eliciting ACTH and corticosterone responses, produced an increase in CRH mRNA expression in the PVN, while IL-6 did not induce an increase in CRH mRNA expression, despite an increase in ACTH and corticosterone (126). Initially, Vallieres et al. (304) reported that a single systemic injection of IL-6 was insufficient by itself to induce c-fos and CRH mRNA in the PVN. However, if IL-6 receptors were first induced in the PVN by prior LPS (iv) exposure (IL-6R was barely expressed in the PVN under basal conditions), then IL-6 (iv) was able to induce an increase in c-fos mRNA and CRH hnRNA in the PVN, concordant with a significant corticosterone response (306). In addition, continuous infusion of IL-6 has been shown to induce Fos expression in the PVN (207). Vallieres et al. (306) also demonstrated that although IL-6 did not appear to be essential during the early phases of endotoxemia, IL-6 was required during the later phases to prolong the activation of neurons throughout the brain and to maintain CRH expression in the PVN. In addition, iv administration of TNFα induces c-fos mRNA and CRH hnRNA expression in the PVN of rats, concordant with elevated plasma corticosterone levels (198). Taken together, these data suggest that IL-1 and TNFα may be responsible for activating CRH-producing neurons of the PVN in response to an acute immunological challenge, while IL-6 may be responsible for the PVN response to chronic inflammation or in instances where the PVN has been previously sensitized to IL-6 by prior exposure to other cytokines (i.e., TNFα, IL-1).
IFN-α is a mainstay therapy used to treat certain chronic viral infections (e.g., hepatitis C) and malignant disorders (e.g., cancer) and has been shown to cause HPA axis stimulation in humans when administered to either of these populations of patients (65,113,195,252). However, when administered to rats, either peripherally or centrally, recombinant human IFN-α inhibits HPA axis activity as indicated by a reduction in the electrical activity of CRH neurons in the PVN and plasma corticosterone levels (260,261). Interestingly, low doses of rat IFN-α (by both ip and icv administration) also decreases corticosterone levels in the rat, while increases in corticosterone concentrations were found following icv administration of high doses of rat IFN-α (260). Saphier et al. (260) suggest that the physiological significance of the IFN-α-induced inhibition of HPA activity, and hence glucocorticoid release, may be to enhance the immunopotentiating effects of IFN-α. Dafny et al. (80) also have shown that both peripheral and central administration of IFN-α in rats suppresses hypothalamic neural activity, while stimulating neural activity in other brain regions, such as the hippocampus. In vitro, IFN-α has been shown to stimulate (113,244) or to have no effect (191,206) on CRH release from rat hypothalamic cultures. IFN-β is a common therapy in patients suffering from multiple sclerosis. The HPA axis–stimulating effects of IFN-β has been less studied than IFN-α. In studying the neuroendocrine effects of this cytokine, Geobel et al. (114) have demonstrated that the administration of IFN-β to healthy subjects elevates plasma cortisol levels, concomitant with increases in plasma TNFα and IL-6 concentrations. Peripheral administration of IFN-γ enhances cortisol secretion when given to healthy men (136), but fails to induce ACTH/corticosterone responses when given to mice or rats (35). Central administration of IFN-γ has been shown to potentiate the LPS-induced increases of TNFα and IL-6 protein in CSF and serum and mRNA in the rat brain (82). In vitro, IFN-γ has failed to exhibit an effect on CRH release from rat hypothalamic explants (206) or superinfused hypothalamic-neurohypophysial complexes (218). Disparities between results within in vivo and in vitro paradigms may be due to species-specificity effects and dose effects of the cytokines tested. In addition, some of the effects of IFN’s on HPA axis activity may be attributed to the induction and action of downstream proinflammatory cytokines (e.g., TNFα and IL-6).
IL-2 also is used in immunotherapy for cancer patients and has been shown to stimulate ACTH and cortisol responses in this population (84,176). When administered to HIV-infected patients, IL-2 induces ACTH and cortisol responses and increases the ACTH/cortisol responses to CRH stimulation as well (332). On the contrary, acute (35) and chronic (201) systemic IL-2 administration to rats fail to induce an increase in ACTH and corticosterone. However, recombinant human IL-2 was used in these studies, and Naito et al. (203) have shown that iv injection of rat IL-2, but not human IL-2, is able to stimulate ACTH release in rats. Although systemic administration of IL-2 in mice induces a corticosterone response (60,69), Zalcman et al. (342) failed to show significant glucocorticoid induction following an ip injection of IL-2 in mice. Icv infusion of IL-2 in rats also is capable of activating an ACTH/corticosterone response (122). Both peripheral and central administration of IL-2 alters hypothalamic electrical activity in mice and rats, however, there is region-specificity, where decreases in activity have been observed in the lateral anterior (LAH) and ventromedial (VMH) areas and increases in the supraoptic (SON) and paraventricular (PVN) nuclei (20,46). Whereas an ip injection of IL-2 into rats does not produce an increase in CRH mRNA expression in the PVN (concordant with an increase in POMC mRNA expression in the anterior pituitary) (125), IL-2 has been shown to increase AVP mRNA in the hypothalamus of mice (227). Moreover, IL-2 stimulates the release of AVP (but not CRH) from the intact rat hypothalamus in vitro and hypothalamic cell cultures (132). IL-2 also failed to have an effect on CRH release from rat hypothalamic explants (132,206). Therefore, IL-2–induced ACTH responses may be driven by AVP rather than CRH. On the other hand, there are studies in which IL-2 has been shown to stimulate CRH release from rat perifused hypothalami (63), hypothalamic explants (150), and hypothalamic slices (244). Again, disparities between results may be due to species-specificity effects of the cytokine used.
Arginine vasopressin (AVP)
Aside from CRH, other hypothalamic secretagogues may contribute to cytokine–HPA axis interactions. Arginine vasopressin (AVP) is one such peptide. Although AVP on its own does not result in increased POMC mRNA expression, in the presence of permissive levels of CRH, it acts synergistically with CRH to stimulate ACTH release from the anterior pituitary. Proinflammatory cytokines have been shown in one study or another to influence AVP synthesis or release (59,300). However, a portion of this AVP response may originate from the magnocellular, rather than the parvocellular, cells of the PVN and be destined for release from the posterior pituitary and ultimately the general circulation (Fig. 2). There is strong evidence that under conditions of chronic inflammation, such as rheumatoid arthritis, cytokine effects on HPA axis activity may be driven primarily by AVP (parvocellular-derived). In arthritic rats, Harbuz et al. (124) have demonstrated an increase in POMC mRNA expression in the anterior pituitary concordant with a paradoxical decrease in CRH mRNA in the PVN and CRH release into the hypophysial portal blood (HPB), while an increase in AVP release into the HPB was detected. In addition, Lewis rats, which are susceptible to developing chronic inflammatory disease due to impaired CRH and corticosterone secretion, have high plasma levels (magnocellular-derived AVP), hypothalamic content, and in vitro release of AVP in comparison with the inflammatory-disease resistant Fisher rats (232). Similar findings have been reported in animal models of other chronic inflammatory disorders, such as lupus (256,269). Taken together, these results suggest that an increase in AVP may attempt to compensate for low CRH drive in HPA axis regulation.
Pituitary Gland
Although strong evidence has been provided in support of cytokine action at the level of the CNS to activate the HPA axis, numerous in vitro studies indicate that cytokines can exert a direct effect on ACTH release from the anterior pituitary. In addition, in vivo work by our lab and others have demonstrated that animals whose CRH or CRH-R1 gene has been eliminated can produce ACTH and corticosterone responses to immune challenges (44,273,301). These data indicate that pathways are in place for direct cytokine–pituitary interactions.
Cytokine network in the pituitary gland
Cytokine receptors
Receptors for cytokines have been identified in various cell types that reside in the pituitary. mRNA for both types of IL-1 receptors, IL-1RI and IL-IRII, have been detected in the mouse anterior pituitary and in the mouse corticotropic tumor cell line AtT-20 (78,231). In addition, IL-1R1 mRNA is constitutively expressed in the rat pituitary gland (328) and is upregulated by LPS in the mouse pituitary gland (241). Furthermore, IL-1RI and IL-1RII immunoreactivity have been localized in somatotrophs in the normal mouse anterior pituitary (103). IL-6 binding sites are detected in the rat anterior pituitary and in human gonadotrophs (219). Moreover, IL-6R protein has been localized to mouse corticotrophs and mRNA’s of IL-6R and gp130 are expressed in rat corticotrophs, which are upregulated in response to LPS (110). TNF binding sites are found at high concentrations in the anterior lobe of both mouse and rat pituitary glands (334). In addition, the ACTH-producing AtT-20 cells express both Type I and II TNF receptor mRNA and the folliculostellate TtT/GF cells (pituitary source of IL-6, see below) have been found to express TNF-R1 mRNA (160). IFN-γR mRNA is constitutively expressed in the mouse pituitary and is upregulated by LPS (303). IL-2R immunoreactivity has been detected in rat pituitary and its expression is upregulated by the pre-exposure to IL-2 (295). IL-2R mRNA expression in the pituitary has been shown to increase upon exposure to LPS in mice (303). More specifically, Arzt et al (10) have detected IL-2R mRNA and protein in AtT-20 cells and corticotrophs of normal rat anterior pituitary cells. Moreover, CRH increases IL-2R mRNA expression in AtT-20 cells (10,237).
Although cytokine receptors have been found on AtT-20 cells, it is unclear to what extent corticotroph tumors accurately represent normal anterior pituitary corticotrophs. The lack of evidence on some cytokine receptor localization to normal corticotrophs may suggest that paracrine interactions are involved between the endocrine cells that express those receptors and the ACTH-producing corticotrophs themselves. Moreover, the presence of cytokine receptors on non-endocrine cell types, suggests that in addition to exerting a direct effect on ACTH release, cytokines (i.e., TNFα) may indirectly modulate ACTH release by first stimulating the release of local cytokines, such as IL-6 from folliculostellate cells.
Cytokine synthesis
Like the brain, the pituitary can synthesize its own set of local cytokines and serve as an integration point for immune and neuroendocrine signals. IL-1β protein and mRNA have been detected in the rat anterior pituitary, and both are increased following ip LPS injections (161,291,328). LPS administration also upregulates IL-1α/β expression in the pituitary gland of mice (105,241). In addition, Koenig et al. (161) showed that IL-1β was localized to cytoplasmic granules in anterior pituitary endocrine cells and colocalized with TSH in thyrotrophs. IL-1α/β mRNA also have been demonstrated in monocytes in both the anterior and posterior lobes of the rat pituitary gland (98). Cultured rat anterior pituitary cells spontaneously secrete large quantities of IL-6, which is enhanced by exposure to LPS (285) and to IL-1α/β (282,283). The source of pituitary IL-6 has been shown to be the folliculostellate cell (313), which is not a hormone-secreting cell, but a cell of monocytic lineage that is thought to be involved in paracrine regulation of hormone secretion from the pituitary (13,138). Moreover, LPS and TNFα have been shown to directly stimulate IL-6 release from mouse pituitary FS TtT/GF tumor cells (160,174), while LPS produces a similar effect on FS cells of mouse pituitary cell cultures (174). IL-6 immunoreactivity also is expressed in ACTH and FSH/LH-secreting cells in normal human pituitary glands (167) and IL-6 mRNA is induced in the rat pituitary by ip administration of LPS (197). TNFα mRNA is constitutively expressed in the mouse pituitary (239) and is upregulated in response to systemic injection of LPS in the mouse and rat (109,170,199,239). IFN-γ mRNA is constitutively expressed in the mouse pituitary and is upregulated by LPS (239). Arzt et al. (10) have detected IL-2 mRNA in human corticotrophic adenoma cells and mouse AtT-20 cells. The role of local, pituitary-derived cytokines may be to amplify and maintain elevated pituitary-adrenal activity during chronic inflammation.
Cytokine induction of ACTH
Numerous in vitro studies support the contention that cytokines can directly stimulate ACTH release from the pituitary gland. IL-1 has been shown to induce POMC transcription and ACTH release from cultured rat anterior pituitary cells (34,57,153), while IL-6 has been demonstrated to stimulate ACTH secretion from cultured hemipituitaries (177). In addition, both IL-1 and IL-6 have been shown to increase POMC mRNA expression or promoter activity and ACTH release from AtT-20 cells (57,104,152,333). Moreover, Fukata et al. (104) have shown that IL-1β can sensitize AtT-20 cells to release ACTH in response to CRH. TNFα also has the ability to induce ACTH release from cultured rat anterior pituitary cells (33) and to stimulate POMC transcription from AtT-20 cells (152,160). Although the acute effects (<4 h) of IL-1, IL-6, and TNFα alone, on POMC promoter activity in AtT-20 cells were minimal (peak responses occurred >12 h), these cytokines significantly potentiated the stimulatory effect of CRH at that time (152). However, according to Gaillard et al. (108), TNFα exerts an inhibitory effect on CRH-stimulated ACTH release from rat anterior pituitary cells, while having no effect on basal ACTH secretion.
It should be noted, however, there are studies that have failed to demonstrate a direct action of IL-1 (32,206,262), IL-6 (206), and TNFα (153,206,270) on cultured anterior pituitary cells to stimulate ACTH release. The discrepancy between these reports may be explained by differences in the incubation times used, in which short incubation periods (<4 h) may not be long enough to observe a direct effect of the cytokine on the pituitary. Therefore, CRH-dependent ACTH release may mediate the rapid onset of an acutely induced glucocorticoid response, whereas a direct action of cytokines on the anterior pituitary may mediate more latent glucocorticoid responses due to chronic inflammation. Another possible reason for the lack of an effect of individual cytokine action on ACTH release at early time points may be the need for synergistic actions between multiple cytokines. For example, Buckingham et al. (59) have demonstrated that rat anterior pituitary cells incubated with conditioned media from macrophages stimulated with LPS (which contains multiple cytokines) increase their ACTH secretion, while no effect was observed when incubated with various cytokines alone.
Although IFN-γ increases ACTH release from AtT-20 cells (152), neither IFN-α nor IFN-γ has been reported to exert a direct effect on ACTH release from rat pituitary cells (113,136,206). Interestingly, Katahira et al. (152) have shown that both IFN-α and IFN-γ exhibit acute stimulatory effects (~4 h), followed by marked inhibitory effects (~12 h) on POMC promoter activity in AtT-20 cells. IFN-γ inhibits CRH-induced ACTH release from rat anterior pituitary cell cultures, although fails to do so when AtT-20 cells are used (312). Vankelecom et al. demonstrate the dependence of this effect on the presence of folliculostellate cells in the normal pituitary cultures (312) and that this immune–endocrine interaction may be mediated by the induction of iNOS in a subpopulation of these FS cells and other types of yet to be identified non-hormone-secreting cells (314). IL-2 has been shown to stimulate ACTH release from rat hemipituitaries (151) and pituitary cells (279). However, Navarra et al. (206) failed to show an effect of IL-2 on ACTH release from rat pituitary cells, which may be attributed to the use of recombinant human IL-2 rather than rat IL-2. Although Brown et al. (57) demonstrated that IL-2 enhances POMC mRNA in both rat primary pituitary and AtT-20 cells, other studies have failed to show an effect of IL-2 on ACTH release (104) and POMC promoter activity (152) in AtT-20 cells. The observation that one can detect an effect of cytokines on POMC transcription or ACTH release in normal pituitary cells and not in AtT-20 cells (mouse corticotrophic cell line) may be due to the presence of other cells types/accessory cells that may be important in mediating the immune-endocrine interaction.
In vivo, ip administration of either IL-1 or IL-6 has been shown to induce Fos expression in the anterior pituitary (62). However, IL-1, but not IL-6, produces an increase in POMC mRNA in the anterior pituitary with a concomitant increase in plasma ACTH (126). Perhaps IL-6 could be exerting a direct effect on the pituitary to increase ACTH release, without increasing its synthesis. Ip administration of TNFα decreases POMC mRNA in the rat anterior pituitary (255). Ip administration of IL-2 in mice induces an increase in POMC mRNA expression in the anterior pituitary along with increases in plasma corticosterone levels (69), and this increase in POMC mRNA expression in rats is seen without a concomitant induction of CRH mRNA in the PVN (125). As mentioned before, IL-2 induction of POMC mRNA may be driven by AVP rather than CRH.
Besides IL-1 being able to increase pituitary sensitivity to CRH, CRH has been shown to play a role in increasing pituitary sensitivity to circulating cytokines. Payne et al. (234) demonstrated that low levels of exogenously administered (or endogenously produced CRH) may be necessary for IL-1 to induce ACTH release via direct action on the anterior pituitary. Once sensitized, IL-1–induced ACTH release was no longer inhibited by a CRH antagonist. CRH also has been shown to increase IL-1R expression in AtT-20 cells (324) and increase IL-1α binding in the anterior pituitary (292). Moreover, PVN-lesioned rats injected (ip) with LPS exhibit a delayed ACTH response compared to CRH-intact animals (95). These data support the idea that while the initial ACTH response to cytokines may be dependent on CRH release from the hypothalamus, a more sustained ACTH response may result from a direct action of cytokines on the pituitary gland that previously has been sensitized by CRH. However, without the prior sensitization by CRH (possibly by the upregulation of cytokine receptor expression), direct cytokine action on the pituitary may occur only after prolonged exposure to circulating cytokines.
Adrenal Gland
There has been a growing body of evidence to support the idea of extrahypothalamic and extrapituitary regulation of adrenal steroidogenesis, whereby circulating cytokines act directly on the adrenal gland. The adrenal cortex is comprised of three histologically distinct layers. From the outermost to innermost (adjacent to the medulla), these layers are the zona granulosa (synthesize mineralocorticoids) and the zonas fasciculata and reticularis (synthesize glucocorticoids and androgens). Although blood flow within the adrenal gland is directed centripetally from the cortex to the medulla, secretory products released by medullary chromaffin cells (catecholamines and various neuropeptides) are able to influence steroidogenesis in the adrenal cortex in a paracrine manner (93). Rays of medullary tissue have been found to extend through the entire cortex of the human adrenal gland, and steroid producing cells occur diffusely within the entire adrenal medulla, with the two cell types located in direct apposition (52,53,330). Systemic cytokine actions at the adrenal gland and their effect on glucocorticoid synthesis/release may be dependent upon intricate intra-adrenal interactions between cortical and medullary cells, the vascular supply, neural input, and locally synthesized cytokines and neuropeptides (e.g., an intra-adrenal CRH-ACTH system) (50,94).
Cytokine network in the adrenal gland
Cytokine receptors
Although some studies have failed to detect the presence of IL-1 receptors (mRNA or binding) in the adrenal gland (78,331), Nussdorfer et al. (215) have demonstrated the presence of IL-1β binding sites in the zona glomerulosa and medulla of the human adrenal gland, and Nazarloo et al. (241) have shown that IL-1RI mRNA is expressed in mouse adrenal glands. Furthermore, the expression of IL-1RI mRNA in the mouse adrenal gland is upregulated by LPS (241). Receptors for both IL-6 and TNFα also have been identified in the adrenal gland of numerous species. IL-6R mRNA has been detected primarily in the zona reticularis and the zona fasciculata (to a lesser extent in the zona glomerulosa and the medulla) of the mouse (44) and the human adrenal gland (233), the zona fasciculata of the bovine adrenal gland (18), and more intensely in the medulla than in the cortical regions of the rat adrenal gland (107). Based on immunohistochemical studies, TNF receptors (types I and II) have been localized to the zona fasciculata of the bovine adrenal gland (18). In addition, TNF binding has been demonstrated throughout the rat and bovine adrenal cortex and in the human adrenocortical tumor cell line H295R (143). IFN-α binding sites have been demonstrated in bovine adrenals (67). IL-2R immunoreactivity has been detected in rat adrenal cells and its expression is upregulated by the pre-exposure to IL-2 (295). In addition, the presence of IL-2 binding sites have been demonstrated in the human (215) and rat (111) adrenal gland.
Cytokine synthesis
Just as the brain and anterior pituitary can produce their own cytokines, cytokine synthesis by multiple cell types of the adrenal gland has been demonstrated. IL-1α is produced by rat chromaffin cells (19). IL-1β, IL-6 and TNFα mRNA’s have been detected in steroid-producing cells and macrophages of the zona reticularis in the human adrenal gland (117–119). Macrophages have been localized in all zones of the human and bovine adrenal gland and lie in direct contact with cortical and chromaffin cells (79,116). LPS administration has been shown to induce an increase in IL-1β mRNA expression (209) and protein concentrations (58) in the rat adrenal gland, and an increase in IL-1α/β mRNA expression in the adrenal glands of mice (241). IL-6 is primarily produced by rat and bovine adrenal zona glomerulosa cells (and to a lesser extent by zona reticularis and fasciculata cells) (61,144). In addition, its release is stimulated by IL-1α/β, LPS, angiotensin II, and ACTH, with IL-1β also potentiating the stimulatory effect of ACTH on IL-6 secretion (144). IL-6 mRNA in the rat adrenal gland also is induced by ip administration of LPS (197). TNFα immunoreactivity is found throughout the bovine adrenal cortex (most prominent in the zona glomerulosa), and while its release is stimulated by IL-1α/β, LPS, and angiotensin II, it is inhibited by ACTH (61). The same has been shown for TNFα release from rat zona glomerulosa cells (145). Rats treated with LPS also exhibit an increase in adrenal IFN-γ concentrations (58). As with the pituitary, the role of local, adrenal-derived cytokines may be to amplify and maintain elevated adrenal activity in response to chronic inflammation.
Cytokine induction of glucocorticoids
Data from numerous in vitro studies have demonstrated that cytokines can directly affect glucocorticoid release from the adrenal gland. IL-1 has been shown to directly stimulate glucocorticoid release. Andreis et al. (6) have demonstrated that IL-1β is capable of stimulating the release of corticosterone from rat adrenal slices including both cortex and medulla (but not in adrenal cortical cells alone), showing its dependence on medullary chromaffin cells. In addition, IL-1 has been shown to stimulate corticosterone release on its own from rat (217,294) and bovine (331) adrenal cells in primary culture and to act synergistically with ACTH (294). Possible mechanisms by which IL-1 stimulates corticosterone release include the involvement of an intra-adrenal CRH/ACTH system (6,182), catecholamines released from medullary chromaffin cells (via α-adrenergic mechanisms) (121,217), and prostaglandins (250,294,331).
IL-6 also has been shown to stimulate the release of corticosterone from primary cultures of rat, human, and bovine adrenal cells alone (18,107,233,257,323) and in synergy with ACTH (18,257). In addition, Barney et al. (18) have shown that ACTH and IL-6 act synergistically to induce corticosterone release from adrenal cell cultures at time points when IL-6 alone is ineffective. Due to the long incubation time required in the majority of the studies for IL-1 and IL-6 to stimulate glucocorticoid release (>12 h), it has been suggested that (CRH-dependent) ACTH may mediate early glucocorticoid responses to an immune challenge, while a direct action of these cytokines on the adrenal gland may mediate long-term glucocorticoid induction. Furthermore, the observation that glucocorticoid levels can remain elevated over several days despite declining plasma ACTH levels, such as it occurs during or after sepsis (24,25,317), major surgery (202,204), burn trauma (208), and chronic treatment with IL-6 (172,173,286) in humans, in response to turpentine-induced inflammation in rats (299), and in a murine model of colitis (101), lends support to the idea that proinflammatory cytokines may contribute to a sustained glucocorticoid response by directly stimulating glucocorticoid release. Nevertheless, there also have been reports in which IL-1 and IL-6 failed to stimulate corticosterone production by cultured normal rat adrenal cells (262) and by mouse adrenal tumor cells (333), which may be due to the lack of long enough incubation periods.
While IL-1 and IL-6 exert a positive influence on glucocorticoid release from the adrenal gland, TNFα has been shown to have an inhibitory effect on glucocorticoid synthesis and release. Barney et al. (18) demonstrate that TNFα inhibits basal cortisol release from bovine adrenal zona fasciculata cells after a 16 h incubation period, while inhibiting ACTH-stimulated cortisol release at 4 h. In a preparation of rat adrenal cells, TNFα also inhibited ACTH-induced corticosterone release at 6 h, but did not have an effect on basal corticosterone release at that time point (311). In addition to attenuating ACTH-stimulated glucocorticoid release, TNFα has been shown to inhibit the ACTH-induced increase in cytochrome P450 oxidase mRNA expression (a group of enzymes involved in glucocorticoid synthesis) in cultured human fetal adrenal cells (140). Interestingly, the direct effect of TNFα on the adrenal gland contradicts the stimulatory effect of circulating TNFα on HPA axis activity seen in an intact animal. Therefore, the role of intra-adrenal TNFα may be different than that of plasma TNFα.
IFN-α induces glucocorticoid secretion from human adrenal slices (66) and rat adrenal cells (113). IL-2 has been shown to stimulate corticosterone release from rat adrenal cells (at 12 h) (294). In addition to stimulating corticosterone release from adrenal cells alone, IL-2 also acts in synergy with submaximal doses of ACTH (294). However, after only a 2-h incubation period, IL-2 failed to stimulate corticosterone release by rat adrenal cells or fragments (309).
Although in vitro studies have demonstrated the ability of cytokines to stimulate glucocorticoid release directly from adrenal cells, numerous in vivo studies have shown that immune stimuli (i.e., IL-1, NDV, and MCMV) given to hypophysectomized animals can not induce a glucocorticoid response independent of pituitary-derived ACTH (88,120,220,273). In contrast, two studies have reported that NDV (277) and IL-1β (6) can induce a corticosterone response in hypophysectomized mice. However, in the NDV study (277), the results were attributed to the possibility of incomplete hypophysectomies, and the animals in the IL-1 study (6) were pre-treated with a low dose of ACTH.
Studies from our lab (273) have shown that MCMV-infected mice administered CRH-antisera are able to produce a robust corticosterone response despite their inability to produce an ACTH response. However, baseline levels of ACTH are still present (unlike hypophysectomized mice) and may serve as a permissive factor to allow for direct cytokine-adrenal interactions. In addition, mice deficient in the CRH or CRH-R1 gene are able to mount a significant corticosterone response to a variety of immunological stressors, such as carageenan (149), turpentine (301,315), LPS (44,149,301), and MCMV (273), even though they exhibit markedly attenuated corticosterone responses to physical stressors such as restraint, ether, fasting and forced swim (193,278,293). Of note, the genetically manipulated animals (and those treated with CRH-antisera) that receive the immune stimulus, exhibit dramatically greater increases in IL-6 compared to immune challenged control animals. This marked increase in IL-6 may compensate for the lack of CRH function in driving the HPA axis, and become a predominant driving force for glucocorticoid induction by acting directly on the adrenal glands.
The mechanism by which ACTH supports the direct action of cytokines, such as IL-1 and IL-6, on the adrenal gland has not been elucidated. One possibility includes an ACTH-induced upregulation of adrenal gland cytokine receptor expression. Such upregulation of cytokine receptors by upstream neuroendocrine factors has been found in the pituitary gland following administration of CRH. On the other hand, the high levels of IL-6 may increase adrenal sensitivity to submaximal doses of ACTH. Adrenal sensitivity to ACTH is regulated by adrenal innervation (e.g., splanchnic nerve stimulation) with the subsequent release of catecholamines and vasoactive neuropeptides that affect adrenal blood flow and hence, increase the presentation of ACTH to the steroidogenic cells of the adrenal cortex and result in an increase of glucocorticoid release (21,51,68,92,96,133,134,184,297, 298,329). However, the role of IL-6 and/or other cytokines, whether circulating or intra-adrenal, in this interplay requires further investigation.
OTHER CYTOKINES IN IMMUNE–HPA INTERACTIONS
Although this review has focused on the stimulatory effects of the macrophage-produced proinflammatory cytokines, TNFα, IL-1, and IL-6, the type I IFN’s α and β, released during the early innate immune response by virus-infected cells, and the proinflammatory Th1–produced cytokines, IL-2 and IFN-γ (type II), numerous other cytokines and their receptors have been identified in HPA axis tissue and have been shown to modulate immune-HPA axis interactions. Some of these cytokines are briefly described below.
Leukemia inhibitory factor (LIF)
Another proinflammatory cytokine that has recently gained attention for its role in neuroendocrine function is leukemia inhibitory factor (LIF). LIF is member of the IL-6 cytokine superfamily, in which the LIF receptor (LIF-R) shares the gp130 receptor subunit for signal transduction. Serum LIF is increased in a variety of inflammatory conditions, concordant with HPA activation, and systemic injection of LIF has been shown to stimulate pituitary POMC transcription and ACTH release alone and in synergy with CRH. In addition, the local expression of mouse LIF and LIF-R in the hypothalamus and the pituitary (corticotrophs) is upregulated by various inflammatory stressors. Moreover, LIF KO mice fail to mount an ACTH response and demonstrate blunted POMC and corticosterone responses to inflammatory stressors (11,70).
Macrophage migration inhibitory factor (MIF)
Like LIF, macrophage migration inhibitory factor (MIF) is another proinflammatory cytokine that is detected in the corticotrophs (and thyrotrophs) of the anterior pituitary and is also expressed (although in lesser amounts) in brain regions such as the hypothalamus, hippocampus, and perivascular epithelial cells. The release of MIF from the pituitary is stimulated by CRH and inflammatory stimuli, such as LPS. Once released into the general circulation, MIF plays a role in opposing the anti-inflammatory actions of glucocorticoids. Interestingly, physiological concentrations of glucocorticoids stimulate macrophage release of MIF, which is in contrast to the typical inhibitory action of glucocorticoids on proinflammatory cytokines (194,238,251).
Interleukin-10
IL-10 is an anti-inflammatory macrophage- and Th2–produced cytokine. Similar to glucocorticoids, it inhibits the production of proinflammatory cytokines from macrophages and Th1 cells. IL-10 is produced in pituitary, hypothalamic, and neural tissue. In addition, IL-10 has been shown to stimulate ME fragments to release CRH and AtT-20 pituitary cells to release ACTH and MIF. However, IL-10 seems to negatively regulate glucocorticoid release from the adrenal gland, since cold-swim and poly I:C-induced glucocorticoid responses are enhanced in IL-10 deficient mice and Y-1 adrenal cells treated with an anti-IL-10 Ab (276).
Interleukin-12
IL-12 is mainly released by activated macrophages and dendritic cells. IL-12 causes NK cells and T cells to secrete IFN-γ; acts as a differentiation factor on CD4+ T cells, promoting their specialization into IFN-γ–producing, Th1 cells; and enhances the cytolytic functions of activated NK cells and CD8+ T cells. Therefore, IL-12 provides an important link between innate and adaptive immunity, favoring the development of cellular immune responses that can better protect the host against viruses. Administration (ip) of IL-12 to mice induces an increase in serum corticosterone levels alone and produces a synergistic increase in the corticosterone response to LCMV infection (223). The corticosterone response in IL-12-treated, LCMV-infected mice was shown to be largely dependent on TNF (223). In addition, IL-12 mRNA has been detected in the brains (microglia) of LPS-treated mice (229), in the pituitary glands of LPS-treated rats (340), and in the adrenal glands of mice undergoing graft-versus-host disease (GVHD) (341).
Interleukin-18
Finally, IL-18 (also known as interferon-γ–inducing factor [IGIF] or IL-1γ) is a recently identified cytokine that has been detected in the brain, pituitary, and adrenal cortex, specifically in the zona reticularis and fasciculata. Microglial IL-18 mRNA expression is induced by LPS and adrenal IL-18 mRNA is upregulated by ACTH treatment (76,77,288). Interestingly, the IL-18 mRNA found in the pituitary and adrenal glands is of a different size than that found in immune tissues, indicating that IL-18 in endocrine tissue may be distinct from that in immune tissues (278). Although HPA axis stimulating activity has not yet been demonstrated for this cytokine, the localization of IL-18 in the three levels of the HPA axis suggests that it may play a modulatory role in immune-neuroendocrine interactions.
THE GLUCOCORTICOID RECEPTOR
There is no question that circulating levels of glucocorticoid hormones are relevant to steroid action, however, at the cellular level, activity of glucocorticoids is determined by local factors that regulate the access of free hormone to its receptor. Such factors include corticosterone binding globulin (CBG), 11β-hydroxysteroid dehydrogenase (11β-HSD), and the multidrug resistance transporter (MDR) (Fig. 5). All of the above have been shown to be altered under conditions of immune activation, whereby proinflammatory cytokines tend to decrease CBG levels, increase 11β-HSD-1 expression and reductase activity, and decrease MDR expression and/or function, thereby favoring an increase in glucocorticoid bioavailability (274).
FIG. 5.
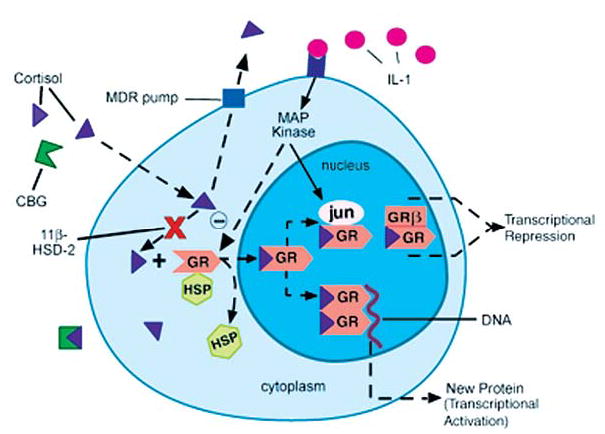
Local factors regulating glucocorticoid bioavailability and action. (1) corticosterone binding globulin (CBG), (2) 11β-hydroxysteroid dehydrogenase (11β-HSD), (3) multidrug resistance transporter (MDR pump), (4) glucocorticoid receptor (GR = GRα) nuclear translocation, (5) GR interaction with other transcription factors (AP-1 [jun/fos], NFκB), and (6) ratio of GRα:GRβ isoform expression. HSP, heat shock protein; MAP kinase, mitogen-activated protein kinase. Reprinted with modifications by permission from Silverman et al. (274).
The ultimate effect of glucocorticoids on immune system regulation is established at the level of the glucocorticoid receptor (GR). Upon glucocorticoid binding to cytosolic GR’s, a conformational change in GR causes heat-shock protein 90 (hsp90) and other ancillary proteins to dissociate from the receptor, and the ligand-bound receptor then translocates into the nucleus. Here, it dimerizes with another ligand-bound GR, thereby allowing it to alter the transcription rates of glucocorticoid–sensitive genes, either through protein-DNA interactions (transcriptional activation; involving GR binding to a glucocorticoid response element [GRE]) or through protein-protein interactions (transcriptional repression; involving GR binding to other transcription factors, e.g., NFκB and AP-1). A tissue’s sensitivity to glucocorticoid activity can be influenced by a change in (a) GR number or affinity, or (b) GR function, including its ability for nuclear translocation, its interaction with other signal transduction pathways, and the expression of particular GR isoforms (Fig. 5). Proinflammatory cytokines have been shown to impact a number of these factors.
GR number and affinity
There is a large body of data on the impact of cytokines on GR number (188). Interestingly, however, the results are split into those studies that find an increase in GR number following cytokine administration and those that find a decrease. The discrepancy in results appears to depend on how the receptors were measured. Studies using whole-cell assay binding techniques tend to find an increase in cytokine-induced GR expression, while those using cytosolic receptor binding techniques tend to find GR number to be decreased. Results are further complicated by the fact that the studies utilizing whole-cell binding techniques tend to use longer incubation times (i.e., 24 h), whereas the studies employing cytosolic receptor binding tend to examine shorter incubation periods (i.e., 4–6 h). Few studies have documented changes in receptor affinity. However, Kam et al. (147) report that lymphocytes simultaneously exposed to Th1 and Th2 cytokines exhibit a reduced affinity of GR for glucocorticoids.
GR function
Several autoimmune/inflammatory disorders have been associated with impaired GR function, possibly contributing to the excessive inflammation characteristic of these illnesses. Proinflammatory cytokines (TNFα, IL-1, and IL-6) and cytokines that mediate lymphocyte growth and differentiation (IL-2, IL-4) have been found to inhibit GR function (188).
Although the mechanism by which cytokines inhibit GR is unknown, several possibilities have been considered. First, cytokines may influence GR function through their effects on GR translocation from the cytoplasm into the nucleus. For example, TNFα and IL-1 have been shown to block dexamethasone-induced nuclear GR translocation (2,228). Another possible mechanism by which cytokines may influence GR is through cross-talk among signal transduction pathways. For example, downstream in the IL-1 and TNFα signal transduction pathways are the transcription factors NFκB and AP-1 (which consists of jun and fos proteins). GR and NFκB/AP-1 have been shown to mutually antagonize each other’s transcriptional activity through multiple mechanisms (3,81,165,280,326). In addition, IL-1 activates mitogen-activated protein kinase (MAPK) signaling pathways (i.e., p38 MAPK and jun N-terminal kinase [JNK]), which have been shown to be inhibitory to GR function (139,249,318,319). Finally, cytokines may affect GR function by altering the ratio of GRα:GRβ isoform expression. Alternative splicing of the human GR primary transcript produces two isoforms (135), GRα and GRβ, the latter of which may negatively regulate GR activity. Oakely et al. (216) have demonstrated that the GRα isoform binds hormone and activates glucocorticoid-responsive genes, while the GRβ isoform fails to bind hormone and activate glucocorticoid-responsive genes, and attenuates the trans-activation of the hormone-bound GRα isoform. These findings suggest that, when GRβ is present in excess in various tissues, it might repress glucocorticoid-mediated GRα function. In fact, Webster et al. (325) have shown that IL-1 and TNFα lead to the selective accumulation of GRβ protein in cells of lymphoid origin and the development of a glucocorticoid resistant state. Moreover, increases in GRβ levels have been found in immune cells of patients with glucocorticoid-resistant asthma (281) and colitis (137).
Although proinflammatory cytokines have been shown to exert inhibitory effects on GR function via downstream signaling molecules, we will see in the next section that GR function has been found to be stimulated during various viral infections. The differential in vitro effects of cytokines on GR versus in vivo viral effects on GR may be due to the presence of more complex interactions in an in vivo system and/or to the presence of a virus. Such in vivo changes in GR expression/function may be attributed to local factors released in immune tissues during infection or to viral components, themselves, that may act directly on brain sites involved in HPA axis regulation or on GR, itself.
HPA AXIS ACTIVATION DURING VIRAL INFECTION
Although considerable data exist describing the pathways by which purified cytokines or LPS (endotoxin) induce glucocorticoid release (274,300), HPA axis activity in response to viral infections, and the pertinent cytokines involved in such interactions, has received much less attention. By studying glucocorticoid release that is induced by a complex inflammatory stimulus, such as a virus, rather than a bolus of purified cytokine, one can study a more natural situation of immune–neuroendocrine interaction. This gives the advantage of eliciting immune and neuroendocrine responses that evolve over time and occur in the physiological milieu of an in vivo immune response (release and action of multiple cytokines at physiologically relevant concentrations). To examine the impact of viral infection on HPA axis function, investigators have employed polyinosinic polcytidilic acid (poly I:C), a synthetic double-stranded RNA used to mimic viral exposure, and Newcastle disease virus (NDV), which does not actively infect the host. As with LPS, both poly I:C and NDV produce a marked HPA response about 1–2 h post-injection. However, it cannot be concluded a priori that the delayed cytokine response usually elicited by a replicating virus (days as opposed to hours) interacts with the HPA axis in a similar manner as an acute challenge such as LPS, poly I:C, or NDV. In addition to poly I:C and NDV, we describe below what is currently known about HPA axis activation during infection with replicating viruses, including murine cytomegalovirus (MCMV), lymphocytic choriomeningitis virus (LCMV), influenza, herpes simplex virus type–1 (HSV-1), and human immunodeficiency virus (HIV). As will be demonstrated, immune–HPA interactions appear to be both virus specific and phase (of the immune response) specific.
Polyinosinic–polycytidylic acid (poly I:C)
Just as LPS has been used to model the cytokine cascade during a bacterial infection, poly I:C has been used to mimic viral infection. When administered systemically to mice or rabbits, poly I:C induces a rapid activation (1–2 h) of the HPA axis (91,190,253) (Fig. 6). This induced glucocorticoid response has been shown to be dependent on IL-6 in mice (253) and on CRH in rabbits (190). The IL-6 requirement for glucocorticoid release appears to be specific for virus-type stimuli, since IL-6 knockout mice treated with poly I:C (as well as MCMV) exhibit profoundly reduced corticosterone responses, as opposed to modest (but significant) decreases in LPS-treated mice and no significant reductions in restraint-stressed mice (253) (Fig. 7).
FIG. 6.
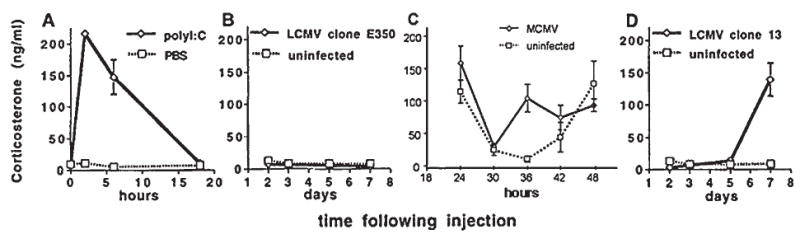
Kinetics and magnitudes of endogenous glucocorticoid responses vary depending on viral challenge. Serum corticosterone responses to polyinosinic-polycytidylic acid (poly I:C) administration and during viral infections are shown. Mice were injected with 100 μg poly I:C (A) or infected with 2 × 104 PFU of lymphocytic choriomeningitis virus (LCMV) clone E350 (B), 5 × 104 PFU murine cytomegalovirus (MCMV) (C), or 1 × 106 PFU LCMV clone 13 (D). Serum samples were collected under low-stress conditions during the morning (36 h post MCMV-infection) and examined for levels of serum corticosterone. Harvests were at indicated times following treatment or infection. Data are presented as means ± SEM. Reprinted with modifications by permission from Miller et al. (188,189).
FIG. 7.
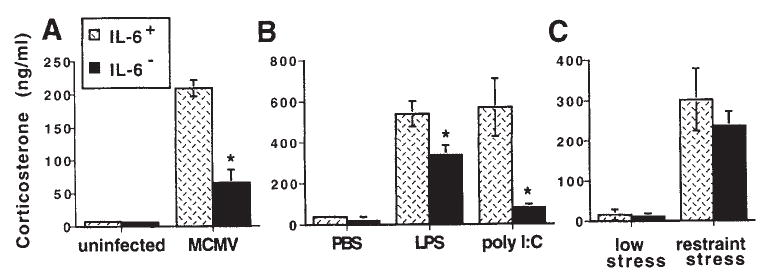
Interleukin-6 (IL-6) plays a pivotal role in the induction of endogenous glucocorticoids in response to viral challenges. Serum corticosterone levels following murine cytomegalovirus (MCMV), lipopolysaccharide (LPS), polyinosinic-polycytidylic acid (poly I:C), or restraint stress administration in IL-6–deficient and wild-type mice. Corticosterone levels were measured in serum collected from mice under low-stress conditions at 36 h following infection with 5 × 104 PFU MCMV (A); 2 h following injections with PBS, 50 μg LPS, or 100 μg poly I:C (B); or after 30 min of restraint (C). Data are presented as means ± SEM. Results are significant at *p < 0.05. Reprinted by permission from Miller et al. (188,253).
Newcastle disease virus (NDV)
NDV is a non-replicating virus, which like LPS and poly I:C, produces marked elevations in ACTH and corticosterone about 2h post ip injection when administered to mice or rats (although some studies report peak ACTH/corticosterone responses at 8 h post-infection) (35,38,88,91,220,248,277). Concomitant with these neuroendocrine changes, are increased brain concentrations of the norepinephrine catabolite, 3-methoxy, 4-hydroxyphenylethyleneglycol (MHPG; at 2 h p.i.) and tryptophan and the serotonin catabolite, 5-hydroxyindoleacetic acid (5-HIAA) (at 8 h p.i.) (90,91). The NDV-induced ACTH/corticosterone response also was observed when animals were injected with virus-free supernatants derived from co-cultures of human peripheral blood leukocytes or mouse spleen cells with NDV, indicating that stimulation of the HPA axis was due to a product released from the NDV-stimulated leukocytes, and not NDV itself (35,38). It was concluded that IL-1 was the most likely mediator of the NDV-induced ACTH/corticosterone response, since pretreatment of the supernatant with IL-1–antisera blocked its stimulatory effect on the HPA axis in the NDV-injected animals (35,38). Moreover, pretreatment of NDV-infected mice with IL-1 receptor antagonist (IL-1ra) prevented the neuroendocrine and neurochemical responses to NDV (91). The NDV-induced corticosterone response also is abolished in hypophysectomized mice (38,88,91,220) and CRH-Ab–treated rats (248), demonstrating its dependence on an intact pituitary and CRH.
Murine cytomegalovirus (MCMV)
MCMV is a cytopathic virus that induces an early natural killer (NK) cell-mediated, anti-viral defense. The anti-viral immune response is characterized by high levels of IL-12 and NK cell–produced IFN-γ (224), and NK cell–mediated liver inflammation (258). In addition to the induction of the aforementioned cytokines, the proinflammatory cytokines, TNFα, IL-1, and IL-6, are induced during the innate immune response to MCMV infection. Peak serum cytokine levels occur around 36–44 h after infection and are paralleled by peak neuroendocrine (ACTH and corticosterone) responses, which occur at 36 h after MCMV infection (253) (Fig. 6). Ruzek et al. (253) have shown that the MCMV-induced corticosterone response is dependent on IL-6, with IL-1α contributing to IL-6 production (Fig. 7). TNFα is required for the development of hepatic necrotic foci and increased levels of liver enzymes in serum, both signs of liver pathology (222). As glucocorticoids have been shown to negatively regulate proinflammatory cytokine production during MCMV infection, if glucocorticoids are removed by adrenalectomy (ADX), IL-12, IFN-γ, TNFα, and IL-6 production, as well as splenic mRNA for a wider range of cytokines (including IL-1α and β), increase and the mice die due to septic shock (254). Corticosterone replacement or the administration of TNF-antisera to MCMV-infected, ADX mice restores survival (254) (Fig. 8). Therefore, the glucocorticoid response during MCMV infection is essential for protection against TNFα-mediated lethality.
FIG. 8.
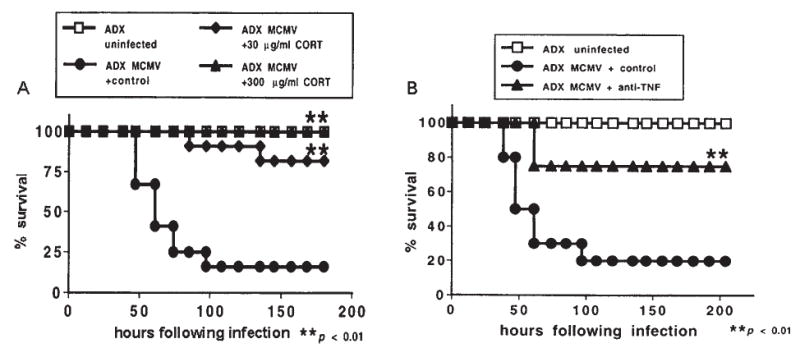
Role of glucocorticoids in protection against cytokine (TNF)–mediated lethality. (A) Adrenalectomized (ADX) mice were vehicle injected (squares) or infected with MCMV (1 × 105 PFU/mouse) 5 days following surgery and either not given (circles) or given corticosterone in 0.9% saline as drinking water at either 30 μg/mL (diamonds) or 300 μg/mL (triangles). Control mice received vehicle-treated water. Denoted p value represents significant differences between corticosterone and vehicle-treated MCMV-infected ADX mice. (B) Control (circles) or anti-TNF (triangles) antibodies (Abs) were administered 8–10 h before infection of ADX mice. Denoted p value represents significant differences between TNF and control Ab-treated MCMV-infected ADX mice. Data are presented as means ± SEM. Results are significant at **p < 0.01. Reprinted by permission from Silverman et al. (274,254).
To determine whether the MCMV-induced ACTH and corticosterone responses are dependent upon CRH, work in our labs has employed both CRH immunoneutralization and CRH-knockout techniques (273). Acute administration of a CRH-antibody (Ab) completely eliminated ACTH responses to both low- and high-dose MCMV. However, corticosterone responses in CRH-Ab-treated animals remained apparent in mice infected with low-dose MCMV and were robust in mice infected with high-dose MCMV (Fig. 9). CRH-knockout (KO) mice exhibited robust corticosterone responses to both MCMV doses, despite reduced baseline and MCMV-induced ACTH. Interestingly, robust corticosterone responses in CRH-Ab-treated and CRH-KO mice were associated with exaggerated IL-6 levels, and IL-6 and corticosterone concentrations in infected CRH-Ab-treated animals were significantly correlated. Neutralization of IL-6 responses in infected CRH-KO mice reduced corticosterone responses by ~70% (273) (Fig. 10). Finally, MCMV-infected mice deprived of ACTH by hypophysectomy failed to elicit glucocorticoid responses, despite elevated plasma IL-6 concentrations (273) (Fig. 11). Taken together, these results suggest that a greater than normal induction of IL-6 compensates for the absence of a normal CRH-dependent ACTH surge during MCMV infection. This enhanced IL-6 response, in turn, may mediate a direct immune-adrenal pathway that can become a predominant driving force for glucocorticoid induction in the absence of CRH. However, the presence of ACTH appears to serve as a necessary permissive factor, enabling direct cytokine actions on the adrenal gland. This requirement of an intact pituitary for corticosterone responses to an immune challenge also has been shown by Besedovsky et al. in rats injected with culture supernatants from mitogen-stimulated peripheral blood or spleen cells (43) and animals injected with NDV (36). However, the MCMV studies were the first to use an actual replicating virus and the first to demonstrate that the presence of low, permissive levels of ACTH were sufficient (along with deficient CRH responses and exaggerated IL-6 responses) to stimulate corticosterone release.
FIG. 9.
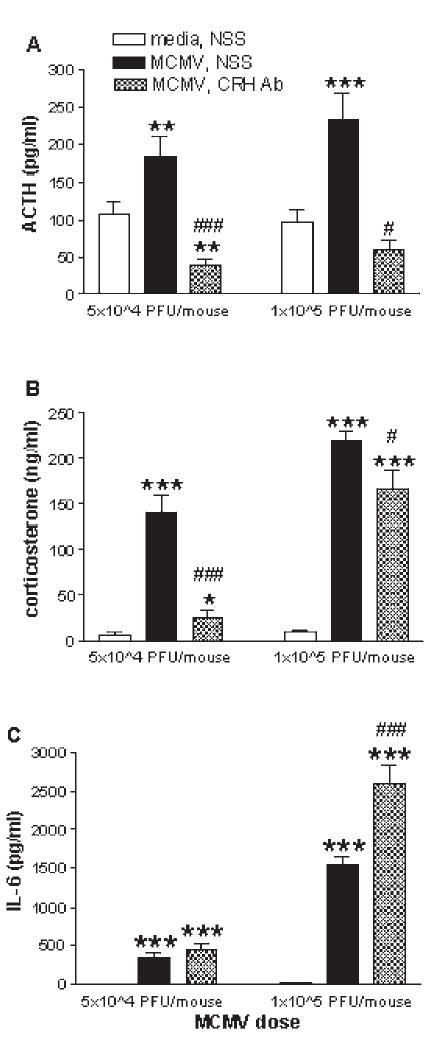
Effect of the administration (ip) of a CRH-Ab on low (5 × 104 PFU/mouse) and high (1 × 105 PFU/mouse) dose MCMV-induced plasma ACTH (A), corticosterone (B), and IL-6 (C) responses. Two hundred microliters of either normal sheep serum (NSS) or CRH-antisera was administered 8 h prior to the peak MCMV-induced corticosterone response (28 h post-infection). Trunk blood was collected from C57BL/6 mice at 36 h post-infection (n = 5–11 animals per group). *p < 0.05; **p < 0.01; ***p < 0.001; compared with media/NSS group within dose. #p < 0.05; ##p < 0.01; ###p < 0.001; compared with MCMV/NSS group within dose. One-way ANOVA was used within dose. Because of limited amounts of Ab, a media/CRH-Ab group was not included in this experiment. Reprinted by permission from Silverman et al. (273).
FIG. 10.
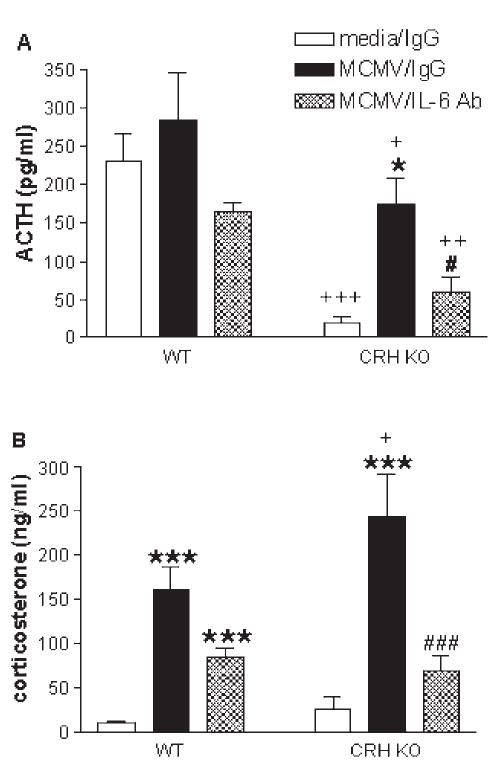
Effect of the administration (ip) of an IL-6–Ab on plasma ACTH (A) and corticosterone (B) levels in WT and CRH-KO mice after injection with MCMV (5 × 104 PFU/mouse) or vehicle. The IL-6-Ab (1 mg/mouse) was administered 16 h prior to the peak MCMV-induced corticosterone response (20 h post-infection). Trunk blood was collected from mice at 36 h post-infection (n = 4–8 animals per group). *p < 0.05; ***p < 0.001 (t test for WT/MCMV/IL-6–Ab vs. WT/media/IgG); compared with media/IgG group within genotype. #p < 0.05; ###p < 0.001; compared with MCMV/IgG group within genotype. +p < 0.05; ++p < 0.01 (t test); +++p < 0.001; compared with respective WT group. Two-way ANOVA was used (unless otherwise indicated). Because of limited amounts of Ab, a media/IL-6–Ab group was not included in this experiment. Reprinted by permission from Silverman et al. (273).
FIG. 11.
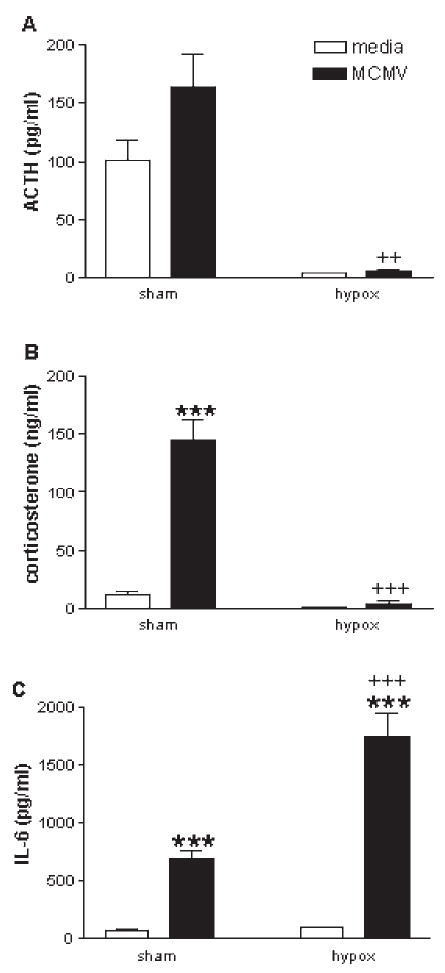
Plasma ACTH (A), corticosterone (B), and IL-6 (C) levels after injection with MCMV (1 × 105 PFU/mouse) or vehicle in sham-operated or hypophysectomized (hypox) C57BL/6 mice. Trunk blood was collected from mice at 36 h post-infection (n = 3–11 animals per group). ***p < 0.001; compared with media group within surgical-manipulation. ++p < 0.01; +++p < 0.001; compared with respective sham group. Two-way ANOVA indicated a significant interaction of surgery and infection on corticosterone and IL-6 responses for MCMV. Reprinted by permission from Silverman et al. (273).
Lymphocytic choriomeningitis virus (LCMV)
LCMV replicates in many organs, including the spleen, thymus, lymph nodes, kidney, and brain (if administered intracranially) (87). Depending on the strain, LCMV can be either cytopathic (clone 13 or WE) or non-cytopathic (clone E350). LCMV induces high levels of early circulating IFN-α/β and of NK cell-mediated cytotoxic activity. However, NK cell IFN-γ production is not induced, and NK cells do not significantly contribute to anti-viral defenses (47). Viral titers in the spleen and blood peak on days 3–5 following infection and decline rapidly thereafter (337). During LCMV infection, late CD8+ T cell responses are prominent, in which CTL activity and IFN-γ production is protective (47). Our labs have worked with three strains of LCMV (systemically injected): LCMV clone 13, which induces peak corticosterone responses around day 7 post-infection, corresponding with a loss of T cell responses and viral persistence; LCMV variant WE, which induces glucocorticoid responses similar to LCMV clone 13, but is not associated with reduced T cell responses or viral persistence; and LCMV clone E350, which fails to induce a glucocorticoid response, along with the lack of significant inflammation or pathology (188) (Fig. 6).
These results suggest that immune activation alone is not sufficient for stimulation of the HPA axis. We hypothesize that (in the absence of pain or other conditions that directly limit physiological function, for example, respiratory distress in influenza virus infection) the presence of significant inflammation or immunopathology with associated release of high serum levels of proinflammatory cytokines (innate or adaptive) may be required for glucocorticoid induction (189). Despite the fact that animals infected with LCMV clone E350 exhibit no significant increase in glucocorticoid release, reduced cytosolic GR binding in immune tissues of infected animals, such as the spleen and thymus, was detected (189). Decreased cytosolic GR binding may be indicative of increased GR activation (may imply an increase in nuclear translocation of the receptor). Indeed, GR activation was enhanced in the spleen of infected animals given an acute injection of corticosterone (189). Such changes in GR expression/function may be attributed to local factors released in immune tissues during infection. Since CD8+ T cells are an actively proliferating cell type in the spleen during LCMV infection, increased GR activation in the spleen might decrease the number and/or activity of this cell type, therefore restraining CD8+ T cell activation at a local level (189). LCMV clone E350–infected mice do, however, exhibit significant corticosterone responses when treated with IL-12 (223). Although, IL-12 alone induces a corticosterone response in mice, the synergistic induction of corticosterone between LCMV and IL-12 has been shown to be dependent on IL-2 and TNF (223).
As mentioned before, LCMV-WE induces a late CD8+ T-cell response that mediates anti-viral defenses and peak corticosterone responses around 7 days post-infection, corresponding to the time of T-cell activation. LCMV-WE–infected ADX mice exhibit increased mortality, along with greater levels of plasma and splenic IFN-γ and TNFα, and a greater number of IFN-γ+ and TNFα+ CD8+ T cells (primed for cytokine expression) relative to sham-operated mice. CD8+ KO mice (sham or ADX) no longer show an LCMV-WE induced increase in plasma and splenic IFNγ and TNFα levels and, therefore, exhibit reduced mortality (ADX). Hence, the CD8+ T cell–mediated corticosterone response is essential for protection against CD8+ T-cell–mediated lethality during LCMV-WE infection (although the cytokines that mediate corticosterone responses and lethality have yet to be determined) (259).
Intra-cranial (ic) inoculation of LCMV (strain Arm-53b–replicates to higher levels in the brain than in the periphery) induces a fatal CD8+ T cell-mediated encephalitis (64). Central LCMV infection produces an early increase in TNFα, IL-1α/β, and IL-6 mRNA expression by resident brain cells and infiltrating monocytes; and a later immune response, characterized by an increase in IFN-γ mRNA expression by infiltrating T cells, concomitant with further increased expression of monokine transcripts (64). In addition to a direct cytolytic action of LCMV-specific CTLs, lymphocytic choriomeningitis may be mediated by the cytotoxic effects of these cytokine, IFN-γ in particular (64). HPA axis activity has not been studied during ic LCMV infection, but one may speculate a biphasic response concomitant with the early peak in proinflammatory cytokines, followed by the later peak in T cell cytokines and may be crucial in controlling the LCMV-induced CNS inflammation and T cell–mediated encephalitis.
Influenza
Influenza virus is cytolytic for epithelial cells of the lung. As the infection progresses, the inflammatory response is dominated by monocyte and T cell infiltration to the site of virus replication (23). Elimination of the virus is dependent on T cell-mediated anti-viral mechanisms. Mice infected with the influenza virus exhibit prolonged corticosterone responses, apparent from about day 2 until about 1 week post-infection (89,130,131). Similar to NDV-injected mice, influenza-infected mice exhibit increases in brain noradrenergic activity (particularly in the hypothalamus) and tryptophan levels that parallel HPA axis activation (89). Interestingly, Hermann et al. (131) demonstrated that mice infected with influenza, actually exhibit two peaks in their corticosterone responses: the first, apparent at 2–3 days post-infection, and the second peak, apparent at 7–8 days post-infection. In the murine model of influenza viral infection, virus titers peak at 48–72 h post-infection, concomitant with the activation of macrophages and the release of proinflammatory cytokines, such as TNFα, IL-1, and IL-6 (128). Therefore, the early corticosterone peak may be due to HPA axis stimulation by these proinflammatory cytokines. The second peak in plasma cortiscoterone levels may be due to HPA axis stimulation by T cell cytokines, such as IL-2 and IFN-γ, since it is around this time that influenza-specific T cell responses occur (4,85). On the other hand, since extensive lung pathology exists at these later times, along with labored respiration, it is possible that the later corticosterone peak may be driven by physical distress (131). Increased GR activation also has been detected in the liver and lung during influenza infection (112).
Herpes simplex virus type–1 (HSV-1)
Herpes simplex virus type-1 (HSV-1) is a neurotropic virus, which when inoculated into various peripheral tissues in animals, invades the peripheral nervous system, travels to the brain, and results in encephalitis, primarily affecting temporal and limbic brain regions (100,296). Upon corneal inoculation of HSV-1 in rats, marked increases in ACTH and corticosterone levels are apparent by day 3 post-infection, peak around day 7, and are still elevated on day 14 post-infection (28). However, no concomitant changes in hypothalamic or ME CRH content are detected (28). On day 7 post-infection, replicating virus is found only in the brainstem (asymptomatic infection) (28). In fact, corneal HSV-1 inoculation leads to the infection of brainstem regions such as the locus coeruleus and the raphe nuclei, resulting in changes in norepinephrine (NE) and serotonin (5-HT), respectively (17,186,226). In addition, corneal inoculation with a neurovirulent, but not with an avirulent strain of HSV-1, increases IL-1β mRNA expression in the brainstem and hypothalamus, coinciding with elevated serum corticosterone levels (26). Since there was no evidence of invasive cellular inflammation in these brain regions, the authors concluded that the IL-1β mRNA was expressed by resident brain cells (26). Moreover, Ben-Hur et al. (26) demonstrated that the HSV-1–induced corticosterone response is dependent upon intact NE innervation of the PVN via the VNAB. Hence, HSV-1 may indirectly affect the hypothalamus and other brain regions involved in HPA axis regulation through stimulation of brainstem monoamine release and/or through the activation of centrally induced cytokines.
The inoculation of virulent HSV-1 strains into the PVN of rats also induces elevated serum ACTH and corticosterone levels around day 3 post-infection (along with overt clinical signs of encephalitis and cellular inflammation of the PVN). However, unlike corneal inoculation, PVN inoculation of virulent HSV-1 strains is accompanied by CRH depletion from the ME (reflecting an increase in release), a more widespread induction of IL-1β mRNA in the brain, and the induction of prostaglandins (PGE2) in various brain regions (including the hypothalamus and brainstem) (30). Because inoculation with a non-virulent HSV-1 strain failed to elicit such responses, it was concluded that activation of the HPA axis was not due to viral replication, but was dependent on neurovirulence (30). Also supporting the contention that HPA activation is not due to viral replication, is the finding that icv inoculation of HSV-1 induces early ACTH and corticosterone responses (at 3.5 h post-infection), before any virus replication could occur, which is dependent on the central induction of IL-1β (27). In addition, brain-derived cytokines and prostaglandins appear to contribute to the clinical syndrome of HSV-1 encephalitis, particularly during early stages of the disease, when virus load is low and cellular inflammation has not yet evolved (30). Nevertheless, the containment of HSV-1 replication, and hence the development of encephalitis, is dependent on a timely (relative to viral spread) and efficient, HSV-1–specific CD8+ T cell response in the brain (7). Of note, immunization with a nonvirulent HSV-1 strain protects animals, not only from the lethal coutcome of infection with a neurovirulent strain, but also from all the clinical and behavioral signs of the disease, including fever, aggressive behavior and motor hyperactivity, brain prostaglandin production, and HPA axis activity (29).
As for HSV-1 effects on GR function, HSV-1 infection of primary human gingival fibroblast (HGF) cells was found to increase nuclear NFκB expression and DNA binding and increase expression and nuclear translocation of GR, while an increase in glucocorticoid-driven reporter gene activity was observed in human embryo kidney cells (99). Erlandsson et al. (99) also showed that dexemethasone (Dex) treatment of HGF cells prior to, but not simultaneously with, HSV-1 infection increased viral yield. Since NFκB activity is necessary for efficient HSV-1 replicaton (5), Erlandsson hypothesized that this phenomenon may be due to reduced cytosolic availability of GR at the time of HSV-1 infection (since GR would already be bound by Dex and translocated to the nucleus), and therefore HSV-1–induced GR activity could no longer counteract NFκB activity (99).
Human immunodeficiency virus (HIV)
Two hallmarks of immunopathogenesis in the progression of HIV-infection to AIDS are the loss of CD4+ T cell function in response to antigens and the reduction in CD4+ T cell numbers (271). In addition, a strong cell-mediated (Th1) immune response to HIV is more critical in preventing the progression of HIV infection to AIDS (73,74). Since glucocorticoids modulate the Th1/Th2 cytokine balance, the level of HPA axis activation in HIV-positive individuals may play a crucial role in the hosts ability to control viral spread. HIV infection is associated with neuroendocrine disturbances, including impaired regulation of the HPA axis. Many studies report that HIV-positive patients, symptomatic as well as asymptomatic, have increased basal ACTH/cortisol levels (12,45,71,75,169,175,187,316), but blunted ACTH and cortisol responses to stress (166) or a CRH challenge (12,45,175). In addition to cytokine-induced stimulation of the HPA axis, these abnormalities may be enhanced further by the release of toxic viral products, such as the envelope protein, gp120, and the structural protein, Vpr. Icv injection of gp120 into rats increases serum levels of ACTH and corticosterone (16,289), which have been shown to be dependent on the central induction of IL-1 (289) and prostaglandins (PGs) (16). In addition, Raber at al. (245) have demonstrated that gp120–overexpressing transgenic mice exhibit elevated plasma ACTH and corticosterone levels, as well as pituitary ACTH content. Gp120-treatment of rat or mouse hypothalamic explants increases CRH mRNA, intrahypothalamic CRH protein, and CRH release (242), as well as AVP release (245). These actions of gp120 on CRH and AVP production are mediated by nitric oxide synthase (NOS). Other activities of viral products include the capacity of Vpr to act as a co-activator of the GR, thereby enhancing GR signaling and the effects of glucocorticoids on HIV replication (156,157,246,272). More specifically, recombinant Vpr can potentiate GR-meditated increases in virion production, downregulation of NFκB and proinflammatory cytokine induction, and programmed cell death (192,272). Therefore, gp120- and Vpr-induced GR activation may be deleterious to the HIV-infected host, by inducing glucocorticoid hypersensitivity and hence, further lowering CD4+ T cell number and function and other required anti-viral immune responses. On the other hand, Kino et al. (158) have shown that glucocorticoids exert a direct suppressive effect on the HIV-1 promoter, thus acting protectively for the host. Interestingly, a subset of HIV-infected patients develop glucocorticoid resistance in the face of hypercortisolism, and exhibit an increased production of proinflammatory/Th1 cytokines, including IFN-α and IL-2, therefore favoring anti-viral defenses and limiting the progression of HIV infection (210–213). Moreover, macrophages from these patients show glucocorticoid receptor (GR) changes, consisting of decreased GR affinity for glucocorticoids and increased GR number (214). According to Norbiato et al. (212), reduced glucocorticoid sensitivity (due to decreased binding affinity of the GR) in HIV disease may be caused by HIV itself, the HIV-1 vpr gene, and/or the simultaneous presence of Th1 (IL-2) and Th2 (IL-4) cytokines.
Summary
Taken together, these observations suggest that the kinetics and magnitude of glucocorticoid production, as well as the changes in GR expression/function induced by systemic viral infection, may be specific to the virus, the kinetics of the immune response to the virus, and the extent of virus-induced pathology. In other words, glucocorticoids will be induced at crucial times to protect against either/both early proinflammatory cytokine-mediated pathology/lethality (e.g., MCMV, influenza, HSV-1) or later T cell–mediated pathology/lethality (e.g., LCMV clones 13 and WE, influenza, HSV-1, HIV). In cases where no significant inflammation/pathology occurs (e.g., LCMV clone E350), glucocorticoid responses may be minimal. As both HSV-1 and influenza virus infections can cause considerable additional physical distress, which may itself stimulate the HPA axis, it is likely that other (non-cytokine) pathways as well are contributing to induction of glucocorticoid responses at later times during infection.
CONCLUSION
During viral infection, the immune system, via early proinflammatory cytokines (TNFα, IL-1, and IL-6) and later T cell cytokines (IL-2 and IFN-γ), stimulates glucocorticoid release by acting at all three levels of the HPA axis. Cytokine receptors have been detected at all HPA axis levels and therefore, each level can serve as an integration point for immune and neuroendocrine signals. It appears that the initial activation of the HPA axis is mediated via CRH dependent pathways, for example, through the traditional CRH to ACTH to glucocorticoid pathway. However, when a sustained glucocorticoid response to an immunological stressor is necessary, complementary, CRH-independent mechanisms of glucocorticoid induction may occur to maintain the initial rise in glucocorticoid levels. These alternative pathways include the direct action of cytokines on the pituitary and adrenal glands. The direct action of cytokines on the pituitary and adrenal glands, however, may not be completely independent of upstream neuroendocrine factors (CRH and ACTH, respectively), where CRH/ACTH may be required as permissive factors to sensitize sub-hypothalamic levels of the HPA axis to circulating cytokines. The local production of cytokines at each level of the HPA axis also may contribute to the amplification and maintenance of elevated HPA activity during chronic inflammation/viral infection. Therefore, each level of the HPA axis contains its own local cytokine network.
Once cytokines have stimulated glucocorticoid release to keep inflammatory responses in check, it is also important to regulate the resultant activity of glucocorticoids themselves. Either too much or too little activity of steroid hormone may result in pathological consequences, due to the inappropriate shaping of the immune response to viral infection. Therefore, several local factors are in place to maintain appropriate glucocorticoid activity at the level of the immune cell (e.g., CBG, 11β-HSD, MDR pump). Moreover, GR number/function can be altered by cytokines and viral products. In conclusion, a closed loop, regulatory feedback circuit exists, with redundant pathways for cytokines to stimulate glucocorticoid release and regulate glucocorticoid activity, which in turn, is crucial to prevent the overshoot of inflammatory responses that may be detrimental to the host organism.
References
- 1.Abbas, A.K., and A.H. Lichtman. 1997. Cellular and Molecular Immunology. Elsevier Science, Philadelphia.
- 2.Adcock IM, Brown CR, Barnes PJ. Tumour necrosis factor alpha causes retention of activated glucocorticoid receptor within the cytoplasm of A549 cells. Biochem Biophys Res Commun. 1996;225:545–550. doi: 10.1006/bbrc.1996.1209. [DOI] [PubMed] [Google Scholar]
- 3.Adcock IM, Caramori G. Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol Cell Biol. 2001;79:376–384. doi: 10.1046/j.1440-1711.2001.01025.x. [DOI] [PubMed] [Google Scholar]
- 4.Allan W, Tabi Z, Cleary A, et al. Cellular events in the lymph node and lung of mice with influenza. Consequences of depleting CD4+ T cells J Immunol. 1990;144:3980–3986. [PubMed] [Google Scholar]
- 5.Amici C, Belardo G, Rossi A, et al. Activation of I kappa b kinase by herpes simplex virus type 1. A novel target for anti-herpetic therapy. J Biol Chem. 2001;276:28759–28766. doi: 10.1074/jbc.M103408200. [DOI] [PubMed] [Google Scholar]
- 6.Andreis PG, Neri G, Belloni AS, et al. Interleukin-1 beta enhances corticosterone secretion by acting directly on the rat adrenal gland. Endocrinology. 1991;129:53–57. doi: 10.1210/endo-129-1-53. [DOI] [PubMed] [Google Scholar]
- 7.Anglen CS, Truckenmiller ME, Schell TD, et al. The dual role of CD8+ T lymphocytes in the development of stress-induced herpes simplex encephalitis. J Neuroimmunol. 2003;140:13–27. doi: 10.1016/s0165-5728(03)00159-0. [DOI] [PubMed] [Google Scholar]
- 8.Araujo DM, Cotman CW. Differential effects of interleukin-1 beta and interleukin-2 on glia and hippocampal neurons in culture. Int J Dev Neurosci. 1995;13:201–212. doi: 10.1016/0736-5748(94)00072-b. [DOI] [PubMed] [Google Scholar]
- 9.Araujo DM, Lapchak PA, Collier B, et al. Localization of interleukin-2 immunoreactivity and interleukin-2 receptors in the rat brain: interaction with the cholinergic system. Brain Res. 1989;498:257–266. doi: 10.1016/0006-8993(89)91104-9. [DOI] [PubMed] [Google Scholar]
- 10.Arzt E, Stelzer G, Renner U, et al. Interleukin-2 and interleukin-2 receptor expression in human corticotrophic adenoma and murine pituitary cell cultures. J Clin Invest. 1992;90:1944–1951. doi: 10.1172/JCI116072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Auernhammer CJ, Melmed S. Leukemia-inhibitory factor-neuroimmune modulator of endocrine function. Endocrinol Rev. 2000;21:313–345. doi: 10.1210/edrv.21.3.0400. [DOI] [PubMed] [Google Scholar]
- 12.Azar ST, Melby JC. Hypothalamic-pituitary-adrenal function in non-AIDS patients with advanced HIV infection. Am J Med Sci. 1993;305:321–325. doi: 10.1097/00000441-199305000-00012. [DOI] [PubMed] [Google Scholar]
- 13.Baes M, Allaerts W, Denef C. Evidence for functional communication between folliculo-stellate cells and hormone-secreting cells in perifused anterior pituitary cell aggregates. Endocrinology. 1987;120:685–691. doi: 10.1210/endo-120-2-685. [DOI] [PubMed] [Google Scholar]
- 14.Bailey M, Engler H, Hunzeker J, et al. The hypothalamic-pituitary-adrenal axis and viral infection. Viral Immunol. 2003;16:141–157. doi: 10.1089/088282403322017884. [DOI] [PubMed] [Google Scholar]
- 15.Ban E, Haour F, Lenstra R. Brain interleukin 1 gene expression induced by peripheral lipopolysaccharide administration. Cytokine. 1992;4:48–54. doi: 10.1016/1043-4666(92)90036-q. [DOI] [PubMed] [Google Scholar]
- 16.Barak O, Weidenfeld J, Goshen I, et al. Intra-cerebral HIV-1 glycoprotein 120 produces sickness behavior and pituitary-adrenal activation in rats: role of prostaglandins. Brain Behav Immunol. 2002;16:720–735. doi: 10.1016/s0889-1591(02)00025-9. [DOI] [PubMed] [Google Scholar]
- 17.Barnett EM, Cassell MD, Perlman S. Two neurotropic viruses, herpes simplex virus type 1 and mouse hepatitis virus, spread along different neural pathways from the main olfactory bulb. Neuroscience. 1993;57:1007–1025. doi: 10.1016/0306-4522(93)90045-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barney M, Call GB, McIlmoil CJ, et al. Stimulation by interleukin-6 and inhibition by tumor necrosis factor of cortisol release from bovine adrenal zona fasciculata cells through their receptors. Endocrinol J Uk. 2000;13:369–377. doi: 10.1385/ENDO:13:3:369. [DOI] [PubMed] [Google Scholar]
- 19.Bartfai T, Andersson C, Bristulf J, et al. Interleukin-1 in the noradrenergic chromaffin cells in the rat adrenal medulla. Ann NY Acad Sci. 1990;594:207–213. doi: 10.1111/j.1749-6632.1990.tb40480.x. [DOI] [PubMed] [Google Scholar]
- 20.Bartholomew SA, Hoffman SA. Effects of peripheral cytokine injections on multiple unit activity in the anterior hypothalamic area of the mouse. Brain Behav Immunol. 1993;7:301–316. doi: 10.1006/brbi.1993.1030. [DOI] [PubMed] [Google Scholar]
- 21.Bassett JR, West SH. Vascularization of the adrenal cortex: its possible involvement in the regulation of steroid hormone release. Microsc Res Tech. 1997;36:546–557. doi: 10.1002/(SICI)1097-0029(19970315)36:6<546::AID-JEMT11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 22.Bebo BF, Linthicum DS. Expression of mRNA for 55-kDa and 75-kDa tumor necrosis factor (TNF) receptors in mouse cerebrovascular endothelium: effects of interleukin-1 beta, interferon-gamma and TNF-alpha on cultured cells. J Neuroimmunol. 1995;62:161–167. doi: 10.1016/0165-5728(95)00113-5. [DOI] [PubMed] [Google Scholar]
- 23.Beck MA, Sheridan JF. Regulation of lymphokine response during reinfection by influenza virus. Production of a factor that inhibits lymphokine activity. J Immunol. 1989;142:3560–3567. [PubMed] [Google Scholar]
- 24.Beishuizen A, Thijs LG, Haanen C, et al. Macrophage migration inhibitory factor and hypothalamo-pituitary-adrenal function during critical illness. J Clin Endocrinol Metab. 2001;86:2811–2816. doi: 10.1210/jcem.86.6.7570. [DOI] [PubMed] [Google Scholar]
- 25.Beishuizen A, Thijs LG, Vermes I. Patterns of corticosteroid-binding globulin and the free cortisol index during septic shock and multitrauma. Intensive Care Med. 2001;27:1584–1591. doi: 10.1007/s001340101073. [DOI] [PubMed] [Google Scholar]
- 26.Ben Hur T, Rosenthal J, Itzik A, et al. Adrenocortical activation by herpes virus: involvement of IL-1 beta and central noradrenergic system. Neuroreport. 1996;7:927–931. [PubMed] [Google Scholar]
- 27.Ben-Hur T, Cialic R, Itzik A, et al. Acute effects of purified and UV-inactivated Herpes simplex virus type 1 on the hypothalamo-pituitary-adrenocortical axis. Neuroendocrinology. 2001;74:160–166. doi: 10.1159/000054682. [DOI] [PubMed] [Google Scholar]
- 28.Ben-Hur T, Conforti N, Itzik A, et al. Effects of HSV-1, a neurotropic virus, on the hypothalamic-pituitary-adrenocortical axis in rats. Brain Res. 1995;702:17–22. doi: 10.1016/0006-8993(95)00806-7. [DOI] [PubMed] [Google Scholar]
- 29.Ben-Hur T, Itzik A, Barak O, et al. Immunization with a nonpathogenic HSV-1 strain prevents clinical and neuroendocrine changes of experimental HSV-1 encephalitis. J Neuroimmunol. 2004;152:5–10. doi: 10.1016/j.jneuroim.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 30.Ben-Hur T, Rosenthal J, Itzik A, et al. Rescue of HSV-1 neurovirulence is associated with induction of brain interleukin-1 expression, prostaglandin synthesis and neuroendocrine responses. J Neurovirol. 1996;2:279–288. doi: 10.3109/13550289609146891. [DOI] [PubMed] [Google Scholar]
- 31.Bentivoglio M, Florenzano F, Peng ZC, et al. Neuronal IFN-gamma in tuberomammillary neurones. Neuroreport. 1994;5:2413–2416. doi: 10.1097/00001756-199412000-00002. [DOI] [PubMed] [Google Scholar]
- 32.Berkenbosch F, van Oers J, del Rey A, et al. Corticotropin-releasing factor-producing neurons in the rat activated by interleukin-1. Science. 1987;238:524–526. doi: 10.1126/science.2443979. [DOI] [PubMed] [Google Scholar]
- 33.Bernardini R, Kamilaris TC, Calogero AE, et al. Interactions between tumor necrosis factor-alpha, hypothalamic corticotropin-releasing hormone, and adrenocorticotropin secretion in the rat. Endocrinology. 1990;126:2876–2881. doi: 10.1210/endo-126-6-2876. [DOI] [PubMed] [Google Scholar]
- 34.Bernton EW, Beach JE, Holaday JW, et al. Release of multiple hormones by a direct action of interleukin-1 on pituitary cells. Science. 1987;238:519–521. doi: 10.1126/science.2821620. [DOI] [PubMed] [Google Scholar]
- 35.Besedovsky H, del Rey A, Sorkin E, et al. Immunoregulatory feedback between interleukin-1 and glucocorticoid hormones. Science. 1986;233:652–654. doi: 10.1126/science.3014662. [DOI] [PubMed] [Google Scholar]
- 36.Besedovsky H, Sorkin E, Felix D, et al. Hypothalamic changes during the immune response. Eur J Immunol. 1977;7:323–325. doi: 10.1002/eji.1830070516. [DOI] [PubMed] [Google Scholar]
- 37.Besedovsky H, Sorkin E, Keller M, et al. Changes in blood hormone levels during the immune response. Proc Soc Exp Biol Med. 1975;150:466–470. doi: 10.3181/00379727-150-39057. [DOI] [PubMed] [Google Scholar]
- 38.Besedovsky HO, del Rey A. Mechanism of virus-induced stimulation of the hypothalamus-pituitary-adrenal axis. J Steroid Biochem. 1989;34:235–239. doi: 10.1016/0022-4731(89)90087-3. [DOI] [PubMed] [Google Scholar]
- 39.Besedovsky HO, del Rey A. Immune-neuroendocrine interactions: facts and hypotheses. Endocrinol Rev. 1996;17:64–102. doi: 10.1210/edrv-17-1-64. [DOI] [PubMed] [Google Scholar]
- 40.Besedovsky HO, Del Rey A, Sorkin E. Antigenic competition between horse and sheep red blood cells as a hormone-dependent phenomenon. Clin Exp Immunol. 1979;37:106–113. [PMC free article] [PubMed] [Google Scholar]
- 41.Besedovsky HO, del Rey A, Sorkin E. Lymphokine-containing supernatants from con A-stimulated cells increase corticosterone blood levels. J Immunol. 1981;126:385–387. [PubMed] [Google Scholar]
- 42.Besedovsky HO, del Rey A, Sorkin E, et al. Immunoregulation mediated by the sympathetic nervous system. Cell Immunol. 1979;48:346–355. doi: 10.1016/0008-8749(79)90129-1. [DOI] [PubMed] [Google Scholar]
- 43.Besedovsky HO, Del Rey A, Sorkin E, et al. Lymphoid cells produce an immunoregulatory glucocorticoid increasing factor (GIF) acting through the pituitary gland. Clin Exp Immunol. 1985;59:622–628. [PMC free article] [PubMed] [Google Scholar]
- 44.Bethin KE, Vogt SK, Muglia LJ. Interleukin-6 is an essential, corticotropin-releasing hormone-independent stimulator of the adrenal axis during immune system activation. Proc Natl Acad Sci USA. 2000;97:9317–9322. doi: 10.1073/pnas.97.16.9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biglino A, Limone P, Forno B, et al. Altered adrenocorticotropin and cortisol response to corticotropin-releasing hormone in HIV-1 infection. Eur J Endocrinol. 1995;133:173–179. doi: 10.1530/eje.0.1330173. [DOI] [PubMed] [Google Scholar]
- 46.Bindoni M, Perciavalle V, Berretta S, et al. Interleukin 2 modifies the bioelectric activity of some neurosecretory nuclei in the rat hypothalamus. Brain Res. 1988;462:10–14. doi: 10.1016/0006-8993(88)90578-1. [DOI] [PubMed] [Google Scholar]
- 47.Biron CA, Cousens LP, Ruzek MC, et al. Early cytokine responses to viral infections and their roles in shaping endogenous cellular immunity. Adv Exp Med Biol. 1998;452:143–149. doi: 10.1007/978-1-4615-5355-7_15. [DOI] [PubMed] [Google Scholar]
- 48.Blalock JE. Shared ligands and receptors as a molecular mechanism for communication between the immune and neuroendocrine systems. Ann NY Acad Sci. 1994;741:292–298. doi: 10.1111/j.1749-6632.1994.tb23112.x. [DOI] [PubMed] [Google Scholar]
- 49.Boka G, Anglade P, Wallach D, et al. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci Lett. 1994;172:151–154. doi: 10.1016/0304-3940(94)90684-x. [DOI] [PubMed] [Google Scholar]
- 50.Bornstein SR, Chrousos GP. Clinical review 104: Adrenocorticotropin (ACTH)– and non-ACTH-mediated regulation of the adrenal cortex: neural and immune inputs. J Clin Endocrinol Metab. 1999;84:1729–1736. doi: 10.1210/jcem.84.5.5631. [DOI] [PubMed] [Google Scholar]
- 51.Bornstein SR, Ehrhart-Bornstein M, Scherbaum WA. Morphological and functional studies of the paracrine interaction between cortex and medulla in the adrenal gland. Microsc Res Tech. 1997;36:520–533. doi: 10.1002/(SICI)1097-0029(19970315)36:6<520::AID-JEMT9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 52.Bornstein SR, Ehrhart-Bornstein M, Scherbaum WA, et al. Effects of splanchnic nerve stimulation on the adrenal cortex may be mediated by chromaffin cells in a paracrine manner. Endocrinology. 1990;127:900–906. doi: 10.1210/endo-127-2-900. [DOI] [PubMed] [Google Scholar]
- 53.Bornstein SR, Gonzalez-Hernandez JA, Ehrhart-Bornstein M, et al. Intimate contact of chromaffin and cortical cells within the human adrenal gland forms the cellular basis for important intraadrenal interactions. J Clin Endocrinol Metab. 1994;78:225–232. doi: 10.1210/jcem.78.1.7507122. [DOI] [PubMed] [Google Scholar]
- 54.Brandt ER, Mackay IR, Hertzog PJ, et al. Molecular detection of interferon-alpha expression in multiple sclerosis brain. J Neuroimmunol. 1993;44:1–5. doi: 10.1016/0165-5728(93)90261-v. [DOI] [PubMed] [Google Scholar]
- 55.Breder CD, Hazuka C, Ghayur T, et al. Regional induction of tumor necrosis factor alpha expression in the mouse brain after systemic lipopolysaccharide administration. Proc Natl Acad Sci USA. 1994;91:11393–11397. doi: 10.1073/pnas.91.24.11393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Breder CD, Tsujimoto M, Terano Y, et al. Distribution and characterization of tumor necrosis factor-alpha-like immunoreactivity in the murine central nervous system. J Comp Neurol. 1993;337:543–567. doi: 10.1002/cne.903370403. [DOI] [PubMed] [Google Scholar]
- 57.Brown SL, Smith LR, Blalock JE. Interleukin 1 and interleukin 2 enhance proopiomelanocortin gene expression in pituitary cells. J Immunol. 1987;139:3181–3183. [PubMed] [Google Scholar]
- 58.Bucher M, Hobbhahn J, Kurtz A. Nitric oxide–dependent down-regulation of angiotensin II type 2 receptors during experimental sepsis. Crit Care Med. 2001;29:1750–1755. doi: 10.1097/00003246-200109000-00016. [DOI] [PubMed] [Google Scholar]
- 59.Buckingham JC, Loxley HD, Christian HC, et al. Activation of the HPA axis by immune insults: roles and interactions of cytokines, eicosanoids, glucocorticoids. Pharmacol Biochem Behav. 1996;54:285–298. doi: 10.1016/0091-3057(95)02127-2. [DOI] [PubMed] [Google Scholar]
- 60.Butler LD, Mohler KM, Layman NK, et al. Interleukin-2 induced systemic toxicity: induction of mediators and immunopharmacologic intervention. Immunopharmacol Immunotoxicol. 1989;11:445–487. doi: 10.3109/08923978909005379. [DOI] [PubMed] [Google Scholar]
- 61.Call GB, Husein OF, McIlmoil CJ, et al. Bovine adrenal cells secrete interleukin-6 and tumor necrosis factor in vitro. Gen Comp Endocrinol. 2000;118:249–261. doi: 10.1006/gcen.2000.7458. [DOI] [PubMed] [Google Scholar]
- 62.Callahan TA, Piekut DT. Differential Fos expression induced by IL-1beta and IL-6 in rat hypothalamus and pituitary gland. J Neuroimmunol. 1997;73:207–211. doi: 10.1016/s0165-5728(96)00193-2. [DOI] [PubMed] [Google Scholar]
- 63.Cambronero JC, Rivas FJ, Borrell J, et al. Interleukin-2 induces corticotropin-releasing hormone release from superfused rat hypothalami: influence of glucocorticoids. Endocrinology. 1992;131:677–683. doi: 10.1210/endo.131.2.1639014. [DOI] [PubMed] [Google Scholar]
- 64.Campbell IL, Hobbs MV, Kemper P, et al. Cerebral expression of multiple cytokine genes in mice with lymphocytic choriomeningitis. J Immunol. 1994;152:716–723. [PubMed] [Google Scholar]
- 65.Capuron L, Raison CL, Musselman DL, et al. Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am J Psychiatry. 2003;160:1342–1345. doi: 10.1176/appi.ajp.160.7.1342. [DOI] [PubMed] [Google Scholar]
- 66.Cardoso E, Arzt E, Coumroglon M, et al. Alpha-interferon induces cortisol release by human adrenals in vitro. Int Arch Allergy Appl Immunol. 1990;93:263–266. doi: 10.1159/000235311. [DOI] [PubMed] [Google Scholar]
- 67.Chambers PJ, Saltis J, Alin P, et al. Receptors for human interferon alpha on bovine cells: specificity and tissue distribution. Immunopharmacol Immunotoxicol. 1990;12:513–525. doi: 10.3109/08923979009006475. [DOI] [PubMed] [Google Scholar]
- 68.Charlton BG. Adrenal cortical innervation and glucocorticoid secretion. J Endocrinol. 1990;126:5–8. doi: 10.1677/joe.0.1260005. [DOI] [PubMed] [Google Scholar]
- 69.Chesnokova V, Busick J, Chesnokov V, et al. Different effects of IL-2 in vivo treatment and emotional stress on POMC gene expression in the pituitary of mice. Horm Metab Res. 1997;29:88–89. doi: 10.1055/s-2007-978994. [DOI] [PubMed] [Google Scholar]
- 70.Chesnokova V, Melmed S. Leukemia inhibitory factor mediates the hypothalamic pituitary adrenal axis response to inflammation. Endocrinology. 2000;141:4032–4040. doi: 10.1210/endo.141.11.7778. [DOI] [PubMed] [Google Scholar]
- 71.Christeff N, Gherbi N, Mammes O, et al. Serum cortisol and DHEA concentrations during HIV infection. Psychoneuroendocrinology. 1997;22(Suppl 1):S11–S18. doi: 10.1016/s0306-4530(97)00015-2. [DOI] [PubMed] [Google Scholar]
- 72.Chrousos GP. The stress response and immune function: clinical implications. The 1999 Novera H Spector Lecture. Ann NY Acad Sci. 2000;917:38–67. doi: 10.1111/j.1749-6632.2000.tb05371.x. [DOI] [PubMed] [Google Scholar]
- 73.Clerici M. New research hypotheses in the immunopathogenesis of human immunodeficiency virus infection. Q J Nucl Med. 1995;39:163–168. [PubMed] [Google Scholar]
- 74.Clerici M, Fusi ML, Ruzzante S, et al. Type 1 and type 2 cytokines in HIV infection—a possible role in apoptosis and disease progression. Ann Med. 1997;29:185–188. doi: 10.3109/07853899708999334. [DOI] [PubMed] [Google Scholar]
- 75.Clerici M, Galli M, Bosis S, et al. Immunoendocrinologic abnormalities in human immunodeficiency virus infection. Ann NY Acad Sci. 2000;917:956–961. doi: 10.1111/j.1749-6632.2000.tb05462.x. [DOI] [PubMed] [Google Scholar]
- 76.Conti B, Jahng JW, Tinti C, et al. Induction of interferon-gamma inducing factor in the adrenal cortex. J Biol Chem. 1997;272:2035–2037. doi: 10.1074/jbc.272.4.2035. [DOI] [PubMed] [Google Scholar]
- 77.Conti B, Park LC, Calingasan NY, et al. Cultures of astrocytes and microglia express interleukin 18. Brain Res Mol Brain Res. 1999;67:46–52. doi: 10.1016/s0169-328x(99)00034-0. [DOI] [PubMed] [Google Scholar]
- 78.Cunningham ET, Wada E, Carter DB, et al. In situ histochemical localization of type I interleukin-1 receptor messenger RNA in the central nervous system, pituitary, and adrenal gland of the mouse. J Neurosci. 1992;12:1101–1114. doi: 10.1523/JNEUROSCI.12-03-01101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Currie KP, Zhou Z, Fox AP. Evidence for paracrine signaling between macrophages and bovine adrenal chromaffin cell Ca2+ channels. J Neurophysiol. 2000;83:280–287. doi: 10.1152/jn.2000.83.1.280. [DOI] [PubMed] [Google Scholar]
- 80.Dafny N, Prieto-Gomez B, Dong WQ, et al. Interferon modulates neuronal activity recorded from the hypothalamus, thalamus, hippocampus, amygdala and the somatosensory cortex. Brain Res. 1996;734:269–274. [PubMed] [Google Scholar]
- 81.De Bosscher K, Vanden Berghe W, Haegeman G. Mechanisms of anti-inflammatory action and of immunosuppression by glucocorticoids: negative interference of activated glucocorticoid receptor with transcription factors. J Neuroimmunol. 2000;109:16–22. doi: 10.1016/s0165-5728(00)00297-6. [DOI] [PubMed] [Google Scholar]
- 82.De Simoni MG, Terreni L, Chiesa R, et al. Interferon-gamma potentiates interleukin (IL)–6 and tumor necrosis factor-alpha but not IL-1beta induced by endotoxin in the brain. Endocrinology. 1997;138:5220–5226. doi: 10.1210/endo.138.12.5616. [DOI] [PubMed] [Google Scholar]
- 83.del Rey A, Besedovsky H, Sorkin E. Endogenous blood levels of corticosterone control the immunologic cell mass and B cell activity in mice. J Immunol. 1984;133:572–575. [PubMed] [Google Scholar]
- 84.Denicoff KD, Durkin TM, Lotze MT, et al. The neuroendocrine effects of interleukin-2 treatment. J Clin Endocrinol Metab. 1989;69:402–410. doi: 10.1210/jcem-69-2-402. [DOI] [PubMed] [Google Scholar]
- 85.Dobbs CM, Feng N, Beck FM, et al. Neuroendocrine regulation of cytokine production during experimental influenza viral infection: effects of restraint stress-induced elevation in endogenous corticosterone. J Immunol. 1996;157:1870–1877. [PubMed] [Google Scholar]
- 86.Dopp JM, Mackenzie-Graham A, Otero GC, et al. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol. 1997;75:104–112. doi: 10.1016/s0165-5728(97)00009-x. [DOI] [PubMed] [Google Scholar]
- 87.Doyle MV, Oldstone MB. Interactions between viruses and lymphocytes. I In vivo replication of lymphocytic choriomeningitis virus in mononuclear cells during both chronic and acute viral infections. J Immunol. 1978;121:1262–1269. [PubMed] [Google Scholar]
- 88.Dunn AJ, Powell ML, Gaskin JM. Virus-induced increases in plasma corticosterone. Science. 1987;238:1423–1425. doi: 10.1126/science.3685987. [DOI] [PubMed] [Google Scholar]
- 89.Dunn AJ, Powell ML, Meitin C, et al. Virus infection as a stressor: influenza virus elevates plasma concentrations of corticosterone, and brain concentrations of MHPG and tryptophan. Physiol Behav. 1989;45:591–594. doi: 10.1016/0031-9384(89)90078-4. [DOI] [PubMed] [Google Scholar]
- 90.Dunn AJ, Powell ML, Moreshead WV, et al. Effects of Newcastle disease virus administration to mice on the metabolism of cerebral biogenic amines, plasma corticosterone, and lymphocyte proliferation. Brain Behav Immunol. 1987;1:216–230. doi: 10.1016/0889-1591(87)90024-9. [DOI] [PubMed] [Google Scholar]
- 91.Dunn AJ, Vickers SL. Neurochemical and neuroendocrine responses to Newcastle disease virus administration in mice. Brain Res. 1994;645:103–112. doi: 10.1016/0006-8993(94)91643-8. [DOI] [PubMed] [Google Scholar]
- 92.Edwards AV. Regulation of adrenal function in the conscious calf. Horm Metab Res. 1998;30:303–310. doi: 10.1055/s-2007-978889. [DOI] [PubMed] [Google Scholar]
- 93.Ehrhart-Bornstein M, Haidan A, Alesci S, et al. Neurotransmitters and neuropeptides in the differential regulation of steroidogenesis in adrenocortical-chromaffin co-cultures. Endocrinol Res. 2000;26:833–842. doi: 10.3109/07435800009048606. [DOI] [PubMed] [Google Scholar]
- 94.Ehrhart-Bornstein M, Hinson JP, Bornstein SR, et al. Intraadrenal interactions in the regulation of adrenocortical steroidogenesis. Endocrinol Rev. 1998;19:101–143. doi: 10.1210/edrv.19.2.0326. [DOI] [PubMed] [Google Scholar]
- 95.Elenkov IJ, Kovacs K, Kiss J, et al. Lipopolysaccharide is able to bypass corticotrophin-releasing factor in affecting plasma ACTH and corticosterone levels: evidence from rats with lesions of the paraventricular nucleus. J Endocrinol. 1992;133:231–236. doi: 10.1677/joe.0.1330231. [DOI] [PubMed] [Google Scholar]
- 96.Engeland WC. Functional innervation of the adrenal cortex by the splanchnic nerve. Horm Metab Res. 1998;30:311–314. doi: 10.1055/s-2007-978890. [DOI] [PubMed] [Google Scholar]
- 97.Ericsson A, Kovacs KJ, Sawchenko PE. A functional anatomical analysis of central pathways subserving the effects of interleukin-1 on stress-related neuroendocrine neurons. J Neurosci. 1994;14:897–913. doi: 10.1523/JNEUROSCI.14-02-00897.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Eriksson C, Nobel S, Winblad B, et al. Expression of interleukin 1 alpha and beta, and interleukin 1 receptor antagonist mRNA in the rat central nervous system after peripheral administration of lipopolysaccharides. Cytokine. 2000;12:423–431. doi: 10.1006/cyto.1999.0582. [DOI] [PubMed] [Google Scholar]
- 99.Erlandsson AC, Bladh LG, Stierna P, et al. Herpes simplex virus type 1 infection and glucocorticoid treatment regulate viral yield, glucocorticoid receptor and NF-kappaB levels. J Endocrinol. 2002;175:165–176. doi: 10.1677/joe.0.1750165. [DOI] [PubMed] [Google Scholar]
- 100.Esiri MM. Herpes simplex encephalitis. An immunohistological study of the distribution of viral antigen within the brain. J Neurol Sci. 1982;54:209–226. doi: 10.1016/0022-510x(82)90183-6. [DOI] [PubMed] [Google Scholar]
- 101.Franchimont D, Bouma G, Galon J, et al. Adrenal cortical activation in murine colitis. Gastroenterology. 2000;119:1560–1568. doi: 10.1053/gast.2000.20235. [DOI] [PubMed] [Google Scholar]
- 102.French RA, VanHoy RW, Chizzonite R, et al. Expression and localization of p80 and p68 interleukin-1 receptor proteins in the brain of adult mice. J Neuroimmunol. 1999;93:194–202. doi: 10.1016/s0165-5728(98)00224-0. [DOI] [PubMed] [Google Scholar]
- 103.French RA, Zachary JF, Dantzer R, et al. Dual expression of p80 type I and p68 type II interleukin-I receptors on anterior pituitary cells synthesizing growth hormone. Endocrinology. 1996;137:4027–4036. doi: 10.1210/endo.137.9.8756580. [DOI] [PubMed] [Google Scholar]
- 104.Fukata J, Usui T, Naitoh Y, et al. Effects of recombinant human interleukin-1 alpha, -1 beta, 2 and 6 on ACTH synthesis and release in the mouse pituitary tumour cell line AtT-20. J Endocrinol. 1989;122:33–39. doi: 10.1677/joe.0.1220033. [DOI] [PubMed] [Google Scholar]
- 105.Gabellec MM, Griffais R, Fillion G, et al. Expression of interleukin 1 alpha, interleukin 1 beta and interleukin 1 receptor antagonist mRNA in mouse brain: regulation by bacterial lipopolysaccharide (LPS) treatment. Brain Res Mol Brain Res. 1995;31:122–130. doi: 10.1016/0169-328x(95)00042-q. [DOI] [PubMed] [Google Scholar]
- 106.Gabellec MM, Griffais R, Fillion G, et al. Interleukin-1 receptors type I and type II in the mouse brain: kinetics of mRNA expressions after peripheral administration of bacterial lipopolysaccharide. J Neuroimmunol. 1996;66:65–70. doi: 10.1016/0165-5728(96)00021-5. [DOI] [PubMed] [Google Scholar]
- 107.Gadient RA, Lachmund A, Unsicker K, et al. Expression of interleukin-6 (IL-6) and IL-6 receptor mRNAs in rat adrenal medulla. Neurosci Lett. 1995;194:17–20. doi: 10.1016/0304-3940(95)11708-5. [DOI] [PubMed] [Google Scholar]
- 108.Gaillard RC, Turnill D, Sappino P, et al. Tumor necrosis factor–alpha inhibits the hormonal response of the pituitary gland to hypothalamic releasing factors. Endocrinology. 1990;127:101–106. doi: 10.1210/endo-127-1-101. [DOI] [PubMed] [Google Scholar]
- 109.Gatti S, Bartfai T. Induction of tumor necrosis factor-alpha mRNA in the brain after peripheral endotoxin treatment: comparison with interleukin-1 family and interleukin-6. Brain Res. 1993;624:291–294. doi: 10.1016/0006-8993(93)90090-a. [DOI] [PubMed] [Google Scholar]
- 110.Gautron L, Lafon P, Tramu G, et al. In vivo activation of the interleukin-6 receptor/gp130 signaling pathway in pituitary corticotropes of lipopolysaccharide-treated rats. Neuroendocrinology. 2003;77:32–43. doi: 10.1159/000068336. [DOI] [PubMed] [Google Scholar]
- 111.Gennuso R, Spigelman MK, Vallabhajosula S, et al. Systemic biodistribution of radioiodinated interleukin-2 in the rat. J Biol Respir Modif. 1989;8:375–384. [PubMed] [Google Scholar]
- 112.Ghoshal K, Majumder S, Zhu Q, et al. Influenza virus infection induces metallothionein gene expression in the mouse liver and lung by overlapping but distinct molecular mechanisms. Mol Cell Biol. 2001;21:8301–8317. doi: 10.1128/MCB.21.24.8301-8317.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gisslinger H, Svoboda T, Clodi M, et al. Interferon-alpha stimulates the hypothalamic-pituitary-adrenal axis in vivo and in vitro. Neuroendocrinology. 1993;57:489–495. doi: 10.1159/000126396. [DOI] [PubMed] [Google Scholar]
- 114.Goebel MU, Baase J, Pithan V, et al. Acute interferon beta-1b administration alters hypothalamic-pituitary-adrenal axis activity, plasma cytokines and leukocyte distribution in healthy subjects. Psychoneuroendocrinology. 2002;27:881–892. doi: 10.1016/s0306-4530(01)00099-3. [DOI] [PubMed] [Google Scholar]
- 115.Goehler LE, Relton JK, Dripps D, et al. Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: a possible mechanism for immune-to-brain communication. Brain Res Bull. 1997;43:357–364. doi: 10.1016/s0361-9230(97)00020-8. [DOI] [PubMed] [Google Scholar]
- 116.Gonzalez-Hernandez JA, Bornstein SR, Ehrhart-Bornstein M, et al. Macrophages within the human adrenal gland. Cell Tissue Res. 1994;278:201–205. doi: 10.1007/BF00414161. [DOI] [PubMed] [Google Scholar]
- 117.Gonzalez-Hernandez JA, Bornstein SR, Ehrhart-Bornstein M, et al. IL-1 is expressed in human adrenal gland in vivo. Possible role in a local immune-adrenal axis. Clin Exp Immunol. 1995;99:137–141. doi: 10.1111/j.1365-2249.1995.tb03484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gonzalez-Hernandez JA, Bornstein SR, Ehrhart-Bornstein M, et al. Interleukin-6 messenger ribonucleic acid expression in human adrenal gland in vivo: new clue to a paracrine or autocrine regulation of adrenal function. J Clin Endocrinol Metab. 1994;79:1492–1497. doi: 10.1210/jcem.79.5.7962348. [DOI] [PubMed] [Google Scholar]
- 119.Gonzalez-Hernandez JA, Ehrhart-Bornstein M, Spath-Schwalbe E, et al. Human adrenal cells express tumor necrosis factor-alpha messenger ribonucleic acid: evidence for paracrine control of adrenal function. J Clin Endocrinol Metab. 1996;81:807–813. doi: 10.1210/jcem.81.2.8636308. [DOI] [PubMed] [Google Scholar]
- 120.Gwosdow AR, Kumar MS, Bode HH. Interleukin 1 stimulation of the hypothalamic-pituitary-adrenal axis. Am J Physiol. 1990;258:E65–70. doi: 10.1152/ajpendo.1990.258.1.E65. [DOI] [PubMed] [Google Scholar]
- 121.Gwosdow AR, O’Connell NA, Spencer JA, et al. Interleukin-1–induced corticosterone release occurs by an adrenergic mechanism from rat adrenal gland. Am J Physiol. 1992;263:E461–E466. doi: 10.1152/ajpendo.1992.263.3.E461. [DOI] [PubMed] [Google Scholar]
- 122.Hanisch UK, Rowe W, Sharma S, et al. Hypothalamic-pituitary-adrenal activity during chronic central administration of interleukin-2. Endocrinology. 1994;135:2465–2472. doi: 10.1210/endo.135.6.7988433. [DOI] [PubMed] [Google Scholar]
- 123.Harbuz MS, Lightman SL. Stress and the hypothalamo-pituitary-adrenal axis: acute, chronic and immunological activation. J Endocrinol. 1992;134:327–339. doi: 10.1677/joe.0.1340327. [DOI] [PubMed] [Google Scholar]
- 124.Harbuz MS, Rees RG, Eckland D, et al. Paradoxical responses of hypothalamic corticotropin-releasing factor (CRF) messenger ribonucleic acid (mRNA) and CRF-41 peptide and adenohypophysial proopiomelanocortin mRNA during chronic inflammatory stress. Endocrinology. 1992;130:1394–1400. doi: 10.1210/endo.130.3.1537299. [DOI] [PubMed] [Google Scholar]
- 125.Harbuz MS, Stephanou A, Knight RA, et al. Action of interleukin-2 and interleukin-4 on CRF mRNA in the hypothalamus and POMC mRNA in the anterior pituitary. Brain Behav Immun. 1992;6:214–222. doi: 10.1016/0889-1591(92)90044-o. [DOI] [PubMed] [Google Scholar]
- 126.Harbuz MS, Stephanou A, Sarlis N, et al. The effects of recombinant human interleukin (IL)-1 alpha, IL-1 beta or IL-6 on hypothalamo-pituitary-adrenal axis activation. J Endocrinol. 1992;133:349–355. doi: 10.1677/joe.0.1330349. [DOI] [PubMed] [Google Scholar]
- 127.Hauger, R.L. and F.M. Dautzenberg. 2000. Regulation of the stress response by corticotropin-releasing factor receptors, pp. 261–286. In: P.M. Conn and M.E. Freeman (eds.), Neuroendocrinology in Physiology and Medicine. Humana Press Inc., Totowa, NJ.
- 128.Hennet T, Ziltener HJ, Frei K, et al. A kinetic study of immune mediators in the lungs of mice infected with influenza A virus. J Immunol. 1992;149:932–939. [PubMed] [Google Scholar]
- 129.Herman JP, Schafer MK, Young EA, et al. Evidence for hippocampal regulation of neuroendocrine neurons of the hypothalamo-pituitary-adrenocortical axis. J Neurosci. 1989;9:3072–3082. doi: 10.1523/JNEUROSCI.09-09-03072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hermann G, Beck FM, Sheridan JF. Stress-induced glucocorticoid response modulates mononuclear cell trafficking during an experimental influenza viral infection. J Neuroimmunol. 1995;56:179–186. doi: 10.1016/0165-5728(94)00145-e. [DOI] [PubMed] [Google Scholar]
- 131.Hermann G, Tovar CA, Beck FM, et al. Kinetics of glucocorticoid response to restraint stress and/or experimental influenza viral infection in two inbred strains of mice. J Neuroimmunol. 1994;49:25–33. doi: 10.1016/0165-5728(94)90177-5. [DOI] [PubMed] [Google Scholar]
- 132.Hillhouse EW. Interleukin-2 stimulates the secretion of arginine vasopressin but not corticotropin-releasing hormone from rat hypothalamic cells in vitro. Brain Res. 1994;650:323–325. doi: 10.1016/0006-8993(94)91799-x. [DOI] [PubMed] [Google Scholar]
- 133.Hinson JP. Paracrine control of adrenocortical function: a new role for the medulla? J Endocrinol. 1990;124:7–9. doi: 10.1677/joe.0.1240007. [DOI] [PubMed] [Google Scholar]
- 134.Holgert H, Dagerlind A, Hokfelt T. Immunohistochemical characterization of the peptidergic innervation of the rat adrenal gland. Horm Metab Res. 1998;30:315–322. doi: 10.1055/s-2007-978891. [DOI] [PubMed] [Google Scholar]
- 135.Hollenberg SM, Weinberger C, Ong ES, et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318:635–641. doi: 10.1038/318635a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Holsboer F, Stalla GK, von Bardeleben U, et al. Acute adrenocortical stimulation by recombinant gamma interferon in human controls. Life Sci. 1988;42:1–5. doi: 10.1016/0024-3205(88)90617-0. [DOI] [PubMed] [Google Scholar]
- 137.Honda M, Orii F, Ayabe T, et al. Expression of glucocorticoid receptor beta in lymphocytes of patients with glucocorticoid-resistant ulcerative colitis. Gastroenterology. 2000;118:859–866. doi: 10.1016/s0016-5085(00)70172-7. [DOI] [PubMed] [Google Scholar]
- 138.Inoue K, Couch EF, Takano K, et al. The structure and function of folliculo-stellate cells in the anterior pituitary gland. Arch Histol Cytol. 1999;62:205–218. doi: 10.1679/aohc.62.205. [DOI] [PubMed] [Google Scholar]
- 139.Irusen E, Matthews JG, Takahashi A, et al. p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: role in steroid-insensitive asthma. J Allergy Clin Immunol. 2002;109:649–657. doi: 10.1067/mai.2002.122465. [DOI] [PubMed] [Google Scholar]
- 140.Jaattela M, Ilvesmaki V, Voutilainen R, et al. Tumor necrosis factor as a potent inhibitor of adrenocorticotropin-induced cortisol production and steroidogenic P450 enzyme gene expression in cultured human fetal adrenal cells. Endocrinology. 1991;128:623–629. doi: 10.1210/endo-128-1-623. [DOI] [PubMed] [Google Scholar]
- 141.Janeway, Jr., C.A., and P. Travers. 1999. Immunobiology: The Immune System in Health and Disease. Current Biology Limited, San Francisco.
- 142.Janicki PK. Binding of human alpha-interferon in the brain tissue membranes of rat. Res Commun Chem Pathol Pharmacol. 1992;75:117–120. [PubMed] [Google Scholar]
- 143.Judd AM, Call GB, Barney M, et al. Possible function of IL-6 and TNF as intraadrenal factors in the regulation of adrenal steroid secretion. Ann NY Acad Sci. 2000;917:628–637. doi: 10.1111/j.1749-6632.2000.tb05428.x. [DOI] [PubMed] [Google Scholar]
- 144.Judd AM, MacLeod RM. Adrenocorti-cotropin increases interleukin-6 release from rat adrenal zona glomerulosa cells. Endocrinology. 1992;130:1245–1254. doi: 10.1210/endo.130.3.1311232. [DOI] [PubMed] [Google Scholar]
- 145.Judd AM, MacLeod RM. Differential release of tumor necrosis factor and IL-6 from adrenal zona glomerulosa cells in vitro. Am J Physiol. 1995;268:E114–E120. doi: 10.1152/ajpendo.1995.268.1.E114. [DOI] [PubMed] [Google Scholar]
- 146.Kageyama K, Watanobe H, Takebe K. In vivo evidence that arginine vasopressin is involved in the adrenocorticotropin response induced by interleukin-6 but not by tumor necrosis factor-alpha in the rat. Neuroimmunomodulation. 1995;2:137–140. doi: 10.1159/000096883. [DOI] [PubMed] [Google Scholar]
- 147.Kam JC, Szefler SJ, Surs W, et al. Combination IL-2 and IL-4 reduces glucocorticoid receptor-binding affinity and T cell response to glucocorticoids. J Immunol. 1993;151:3460–3466. [PubMed] [Google Scholar]
- 148.Kapcala LP, Chautard T, Eskay RL. The protective role of the hypothalamic-pituitary-adrenal axis against lethality produced by immune, infectious, and inflammatory stress. Ann NY Acad Sci. 1995;771:419–437. doi: 10.1111/j.1749-6632.1995.tb44699.x. [DOI] [PubMed] [Google Scholar]
- 149.Karalis K, Muglia LJ, Bae D, et al. CRH and the immune system. J Neuroimmunol. 1997;72:131–136. doi: 10.1016/s0165-5728(96)00178-6. [DOI] [PubMed] [Google Scholar]
- 150.Karanth S, Lyson K, McCann SM. Effects of cholinergic agonists and antagonists on interleukin-2-induced corticotropin-releasing hormone release from the mediobasal hypothalamus. Neuroimmunomodulation. 1999;6:168–174. doi: 10.1159/000026378. [DOI] [PubMed] [Google Scholar]
- 151.Karanth S, McCann SM. Anterior pituitary hormone control by interleukin 2. Proc Natl Acad Sci USA. 1991;88:2961–2965. doi: 10.1073/pnas.88.7.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Katahira M, Iwasaki Y, Aoki Y, et al. Cytokine regulation of the rat proopiomelanocortin gene expression in AtT-20 cells. Endocrinology. 1998;139:2414–2422. doi: 10.1210/endo.139.5.6005. [DOI] [PubMed] [Google Scholar]
- 153.Kehrer P, Turnill D, Dayer JM, et al. Human recombinant interleukin-1 beta and -alpha, but not recombinant tumor necrosis factor–alpha stimulate ACTH release from rat anterior pituitary cells in vitro in a prostaglandin E2 and cAMP-independent manner. Neuroendocrinology. 1988;48:160–166. doi: 10.1159/000125004. [DOI] [PubMed] [Google Scholar]
- 154.Keller-Wood ME, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocrinol Rev. 1984;5:1–24. doi: 10.1210/edrv-5-1-1. [DOI] [PubMed] [Google Scholar]
- 155.Kiefer R, Kreutzberg GW. Gamma interferon–like immunoreactivity in the rat nervous system. Neuroscience. 1990;37:725–734. doi: 10.1016/0306-4522(90)90103-b. [DOI] [PubMed] [Google Scholar]
- 156.Kino T, Gragerov A, Kopp JB, et al. The HIV-1 virion-associated protein vpr is a coactivator of the human glucocorticoid receptor. J Exp Med. 1999;189:51–62. doi: 10.1084/jem.189.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kino T, Gragerov A, Slobodskaya O, et al. Human immunodeficiency virus type 1 (HIV-1) accessory protein Vpr induces transcription of the HIV-1 and glucocorticoid-responsive promoters by binding directly to p300/CBP coactivators. J Virol. 2002;76:9724–9734. doi: 10.1128/JVI.76.19.9724-9734.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kino T, Kopp JB, Chrousos GP. Glucocorticoids suppress human immunodeficiency virus type-1 long terminal repeat activity in a cell type-specific, glucocorticoid receptor-mediated fashion: direct protective effects at variance with clinical phenomenology. J Steroid Biochem Mol Biol. 2000;75:283–290. doi: 10.1016/s0960-0760(00)00187-4. [DOI] [PubMed] [Google Scholar]
- 159.Kinouchi K, Brown G, Pasternak G, et al. Identification and characterization of receptors for tumor necrosis factor–alpha in the brain. Biochem Biophys Res Commun. 1991;181:1532–1538. doi: 10.1016/0006-291x(91)92113-x. [DOI] [PubMed] [Google Scholar]
- 160.Kobayashi H, Fukata J, Murakami N, et al. Tumor necrosis factor receptors in the pituitary cells. Brain Res. 1997;758:45–50. doi: 10.1016/s0006-8993(96)01437-0. [DOI] [PubMed] [Google Scholar]
- 161.Koenig JI, Snow K, Clark BD, et al. Intrinsic pituitary interleukin-1 beta is induced by bacterial lipopolysaccharide. Endocrinology. 1990;126:3053–3058. doi: 10.1210/endo-126-6-3053. [DOI] [PubMed] [Google Scholar]
- 162.Korneva EA, Barabanova SV, Golovko OI, et al. C-fos and IL-2 gene expression in rat brain cells and splenic lymphocytes after nonantigenic and antigenic stimuli. Ann NY Acad Sci. 2000;917:197–209. doi: 10.1111/j.1749-6632.2000.tb05384.x. [DOI] [PubMed] [Google Scholar]
- 163.Kovacs KJ. Functional neuroanatomy of the parvocellular vasopressinergic system: transcriptional responses to stress and glucocorticoid feedback. Prog Brain Res. 1998;119:31–43. doi: 10.1016/s0079-6123(08)61560-5. [DOI] [PubMed] [Google Scholar]
- 164.Kovacs KJ, Elenkov IJ. Differential dependence of ACTH secretion induced by various cytokines on the integrity of the paraventricular nucleus. J Neuroendocrinol. 1995;7:15–23. doi: 10.1111/j.1365-2826.1995.tb00662.x. [DOI] [PubMed] [Google Scholar]
- 165.Kovalovsky D, Refojo D, Holsboer F, et al. Molecular mechanisms and Th1/Th2 pathways in corticosteroid regulation of cytokine production. J Neuroimmunol. 2000;109:23–29. doi: 10.1016/s0165-5728(00)00298-8. [DOI] [PubMed] [Google Scholar]
- 166.Kumar M, Kumar AM, Morgan R, et al. Abnormal pituitary-adrenocortical response in early HIV-1 infection. J Acquir Immune Defic Syndr. 1993;6:61–65. [PubMed] [Google Scholar]
- 167.Kurotani R, Yasuda M, Oyama K, et al. Expression of interleukin-6, interleukin-6 receptor (gp80), and the receptor’s signal-transducing subunit (gp130) in human normal pituitary glands and pituitary adenomas. Mod Pathol. 2001;14:791–797. doi: 10.1038/modpathol.3880392. [DOI] [PubMed] [Google Scholar]
- 168.Lapchak PA, Araujo DM. Interleukin-2 regulates monoamine and opioid peptide release from the hypothalamus. Neuroreport. 1993;4:303–306. doi: 10.1097/00001756-199303000-00019. [DOI] [PubMed] [Google Scholar]
- 169.Laudat A, Blum L, Guechot J, et al. Changes in systemic gonadal and adrenal steroids in asymptomatic human immunodeficiency virus–infected men: relationship with the CD4 cell counts. Eur J Endocrinol. 1995;133:418–424. doi: 10.1530/eje.0.1330418. [DOI] [PubMed] [Google Scholar]
- 170.Laye S, Parnet P, Goujon E, et al. Peripheral administration of lipopolysaccharide induces the expression of cytokine transcripts in the brain and pituitary of mice. Brain Res Mol Brain Res. 1994;27:157–162. doi: 10.1016/0169-328x(94)90197-x. [DOI] [PubMed] [Google Scholar]
- 171.Lechan RM, Toni R, Clark BD, et al. Immunoreactive interleukin-1 beta localization in the rat forebrain. Brain Res. 1990;514:135–140. doi: 10.1016/0006-8993(90)90445-h. [DOI] [PubMed] [Google Scholar]
- 172.Ljungdahl A, Olsson T, Van der Meide PH, et al. Interferon-gamma-like immunoreactivity in certain neurons of the central and peripheral nervous system. J Neurosci Res. 1989;24:451–456. doi: 10.1002/jnr.490240316. [DOI] [PubMed] [Google Scholar]
- 173.Loddick SA, Liu C, Takao T, et al. Interleukin-1 receptors: cloning studies and role in central nervous system disorders. Brain Res Brain Res Rev. 1998;26:306–319. doi: 10.1016/s0165-0173(97)00037-4. [DOI] [PubMed] [Google Scholar]
- 174.Lohrer P, Gloddek J, Nagashima AC, et al. Lipopolysaccharide directly stimulates the intrapituitary interleukin-6 production by folliculostellate cells via specific receptors and the p38alpha mitogen-activated protein kinase/nuclear factor-kappaB pathway. Endocrinology. 2000;141:4457–4465. doi: 10.1210/endo.141.12.7811. [DOI] [PubMed] [Google Scholar]
- 175.Lortholary O, Christeff N, Casassus P, et al. Hypothalamo-pituitary-adrenal function in human immunodeficiency virus–infected men. J Clin Endocrinol Metab. 1996;81:791–796. doi: 10.1210/jcem.81.2.8636305. [DOI] [PubMed] [Google Scholar]
- 176.Lotze MT, Frana LW, Sharrow SO, et al. In vivo administration of purified human interleukin 2. I Half-life and immunologic effects of the Jurkat cell line–derived interleukin 2. J Immunol. 1985;134:157–166. [PubMed] [Google Scholar]
- 177.Lyson K, McCann SM. The effect of interleukin-6 on pituitary hormone release in vivo and in vitro. Neuroendocrinology. 1991;54:262–266. doi: 10.1159/000125884. [DOI] [PubMed] [Google Scholar]
- 178.Marquette C, Van Dam AM, Van Rooijen N, et al. Peripheral macrophage depletion prevents down regulation of central interleukin-1 receptors in mice after endotoxin administration. Psychoneuroendocrinology. 1994;19:189–196. doi: 10.1016/0306-4530(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 179.Mastorakos G, Chrousos GP, Weber JS. Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in humans. J Clin Endocrinol Metab. 1993;77:1690–1694. doi: 10.1210/jcem.77.6.8263159. [DOI] [PubMed] [Google Scholar]
- 180.Mastorakos G, Weber JS, Magiakou MA, et al. Hypothalamic-pituitary-adrenal axis activation and stimulation of systemic vasopressin secretion by recombinant interleukin-6 in humans: potential implications for the syndrome of inappropriate vasopressin secretion. J Clin Endocrinol Metab. 1994;79:934–939. doi: 10.1210/jcem.79.4.7962300. [DOI] [PubMed] [Google Scholar]
- 181.Matta SG, Weatherbee J, Sharp BM. A central mechanism is involved in the secretion of ACTH in response to IL-6 in rats: comparison to and interaction with IL-1 beta. Neuroendocrinology. 1992;56:516–525. doi: 10.1159/000126269. [DOI] [PubMed] [Google Scholar]
- 182.Mazzocchi G, Malendowicz LK, Markowska A, et al. Effect of hypophysectomy on corticotropin-releasing hormone and adrenocorticotropin immunoreactivities in the rat adrenal gland. Mol Cell Neurosci. 1994;5:345–349. doi: 10.1006/mcne.1994.1041. [DOI] [PubMed] [Google Scholar]
- 183.McCoy JG, Matta SG, Sharp BM. Prostaglandins mediate the ACTH response to interleukin-1–beta instilled into the hypothalamic median eminence. Neuroendocrinology. 1994;60:426–435. doi: 10.1159/000126777. [DOI] [PubMed] [Google Scholar]
- 184.McDonald TJ, Nathanielsz PW. The involvement of innervation in the regulation of fetal adrenal steroidogenesis. Horm Metab Res. 1998;30:297–302. doi: 10.1055/s-2007-978888. [DOI] [PubMed] [Google Scholar]
- 185.McEwen BS, Biron CA, Brunson KW, et al. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Brain Res Rev. 1997;23:79–133. doi: 10.1016/s0165-0173(96)00012-4. [DOI] [PubMed] [Google Scholar]
- 186.McLean JH, Shipley MT, Bernstein DI, et al. Selective lesions of neural pathways following viral inoculation of the olfactory bulb. Exp Neurol. 1993;122:209–222. doi: 10.1006/exnr.1993.1121. [DOI] [PubMed] [Google Scholar]
- 187.Membreno L, Irony I, Dere W, et al. Adrenocortical function in acquired immunodeficiency syndrome. J Clin Endocrinol Metab. 1987;65:482–487. doi: 10.1210/jcem-65-3-482. [DOI] [PubMed] [Google Scholar]
- 188.Miller, A.H., B.D. Pearce, M.C. Ruzek, et al. 2001. Interactions between the hypothalamic-pituitary-adrenal axis and immune system during viral infection: pathways for environmental effects on disease expression. pp. 425–450. In: B.S. McEwen (ed.), Handbook of Physiology. Section 7: The Endocrine System, IV: Coping with the Environment: Neural and Endocrine Mechanisms Interactions. Oxford University Press, New York.
- 189.Miller AH, Spencer RL, Pearce BD, et al. 1996 Curt P. Richter Award. Effects of viral infection on corticosterone secretion and glucocorticoid receptor binding in immune tissues. Psychoneuroendocrinology. 1997;22:455–474. doi: 10.1016/s0306-4530(97)00028-0. [DOI] [PubMed] [Google Scholar]
- 190.Milton NG, Hillhouse EW, Milton AS. Activation of the hypothalamo-pituitary-adrenocortical axis in the conscious rabbit by the pyrogen polyinosinic:polycytidylic acid is dependent on corticotrophin-releasing factor–41. J Endocrinol. 1992;135:69–75. doi: 10.1677/joe.0.1350069. [DOI] [PubMed] [Google Scholar]
- 191.Milton NG, Self CH, Hillhouse EW. Effects of pyrogenic immunomodulators on the release of corticotrophin-releasing factor-41 and prostaglandin E2 from the intact rat hypothalamus in vitro. Br J Pharmacol. 1993;109:88–93. doi: 10.1111/j.1476-5381.1993.tb13535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mirani M, Elenkov I, Volpi S, et al. HIV-1 protein Vpr suppresses IL-12 production from human monocytes by enhancing glucocorticoid action: potential implications of Vpr coactivator activity for the innate and cellular immunity deficits observed in HIV-1 infection. J Immunol. 2002;169:6361–6368. doi: 10.4049/jimmunol.169.11.6361. [DOI] [PubMed] [Google Scholar]
- 193.Muglia L, Jacobson L, Dikkes P, et al. Corticotropin-releasing hormone deficiency reveals major fetal but not adult glucocorticoid need. Nature. 1995;373:427–432. doi: 10.1038/373427a0. [DOI] [PubMed] [Google Scholar]
- 194.Mulla A, Buckingham JC. Regulation of the hypothalamo-pituitary-adrenal axis by cytokines. Baillieres Clin Endocrinol Metab. 1999;13:503–521. doi: 10.1053/beem.1999.0041. [DOI] [PubMed] [Google Scholar]
- 195.Muller H, Hiemke C, Hammes E, et al. Sub-acute effects of interferon-alpha 2 on adrenocorticotrophic hormone, cortisol, growth hormone and prolactin in humans. Psychoneuroendocrinology. 1992;17:459–465. doi: 10.1016/0306-4530(92)90004-q. [DOI] [PubMed] [Google Scholar]
- 196.Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocrinol Rev. 1984;5:25–44. doi: 10.1210/edrv-5-1-25. [DOI] [PubMed] [Google Scholar]
- 197.Muramami N, Fukata J, Tsukada T, et al. Bacterial lipopolysaccharide-induced expression of interleukin-6 messenger ribonucleic acid in the rat hypothalamus, pituitary, adrenal gland, and spleen. Endocrinology. 1993;133:2574–2578. doi: 10.1210/endo.133.6.8243280. [DOI] [PubMed] [Google Scholar]
- 198.Nadeau S, Rivest S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience. 1999;93:1449–1464. doi: 10.1016/s0306-4522(99)00225-0. [DOI] [PubMed] [Google Scholar]
- 199.Nadeau S, Rivest S. Regulation of the gene encoding tumor necrosis factor–alpha (TNF-alpha) in the rat brain and pituitary in response in different models of systemic immune challenge. J Neuropathol Exp Neurol. 1999;58:61–77. doi: 10.1097/00005072-199901000-00008. [DOI] [PubMed] [Google Scholar]
- 200.Nagano I, Takao T, Nanamiya W, et al. Modulation of type I interleukin-1 receptor messenger RNA followed by one and repeated endotoxin treatment in the mouse. J Neuroimmunol. 2000;105:154–160. doi: 10.1016/s0165-5728(00)00206-x. [DOI] [PubMed] [Google Scholar]
- 201.Naito Y, Fukata J, Nakaishi S, et al. Chronic effects of interleukin-1 on hypothalamus, pituitary and adrenal glands in rat. Neuroendocrinology. 1990;51:637–641. doi: 10.1159/000125404. [DOI] [PubMed] [Google Scholar]
- 202.Naito Y, Fukata J, Tamai S, et al. Biphasic changes in hypothalamo-pituitary-adrenal function during the early recovery period after major abdominal surgery. J Clin Endocrinol Metab. 1991;73:111–117. doi: 10.1210/jcem-73-1-111. [DOI] [PubMed] [Google Scholar]
- 203.Naito Y, Fukata J, Tominaga T, et al. Adrenocorticotropic hormone-releasing activities of interleukins in a homologous in vivo system. Biochem Biophys Res Commun. 1989;164:1262–1267. doi: 10.1016/0006-291x(89)91805-6. [DOI] [PubMed] [Google Scholar]
- 204.Naito Y, Tamai S, Shingu K, et al. Responses of plasma adrenocorticotropic hormone, cortisol, and cytokines during and after upper abdominal surgery. Anesthesiology. 1992;77:426–431. doi: 10.1097/00000542-199209000-00004. [DOI] [PubMed] [Google Scholar]
- 205.Naitoh Y, Fukata J, Tominaga T, et al. Interleukin-6 stimulates the secretion of adrenocorticotropic hormone in conscious, freely-moving rats. Biochem Biophys Res Commun. 1988;155:1459–1463. doi: 10.1016/s0006-291x(88)81305-6. [DOI] [PubMed] [Google Scholar]
- 206.Navarra P, Tsagarakis S, Faria MS, et al. Interleukins-1 and -6 stimulate the release of corticotropin-releasing hormone-41 from rat hypothalamus in vitro via the eicosanoid cyclooxygenase pathway. Endocrinology. 1991;128:37–44. doi: 10.1210/endo-128-1-37. [DOI] [PubMed] [Google Scholar]
- 207.Niimi M, Wada Y, Sato M, et al. Effect of continuous intravenous injection of interleukin-6 and pretreatment with cyclooxygenase inhibitor on brain c-fos expression in the rat. Neuroendocrinology. 1997;66:47–53. doi: 10.1159/000127218. [DOI] [PubMed] [Google Scholar]
- 208.Nijsten MW, de Groot ER, ten Duis HJ, et al. Serum levels of interleukin-6 and acute phase responses. Lancet. 1987;2:921. doi: 10.1016/s0140-6736(87)91413-9. [DOI] [PubMed] [Google Scholar]
- 209.Nobel CS, Schultzberg M. Induction of interleukin-1 beta mRNA and enkephalin mRNA in the rat adrenal gland by lipopolysaccharides studied by in situ hybridization histochemistry. Neuroimmunomodulation. 1995;2:61–73. doi: 10.1159/000096873. [DOI] [PubMed] [Google Scholar]
- 210.Norbiato G, Bevilacqua M, Vago T. Glucocorticoids and the immune system in AIDS. Psychoneuroendocrinology. 1997;22(Suppl 1):S19–S25. doi: 10.1016/s0306-4530(97)00011-5. [DOI] [PubMed] [Google Scholar]
- 211.Norbiato G, Bevilacqua M, Vago T, et al. Glucocorticoids and interferon-alpha in the acquired immunodeficiency syndrome. J Clin Endocrinol Metab. 1996;81:2601–2606. doi: 10.1210/jcem.81.7.8675584. [DOI] [PubMed] [Google Scholar]
- 212.Norbiato G, Bevilacqua M, Vago T, et al. Glucocorticoid resistance and the immune function in the immunodeficiency syndrome. Ann NY Acad Sci. 1998;840:835–847. doi: 10.1111/j.1749-6632.1998.tb09621.x. [DOI] [PubMed] [Google Scholar]
- 213.Norbiato G, Bevilacqua M, Vago T, et al. Glucocorticoids and the immune function in the human immunodeficiency virus infection: a study in hypercortisolemic and cortisol-resistant patients. J Clin Endocrinol Metab. 1997;82:3260–3263. doi: 10.1210/jcem.82.10.4304. [DOI] [PubMed] [Google Scholar]
- 214.Norbiato G, Galli M, Righini V, et al. The syndrome of acquired glucocorticoid resistance in HIV infection. Baillieres Clin Endocrinol Metab. 1994;8:777–787. doi: 10.1016/s0950-351x(05)80300-3. [DOI] [PubMed] [Google Scholar]
- 215.Nussdorfer GG, Mazzocchi G. Immune-endocrine interactions in the mammalian adrenal gland: facts and hypotheses. Int Rev Cytol. 1998;183:143–184. doi: 10.1016/s0074-7696(08)60144-8. [DOI] [PubMed] [Google Scholar]
- 216.Oakley RH, Sar M, Cidlowski JA. The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. J Biol Chem. 1996;271:9550–9559. doi: 10.1074/jbc.271.16.9550. [DOI] [PubMed] [Google Scholar]
- 217.O’Connell NA, Kumar A, Chatzipanteli K, et al. Interleukin-1 regulates corticosterone secretion from the rat adrenal gland through a catecholamine-dependent and prostaglandin E2–independent mechanism. Endocrinology. 1994;135:460–467. doi: 10.1210/endo.135.1.8013385. [DOI] [PubMed] [Google Scholar]
- 218.Ohgo S, Nakatsuru K, Oki Y, et al. Stimulation by interleukin-1 (IL-1) of the release of rat corticotropin-releasing factor (CRF), which is independent of the cholinergic mechanism, from superfused rat hypothalamo-neurohypophysial complexes. Brain Res. 1991;550:213–219. doi: 10.1016/0006-8993(91)91320-z. [DOI] [PubMed] [Google Scholar]
- 219.Ohmichi M, Hirota K, Koike K, et al. Binding sites for interleukin-6 in the anterior pituitary gland. Neuroendocrinology. 1992;55:199–203. doi: 10.1159/000126115. [DOI] [PubMed] [Google Scholar]
- 220.Olsen NJ, Nicholson WE, DeBold CR, et al. Lymphocyte-derived adrenocorticotropin is insufficient to stimulate adrenal steroidogenesis in hypophysectomized rats. Endocrinology. 1992;130:2113–2119. doi: 10.1210/endo.130.4.1312443. [DOI] [PubMed] [Google Scholar]
- 221.Olsson T, Kelic S, Edlund C, et al. Neuronal interferon-gamma immunoreactive molecule: bioactivities and purification. Eur J Immunol. 1994;24:308–314. doi: 10.1002/eji.1830240205. [DOI] [PubMed] [Google Scholar]
- 222.Orange JS, Salazar-Mather TP, Opal SM, et al. Mechanisms for virus-induced liver disease: tumor necrosis factor–mediated pathology independent of natural killer and T cells during murine cytomegalovirus infection. J Virol. 1997;71:9248–9258. doi: 10.1128/jvi.71.12.9248-9258.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Orange JS, Salazar-Mather TP, Opal SM, et al. Mechanism of interleukin 12–mediated toxicities during experimental viral infections: role of tumor necrosis factor and glucocorticoids. J Exp Med. 1995;181:901–914. doi: 10.1084/jem.181.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Orange JS, Wang B, Terhorst C, et al. Requirement for natural killer cell-produced interferon gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J Exp Med. 1995;182:1045–1056. doi: 10.1084/jem.182.4.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Padgett DA, Loria RM, Sheridan JF. Steroid hormone regulation of antiviral immunity. Ann NY Acad Sci. 2000;917:935–943. doi: 10.1111/j.1749-6632.2000.tb05459.x. [DOI] [PubMed] [Google Scholar]
- 226.Paivarinta MA, Marttila RJ, Lonnberg P, et al. Decreased raphe serotonin in rabbits with experimental herpes simplex encephalitis. Neurosci Lett. 1993;156:1–4. doi: 10.1016/0304-3940(93)90424-j. [DOI] [PubMed] [Google Scholar]
- 227.Pardy K, Murphy D, Carter D, et al. The influence of interleukin-2 on vasopressin and oxytocin gene expression in the rodent hypothalamus. J Neuroimmunol. 1993;42:131–138. doi: 10.1016/0165-5728(93)90002-g. [DOI] [PubMed] [Google Scholar]
- 228.Pariante CM, Pearce BD, Pisell TL, et al. The proinflammatory cytokine, interleukin-1alpha, reduces glucocorticoid receptor translocation and function. Endocrinology. 1999;140:4359–4366. doi: 10.1210/endo.140.9.6986. [DOI] [PubMed] [Google Scholar]
- 229.Park JH, Shin SH. Induction of IL-12 gene expression in the brain in septic shock. Biochem Biophys Res Commun. 1996;224:391–396. doi: 10.1006/bbrc.1996.1038. [DOI] [PubMed] [Google Scholar]
- 230.Parnet P, Amindari S, Wu C, et al. Expression of type I and type II interleukin-1 receptors in mouse brain. Brain Res Mol Brain Res. 1994;27:63–70. doi: 10.1016/0169-328x(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 231.Parnet P, Brunke DL, Goujon E, et al. Molecular identification of two types of interleukin-1 receptors in the murine pituitary gland. J Neuroendocrinol. 1993;5:213–219. doi: 10.1111/j.1365-2826.1993.tb00384.x. [DOI] [PubMed] [Google Scholar]
- 232.Patchev VK, Kalogeras KT, Zelazowski P, et al. Increased plasma concentrations, hypothalamic content, and in vitro release of arginine vasopressin in inflammatory disease-prone, hypothalamic corticotropin-releasing hormone-deficient Lewis rats. Endocrinology. 1992;131:1453–1457. doi: 10.1210/endo.131.3.1505475. [DOI] [PubMed] [Google Scholar]
- 233.Path G, Scherbaum WA, Bornstein SR. The role of interleukin-6 in the human adrenal gland. Eur J Clin Invest. 2000;30(Suppl 3):91–95. doi: 10.1046/j.1365-2362.2000.0300s3091.x. [DOI] [PubMed] [Google Scholar]
- 234.Payne LC, Weigent DA, Blalock JE. Induction of pituitary sensitivity to interleukin-1: a new function for corticotropin-releasing hormone. Biochem Biophys Res Commun. 1994;198:480–484. doi: 10.1006/bbrc.1994.1070. [DOI] [PubMed] [Google Scholar]
- 235.Pearce BD, Biron CA, Miller AH. Neuroendocrine-immune interactions during viral infections. Adv Virus Res. 2001;56:469–513. doi: 10.1016/s0065-3527(01)56036-4. [DOI] [PubMed] [Google Scholar]
- 236.Perlstein RS, Whitnall MH, Abrams JS, et al. Synergistic roles of interleukin-6, interleukin-1, and tumor necrosis factor in the adrenocorticotropin response to bacterial lipopolysaccharide in vivo. Endocrinology. 1993;132:946–952. doi: 10.1210/endo.132.3.8382602. [DOI] [PubMed] [Google Scholar]
- 237.Petitto JM, Huang Z, Rinker CM, et al. Isolation of IL-2 receptor-beta cDNA clones from AtT-20 pituitary cells: constitutive expression and role in signal transduction. Neuropsychopharmacology. 1997;17:57–66. doi: 10.1016/S0893-133X(97)00003-1. [DOI] [PubMed] [Google Scholar]
- 238.Petrovsky N, Bucala R. Macrophage migration inhibitory factor (MIF). A critical neurohumoral mediator. Ann NY Acad Sci. 2000;917:665–671. doi: 10.1111/j.1749-6632.2000.tb05432.x. [DOI] [PubMed] [Google Scholar]
- 239.Pitossi F, del Rey A, Kabiersch A, et al. Induction of cytokine transcripts in the central nervous system and pituitary following peripheral administration of endotoxin to mice. J Neurosci Res. 1997;48:287–298. doi: 10.1002/(sici)1097-4547(19970515)48:4<287::aid-jnr1>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 240.Plotsky PM. Pathways to the secretion of adrenocorticotropin: a view from the portal. J Neuroendocrinol. 1991;3:1–9. doi: 10.1111/j.1365-2826.1991.tb00231.x. [DOI] [PubMed] [Google Scholar]
- 241.Pournajafi Nazarloo H, Takao T, Nanamiya W, et al. Effect of non-peptide corticotropin-releasing factor receptor type 1 antagonist on adrenocorticotropic hormone release and interleukin-1 receptors followed by stress. Brain Res. 2001;902:119–126. doi: 10.1016/s0006-8993(01)02383-6. [DOI] [PubMed] [Google Scholar]
- 242.Pozzoli G, Tringali G, Dello Russo C, et al. HIV-1 Gp120 protein modulates corticotropin releasing factor synthesis and release via the stimulation of its mRNA from the rat hypothalamus in vitro: involvement of inducible nitric oxide synthase. J Neuroimmunol. 2001;118:268–276. doi: 10.1016/s0165-5728(01)00351-4. [DOI] [PubMed] [Google Scholar]
- 243.Price P, Olver SD, Silich M, et al. Adrenalitis and the adrenocortical response of resistant and susceptible mice to acute murine cytomegalovirus infection. Eur J Clin Invest. 1996;26:811–819. doi: 10.1046/j.1365-2362.1996.2210562.x. [DOI] [PubMed] [Google Scholar]
- 244.Raber J, Sorg O, Horn TF, et al. Inflammatory cytokines: putative regulators of neuronal and neuroendocrine function. Brain Res Brain Res Rev. 1998;26:320–326. doi: 10.1016/s0165-0173(97)00041-6. [DOI] [PubMed] [Google Scholar]
- 245.Raber J, Toggas SM, Lee S, et al. Central nervous system expression of HIV-1 Gp120 activates the hypothalamic-pituitary-adrenal axis: evidence for involvement of NMDA receptors and nitric oxide synthase. Virology. 1996;226:362–373. doi: 10.1006/viro.1996.0664. [DOI] [PubMed] [Google Scholar]
- 246.Refaeli Y, Levy DN, Weiner DB. The glucocorticoid receptor type II complex is a target of the HIV-1 vpr gene product. Proc Natl Acad Sci USA. 1995;92:3621–3625. doi: 10.1073/pnas.92.8.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Rivest S, Lacroix S, Vallieres L, et al. How the blood talks to the brain parenchyma and the paraventricular nucleus of the hypothalamus during systemic inflammatory and infectious stimuli. Proc Soc Exp Biol Med. 2000;223:22–38. doi: 10.1046/j.1525-1373.2000.22304.x. [DOI] [PubMed] [Google Scholar]
- 248.Rivier C. Effect of peripheral and central cytokines on the hypothalamic-pituitary-adrenal axis of the rat. Ann NY Acad Sci. 1993;697:97–105. doi: 10.1111/j.1749-6632.1993.tb49926.x. [DOI] [PubMed] [Google Scholar]
- 249.Rogatsky I, Logan SK, Garabedian MJ. Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc Natl Acad Sci USA. 1998;95:2050–2055. doi: 10.1073/pnas.95.5.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Roh MS, Drazenovich KA, Barbose JJ, et al. Direct stimulation of the adrenal cortex by interleukin-1. Surgery. 1987;102:140–146. [PubMed] [Google Scholar]
- 251.Rook GA. Glucocorticoids and immune function. Baillieres Clin Endocrinol Metab. 1999;13:567–581. doi: 10.1053/beem.1999.0044. [DOI] [PubMed] [Google Scholar]
- 252.Roosth J, Pollard RB, Brown SL, et al. Cortisol stimulation by recombinant interferon-alpha 2. J Neuroimmunol. 1986;12:311–316. doi: 10.1016/0165-5728(86)90037-8. [DOI] [PubMed] [Google Scholar]
- 253.Ruzek MC, Miller AH, Opal SM, et al. Characterization of early cytokine responses and an interleukin (IL)-6–dependent pathway of endogenous glucocorticoid induction during murine cytomegalovirus infection. J Exp Med. 1997;185:1185–1192. doi: 10.1084/jem.185.7.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Ruzek MC, Pearce BD, Miller AH, et al. Endogenous glucocorticoids protect against cytokine-mediated lethality during viral infection. J Immunol. 1999;162:3527–3533. [PubMed] [Google Scholar]
- 255.Sacerdote P, Brini AT, Locatelli L, et al. Tumor necrosis factor alpha differentially regulates beta-endorphin concentrations and proopiomelanocortin RNA in the anterior and neurointermediate pituitary in vivo. Neuroimmunomodulation. 1994;1:357–360. doi: 10.1159/000097188. [DOI] [PubMed] [Google Scholar]
- 256.Sakic B, Laflamme N, Crnic LS, et al. Reduced corticotropin-releasing factor and enhanced vasopressin gene expression in brains of mice with autoimmunity-induced behavioral dysfunction. J Neuroimmunol. 1999;96:80–91. doi: 10.1016/s0165-5728(99)00021-1. [DOI] [PubMed] [Google Scholar]
- 257.Salas MA, Evans SW, Levell MJ, et al. Interleukin-6 and ACTH act synergistically to stimulate the release of corticosterone from adrenal gland cells. Clin Exp Immunol. 1990;79:470–473. doi: 10.1111/j.1365-2249.1990.tb08114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Salazar-Mather TP, Orange JS, Biron CA. Early murine cytomegalovirus (MCMV) infection induces liver natural killer (NK) cell inflammation and protection through macrophage inflammatory protein 1alpha (MIP-1alpha)–dependent pathways. J Exp Med. 1998;187:1–14. doi: 10.1084/jem.187.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Salomon R, Ruzek MC, Pearce BD, et al. Neuroendocrine and immune system interactions: glucocorticoids and protection against T-cell–dependent disease. Brain Behav Immun. 2002;16:212. [Google Scholar]
- 260.Saphier D. Neuroendocrine effects of interferon-alpha in the rat. Adv Exp Med Biol. 1995;373:209–218. doi: 10.1007/978-1-4615-1951-5_29. [DOI] [PubMed] [Google Scholar]
- 261.Saphier D, Welch JE, Chuluyan HE. Alpha-interferon inhibits adrenocortical secretion via mu 1–opioid receptors in the rat. Eur J Pharmacol. 1993;236:183–191. doi: 10.1016/0014-2999(93)90588-9. [DOI] [PubMed] [Google Scholar]
- 262.Sapolsky R, Rivier C, Yamamoto G, et al. Interleukin-1 stimulates the secretion of hypothalamic corticotropin-releasing factor. Science. 1987;238:522–524. doi: 10.1126/science.2821621. [DOI] [PubMed] [Google Scholar]
- 263.Sawada M, Itoh Y, Suzumura A, et al. Expression of cytokine receptors in cultured neuronal and glial cells. Neurosci Lett. 1993;160:131–134. doi: 10.1016/0304-3940(93)90396-3. [DOI] [PubMed] [Google Scholar]
- 264.Sawchenko PE, Brown ER, Chan RK, et al. The paraventricular nucleus of the hypothalamus and the functional neuroanatomy of visceromotor responses to stress. Prog Brain Res. 1996;107:201–222. doi: 10.1016/s0079-6123(08)61866-x. [DOI] [PubMed] [Google Scholar]
- 265.Schobitz B, De Kloet ER, Holsboer F. Gene expression and function of interleukin 1, interleukin 6 and tumor necrosis factor in the brain. Prog Neurobiol. 1994;44:397–432. doi: 10.1016/0301-0082(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 266.Schobitz B, de Kloet ER, Sutanto W, et al. Cellular localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Eur J Neurosci. 1993;5:1426–1435. doi: 10.1111/j.1460-9568.1993.tb00210.x. [DOI] [PubMed] [Google Scholar]
- 267.Schobitz B, Reul JM, Holsboer F. The role of the hypothalamic-pituitary-adrenocortical system during inflammatory conditions. Crit Rev Neurobiol. 1994;8:263–291. [PubMed] [Google Scholar]
- 268.Semenkov VF, Levina LV, Lavrov VF, et al. The influence of endogenous hypocorticism on the replication of influenza virus in the mouse lungs and the production of specific antibodies. Acta Virol. 1990;34:37–43. [PubMed] [Google Scholar]
- 269.Shanks N, Moore PM, Perks P, et al. Alterations in hypothalamic-pituitary-adrenal function correlated with the onset of murine SLE in MRL +/+ and lpr/lpr mice. Brain Behav Immun. 1999;13:348–360. doi: 10.1006/brbi.1998.0535. [DOI] [PubMed] [Google Scholar]
- 270.Sharp BM, Matta SG, Peterson PK, et al. Tumor necrosis factor-alpha is a potent ACTH secretagogue: comparison to interleukin-1 beta. Endocrinology. 1989;124:3131–3133. doi: 10.1210/endo-124-6-3131. [DOI] [PubMed] [Google Scholar]
- 271.Shearer GM, Clerici M, Sarin A, et al. Cytokines in immune regulation/pathogenesis in HIV infection. Ciba Found Symp. 1995;195:142–153. doi: 10.1002/9780470514849.ch10. [DOI] [PubMed] [Google Scholar]
- 272.Sherman MP, de Noronha CM, Pearce D, et al. Human immunodeficiency virus type 1 Vpr contains two leucine-rich helices that mediate glucocorticoid receptor coactivation independently of its effects on G(2) cell cycle arrest. J Virol. 2000;74:8159–8165. doi: 10.1128/jvi.74.17.8159-8165.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Silverman MN, Miller AH, Biron CA, et al. Characterization of an interleukin-6- and adrenocorticotropin-dependent, immune-to-adrenal pathway during viral infection. Endocrinology. 2004;145:3580–3589. doi: 10.1210/en.2003-1421. [DOI] [PubMed] [Google Scholar]
- 274.Silverman, M.N., B.D. Pearce, and A.H. Miller. 2003. Cytokines and HPA axis regulation, pp. 85–122. In: Z. Kronfol (ed.), Cytokines and Mental Health. Kluwer Adademic Publishers, Norwell, MA.
- 275.Smith EM, Blalock JE. Human lymphocyte production of corticotropin and endorphin-like substances: association with leukocyte interferon. Proc Natl Acad Sci USA. 1981;78:7530–7534. doi: 10.1073/pnas.78.12.7530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Smith EM, Cadet P, Stefano GB, et al. IL-10 as a mediator in the HPA axis and brain. J Neuroimmunol. 1999;100:140–148. doi: 10.1016/s0165-5728(99)00206-4. [DOI] [PubMed] [Google Scholar]
- 277.Smith EM, Meyer WJ, Blalock JE. Virus-induced corticosterone in hypophysectomized mice: a possible lymphoid adrenal axis. Science. 1982;218:1311–1312. doi: 10.1126/science.6183748. [DOI] [PubMed] [Google Scholar]
- 278.Smith GW, Aubry JM, Dellu F, et al. Corticotropin releasing factor receptor 1–deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–1102. doi: 10.1016/s0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- 279.Smith LR, Brown SL, Blalock JE. Interleukin-2 induction of ACTH secretion: presence of an interleukin-2 receptor alpha-chain-like molecule on pituitary cells. J Neuroimmunol. 1989;21:249–254. doi: 10.1016/0165-5728(89)90181-1. [DOI] [PubMed] [Google Scholar]
- 280.Smoak KA, Cidlowski JA. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev. 2004;125:697–706. doi: 10.1016/j.mad.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 281.Sousa AR, Lane SJ, Cidlowski JA, et al. Glucocorticoid resistance in asthma is associated with elevated in vivo expression of the glucocorticoid receptor beta-isoform. J Allergy Clin Immunol. 2000;105:943–950. doi: 10.1067/mai.2000.106486. [DOI] [PubMed] [Google Scholar]
- 282.Spangelo BL, Jarvis WD, Judd AM, et al. Induction of interleukin-6 release by interleukin-1 in rat anterior pituitary cells in vitro: evidence for an eicosanoid-dependent mechanism. Endocrinology. 1991;129:2886–2894. doi: 10.1210/endo-129-6-2886. [DOI] [PubMed] [Google Scholar]
- 283.Spangelo BL, Judd AM, Isakson PC, et al. Interleukin-1 stimulates interleukin-6 release from rat anterior pituitary cells in vitro. Endocrinology. 1991;128:2685–2692. doi: 10.1210/endo-128-6-2685. [DOI] [PubMed] [Google Scholar]
- 284.Spangelo BL, Judd AM, MacLeod RM, et al. Endotoxin-induced release of interleukin-6 from rat medial basal hypothalami. Endocrinology. 1990;127:1779–1785. doi: 10.1210/endo-127-4-1779. [DOI] [PubMed] [Google Scholar]
- 285.Spangelo BL, MacLeod RM, Isakson PC. Production of interleukin-6 by anterior pituitary cells in vitro. Endocrinology. 1990;126:582–586. doi: 10.1210/endo-126-1-582. [DOI] [PubMed] [Google Scholar]
- 286.Spath-Schwalbe E, Born J, Schrezenmeier H, et al. Interleukin-6 stimulates the hypothalamus-pituitary-adrenocortical axis in man. J Clin Endocrinol Metab. 1994;79:1212–1214. doi: 10.1210/jcem.79.4.7962296. [DOI] [PubMed] [Google Scholar]
- 287.Spinedi E, Hadid R, Daneva T, et al. Cytokines stimulate the CRH but not the vasopressin neuronal system: evidence for a median eminence site of interleukin-6 action. Neuroendocrinology. 1992;56:46–53. doi: 10.1159/000126207. [DOI] [PubMed] [Google Scholar]
- 288.Sugama S, Kim Y, Baker H, et al. Tissue-specific expression of rat IL-18 gene and response to adrenocorticotropic hormone treatment. J Immunol. 2000;165:6287–6292. doi: 10.4049/jimmunol.165.11.6287. [DOI] [PubMed] [Google Scholar]
- 289.Sundar SK, Cierpial MA, Kamaraju LS, et al. Human immunodeficiency virus glycoprotein (gp120) infused into rat brain induces interleukin 1 to elevate pituitary-adrenal activity and decrease peripheral cellular immune responses. Proc Natl Acad Sci USA. 1991;88:11246–11250. doi: 10.1073/pnas.88.24.11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Tada M, Diserens AC, Desbaillets I, et al. Analysis of cytokine receptor messenger RNA expression in human glioblastoma cells and normal astrocytes by reverse-transcription polymerase chain reaction. J Neurosurg. 1994;80:1063–1073. doi: 10.3171/jns.1994.80.6.1063. [DOI] [PubMed] [Google Scholar]
- 291.Takao T, Culp SG, De Souza EB. Reciprocal modulation of interleukin-1 beta (IL-1 beta) and IL-1 receptors by lipopolysaccharide (endotoxin) treatment in the mouse brain-endocrine-immune axis. Endocrinology. 1993;132:1497–1504. doi: 10.1210/endo.132.4.8462448. [DOI] [PubMed] [Google Scholar]
- 292.Takao T, Tojo C, Nishioka T, et al. Corticotropin-releasing factor treatment upregulates interleukin-1 receptors in the mouse pituitary: reversal by dexamethasone. Brain Res. 1995;688:219–222. doi: 10.1016/0006-8993(95)00561-4. [DOI] [PubMed] [Google Scholar]
- 293.Timpl P, Spanagel R, Sillaber I, et al. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor. Nat Genet. 1998;19:162–166. doi: 10.1038/520. [DOI] [PubMed] [Google Scholar]
- 294.Tominaga T, Fukata J, Naito Y, et al. Prostaglandin-dependent in vitro stimulation of adrenocortical steroidogenesis by interleukins. Endocrinology. 1991;128:526–531. doi: 10.1210/endo-128-1-526. [DOI] [PubMed] [Google Scholar]
- 295.Tominaga T, Fukata J, Fujimoto T, et al. Immunocytochemical evidence for interleukin-2 receptor regulation on rat anterior pituitary and adrenal cells. Acta Histochem Cytochem. 1996;29:255–260. [Google Scholar]
- 296.Tomlinson AH, Esiri MM. Herpes simplex encephalitis. Immunohistological demonstration of spread of virus via olfactory pathways in mice. J Neurol Sci. 1983;60:473–484. doi: 10.1016/0022-510x(83)90158-2. [DOI] [PubMed] [Google Scholar]
- 297.Toth IE. Transneuronal labeling of CNS neurons involved in the innervation of the adrenal gland. Horm Metab Res. 1998;30:329–333. doi: 10.1055/s-2007-978893. [DOI] [PubMed] [Google Scholar]
- 298.Toth IE, Vizi ES, Hinson JP, et al. Innervation of the adrenal cortex, its physiological relevance, with primary focus on the noradrenergic transmission. Microsc Res Tech. 1997;36:534–545. doi: 10.1002/(SICI)1097-0029(19970315)36:6<534::AID-JEMT10>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 299.Turnbull AV, Dow RC, Hopkins SJ, et al. Mechanisms of activation of the pituitary-adrenal axis by tissue injury in the rat. Psychoneuroendocrinology. 1994;19:165–178. doi: 10.1016/0306-4530(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 300.Turnbull AV, Rivier CL. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action. Physiol Rev. 1999;79:1–71. doi: 10.1152/physrev.1999.79.1.1. [DOI] [PubMed] [Google Scholar]
- 301.Turnbull AV, Smith GW, Lee S, et al. CRF type I receptor–deficient mice exhibit a pronounced pituitary-adrenal response to local inflammation. Endocrinology. 1999;140:1013–1017. doi: 10.1210/endo.140.2.6675. [DOI] [PubMed] [Google Scholar]
- 302.Turrin NP, Rivest S. Unraveling the molecular details involved in the intimate link between the immune and neuroendocrine systems. Exp Biol Med. 2004;229:996–1006. doi: 10.1177/153537020422901003. [DOI] [PubMed] [Google Scholar]
- 303.Utsuyama M, Hirokawa K. Differential expression of various cytokine receptors in the brain after stimulation with LPS in young and old mice. Exp Gerontol. 2002;37:411–420. doi: 10.1016/s0531-5565(01)00208-x. [DOI] [PubMed] [Google Scholar]
- 304.Vallieres L, Lacroix S, Rivest S. Influence of interleukin-6 on neural activity and transcription of the gene encoding corticotrophin-releasing factor in the rat brain: an effect depending upon the route of administration. Eur J Neurosci. 1997;9:1461–1472. doi: 10.1111/j.1460-9568.1997.tb01500.x. [DOI] [PubMed] [Google Scholar]
- 305.Vallieres L, Rivest S. Regulation of the genes encoding interleukin-6, its receptor, and gp130 in the rat brain in response to the immune activator lipopolysaccharide and the proinflammatory cytokine interleukin-1beta. J Neurochem. 1997;69:1668–1683. doi: 10.1046/j.1471-4159.1997.69041668.x. [DOI] [PubMed] [Google Scholar]
- 306.Vallieres L, Rivest S. Interleukin-6 is a needed proinflammatory cytokine in the prolonged neural activity and transcriptional activation of corticotropin-releasing factor during endotoxemia. Endocrinology. 1999;140:3890–3903. doi: 10.1210/endo.140.9.6983. [DOI] [PubMed] [Google Scholar]
- 307.Van Dam AM, Bauer J, Tilders FJ, et al. Endotoxin-induced appearance of immunoreactive interleukin-1 beta in ramified microglia in rat brain: a light and electron microscopic study. Neuroscience. 1995;65:815–826. doi: 10.1016/0306-4522(94)00549-k. [DOI] [PubMed] [Google Scholar]
- 308.Van Dam AM, De Vries HE, Kuiper J, et al. Interleukin-1 receptors on rat brain endothelial cells: a role in neuroimmune interaction? FASEB J. 1996;10:351–356. doi: 10.1096/fasebj.10.2.8641570. [DOI] [PubMed] [Google Scholar]
- 309.van der Meer MJ, Hermus AR, Pesman GJ, et al. Effects of cytokines on pituitary beta-endorphin and adrenal corticosterone release in vitro. Cytokine. 1996;8:238–247. doi: 10.1006/cyto.1996.0033. [DOI] [PubMed] [Google Scholar]
- 310.Van der Meer MJ, Sweep CG, Pesman GJ, et al. Synergism between IL-1 beta and TNF-alpha on the activity of the pituitary-adrenal axis and on food intake of rats. Am J Physiol. 1995;268:E551–557. doi: 10.1152/ajpendo.1995.268.4.E551. [DOI] [PubMed] [Google Scholar]
- 311.van der Meer MJ, Sweep CG, Rijnkels CE, et al. Acute stimulation of the hypothalamic-pituitary-adrenal axis by IL-1 beta, TNF alpha and IL-6: a dose response study. J Endocrinol Invest. 1996;19:175–182. doi: 10.1007/BF03349862. [DOI] [PubMed] [Google Scholar]
- 312.Vankelecom H, Andries M, Billiau A, et al. Evidence that folliculo-stellate cells mediate the inhibitory effect of interferon-gamma on hormone secretion in rat anterior pituitary cell cultures. Endocrinology. 1992;130:3537–3546. doi: 10.1210/endo.130.6.1317788. [DOI] [PubMed] [Google Scholar]
- 313.Vankelecom H, Carmeliet P, Van Damme J, et al. Production of interleukin-6 by folliculo-stellate cells of the anterior pituitary gland in a histiotypic cell aggregate culture system. Neuroendocrinology. 1989;49:102–106. doi: 10.1159/000125097. [DOI] [PubMed] [Google Scholar]
- 314.Vankelecom H, Matthys P, Denef C. Inducible nitric oxide synthase in the anterior pituitary gland: induction by interferon-gamma in a subpopulation of folliculostellate cells and in an unidentifiable population of non-hormone-secreting cells. J Histochem Cytochem. 1997;45:847–857. doi: 10.1177/002215549704500609. [DOI] [PubMed] [Google Scholar]
- 315.Venihaki M, Dikkes P, Carrigan A, et al. Corticotropin-releasing hormone regulates IL-6 expression during inflammation. J Clin Invest. 2001;108:1159–1166. doi: 10.1172/JCI12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Verges B, Chavanet P, Desgres J, et al. Adrenal function in HIV-infected patients. Acta Endocrinol. 1989;121:633–637. doi: 10.1530/acta.0.1210633. [DOI] [PubMed] [Google Scholar]
- 317.Vermes I, Beishuizen A, Hampsink RM, et al. Dissociation of plasma adrenocorticotropin and cortisol levels in critically ill patients: possible role of endothelin and atrial natriuretic hormone. J Clin Endocrinol Metab. 1995;80:1238–1242. doi: 10.1210/jcem.80.4.7714094. [DOI] [PubMed] [Google Scholar]
- 318.Wang X, Wu H, Miller AH. Interleukin 1alpha (IL-1alpha) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Mol Psychiatry. 2004;9:65–75. doi: 10.1038/sj.mp.4001339. [DOI] [PubMed] [Google Scholar]
- 319.Wang X, Wu H, Lakdawala VS, et al. Inhibition of Jun N-terminal kinase (JNK) enhances glucocorticoid receptor-mediated function in mouse hippocampal HT22 cells. Neuropsychopharmacology. 2005;30:242–249. doi: 10.1038/sj.npp.1300606. [DOI] [PubMed] [Google Scholar]
- 320.Watanabe D, Yoshimura R, Khalil M, et al. Characteristic localization of gp130 (the signal-transducing receptor component used in common for IL-6/IL-11/CNTF/LIF/OSM) in the rat brain. Eur J Neurosci. 1996;8:1630–1640. doi: 10.1111/j.1460-9568.1996.tb01307.x. [DOI] [PubMed] [Google Scholar]
- 321.Watanobe H, Takebe K. Intravenous administration of tumor necrosis factor-alpha stimulates corticotropin releasing hormone secretion in the push-pull cannulated median eminence of freely moving rats. Neuropeptides. 1992;22:81–84. doi: 10.1016/0143-4179(92)90058-5. [DOI] [PubMed] [Google Scholar]
- 322.Watkins LR, Maier SF, Goehler LE. Cytokine-to-brain communication: a review & analysis of alternative mechanisms. Life Sci. 1995;57:1011–1026. doi: 10.1016/0024-3205(95)02047-m. [DOI] [PubMed] [Google Scholar]
- 323.Weber MM, Michl P, Auernhammer CJ, et al. Interleukin-3 and interleukin-6 stimulate cortisol secretion from adult human adrenocortical cells. Endocrinology. 1997;138:2207–2210. doi: 10.1210/endo.138.5.5239. [DOI] [PubMed] [Google Scholar]
- 324.Webster EL, Tracey DE, De Souza EB. Upregulation of interleukin-1 receptors in mouse AtT-20 pituitary tumor cells following treatment with corticotropin-releasing factor. Endocrinology. 1991;129:2796–2798. doi: 10.1210/endo-129-5-2796. [DOI] [PubMed] [Google Scholar]
- 325.Webster JC, Oakley RH, Jewell CM, et al. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci USA. 2001;98:6865–6870. doi: 10.1073/pnas.121455098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Webster JC, Cidlowski JA. Mechanisms of glucocorticoid-receptor–mediated repression of gene transcription. Trends Endocrinol Metab. 1999;10:396–402. doi: 10.1016/s1043-2760(99)00186-1. [DOI] [PubMed] [Google Scholar]
- 327.Weigent DA, Blalock JE. Associations between the neuroendocrine and immune systems. J Leukoc Biol. 1995;58:137–150. doi: 10.1002/jlb.58.2.137. [DOI] [PubMed] [Google Scholar]
- 328.Whiteside MB, Quan N, Herkenharn M. Induction of pituitary cytokine transcripts by peripheral lipopolysaccharide. J Neuroendocrinol. 1999;11:115–120. doi: 10.1046/j.1365-2826.1999.00297.x. [DOI] [PubMed] [Google Scholar]
- 329.Whitworth EJ, Kosti O, Renshaw D, et al. Adrenal neuropeptides: regulation and interaction with ACTH and other adrenal regulators. Microsc Res Tech. 2003;61:259–267. doi: 10.1002/jemt.10335. [DOI] [PubMed] [Google Scholar]
- 330.Willenberg HS, Bornstein SR, Hiroi N, et al. Effects of a novel corticotropin-releasing-hormone receptor type I antagonist on human adrenal function. Mol Psychiatry. 2000;5:137–141. doi: 10.1038/sj.mp.4000720. [DOI] [PubMed] [Google Scholar]
- 331.Winter JS, Gow KW, Perry YS, et al. A stimulatory effect of interleukin-1 on adrenocortical cortisol secretion mediated by prostaglandins. Endocrinology. 1990;127:1904–1909. doi: 10.1210/endo-127-4-1904. [DOI] [PubMed] [Google Scholar]
- 332.Witzke O, Winterhagen T, Kribben A, et al. Interleukin-2 given to asymptomatic HIV-infected individuals leads to an exaggerated response of the pituitary gland to the action of CRH. Clin Endocrinol. 2003;59:104–109. doi: 10.1046/j.1365-2265.2003.01803.x. [DOI] [PubMed] [Google Scholar]
- 333.Woloski BM, Smith EM, Meyer WJ, et al. Corticotropin-releasing activity of monokines. Science. 1985;230:1035–1037. doi: 10.1126/science.2997929. [DOI] [PubMed] [Google Scholar]
- 334.Wolvers DA, Marquette C, Berkenbosch F, et al. Tumor necrosis factor–alpha: specific binding sites in rodent brain and pituitary gland. Eur Cytokine Netw. 1993;4:377–381. [PubMed] [Google Scholar]
- 335.Wong ML, Bongiorno PB, al-Shekhlee A, et al. IL-1 beta, IL-1 receptor type I and iNOS gene expression in rat brain vasculature and perivascular areas. Neuroreport. 1996;7:2445–2448. doi: 10.1097/00001756-199611040-00008. [DOI] [PubMed] [Google Scholar]
- 336.Wong ML, Bongiorno PB, Rettori V, et al. Interleukin (IL) 1beta, IL-1 receptor antagonist, IL-10, and IL-13 gene expression in the central nervous system and anterior pituitary during systemic inflammation: pathophysiological implications. Proc Natl Acad Sci USA. 1997;94:227–232. doi: 10.1073/pnas.94.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Wright, K.E. 1989. Arenaviruses, pp. 235–251. In: S. Specter, M. Bendinelli, and H. Friedman (eds.), Virus-Induced Immunosuppression. Plenum, New York.
- 338.Yamada T, Horisberger MA, Kawaguchi N, et al. Immunohistochemistry using antibodies to alpha-interferon and its induced protein, MxA, in Alzheimer’s and Parkinson’s disease brain tissues. Neurosci Lett. 1994;181:61–64. doi: 10.1016/0304-3940(94)90560-6. [DOI] [PubMed] [Google Scholar]
- 339.Yamada T, Yamanaka I. Microglial localization of alpha-interferon receptor in human brain tissues. Neurosci Lett. 1995;189:73–76. doi: 10.1016/0304-3940(95)11452-3. [DOI] [PubMed] [Google Scholar]
- 340.Yang Y, Han SH, Kim H, et al. Interleukin-12 p40 gene expression is induced in lipopolysaccharide-activated pituitary glands in vivo. Neuroendocrinology. 2002;75:347–357. doi: 10.1159/000059431. [DOI] [PubMed] [Google Scholar]
- 341.You-Ten KE, Itie A, Seemayer TA, et al. Increased expression of proopiomelanocortin (POMC) mRNA in adrenal glands of mice undergoing graft-versus-host disease (GVHD): association with persistent elevated plasma corticosterone levels. Clin Exp Immunol. 1995;102:596–602. doi: 10.1111/j.1365-2249.1995.tb03858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Zalcman S, Green-Johnson JM, Murray L, et al. Cytokine-specific central monoamine alterations induced by interleukin-1, -2 and -6. Brain Res. 1994;643:40–49. doi: 10.1016/0006-8993(94)90006-x. [DOI] [PubMed] [Google Scholar]