

Abstract
Studies are presented characterizing platelet CDCrel-1, a protein expressed to high levels by megakaryocytes and belonging to a family of conserved proteins, termed septin. Septin filaments originally were identified in yeast as essential for budding but have become increasingly associated with processes in higher eukaryotic cells involving active membrane movement such as cytokinesis and vesicle trafficking. Direct proof of an in vivo function for septins in higher eukaryotes is limited to the characterization of the Drosophila septin, termed PNUT. We present studies identifying platelet CDCrel-1 as a protein kinase substrate in the presence of known platelet agonists. The immunopurification of CDCrel-1 revealed it to be part of a macromolecular complex containing a protein involved in platelet secretion, syntaxin 4. Moreover, CDCrel-1 was localized in situ to areas surrounding platelet-storage granules. The relevance of CDCrel-1 to normal platelet function was established with the characterization of platelets from a CDCrel-1Null mouse. As compared with platelets from wild-type littermates, CDCrel-1Null platelets aggregate and release stored [14C]serotonin in the presence of subthreshold levels of collagen. These results provide new insights into the mechanisms regulating platelet secretion and identify platelet septins as a protein family contributing to membrane trafficking within the megakaryocyte and platelet.
Septins are a family of proteins originally identified in yeast as essential for budding (1). For years, the presence of septins in higher eukaryotic cells was dismissed, because budding, or asymmetric cell division, is a process unique to yeast (2). However, recent studies have demonstrated that septins do exist in higher eukaryotes and in those cases examined are commonly associated with events in which dynamic membrane movement or cytoplasmic partitioning such as in cytokinesis or vesicle trafficking occurs (3). In characterizing transcripts from the human chromosome 22q11.2 locus, we identified and characterized a human septin gene, termed CDCrel-1 (cell division control-related-1), located immediately 5′ to the gene encoding the β-subunit of the platelet glycoprotein Ib receptor (4). The nomenclature for CDCrel-1 was chosen because the conceptual translation product displayed a striking similarity to a Drosophila septin termed PNUT, which is essential for cytokinesis during embryo growth (5). However, this choice would prove later to be less than perfect, because we observed the highest levels of CDCrel-1 mRNA in organs and cells no longer undergoing active cell division, namely brain, heart, and megakaryocytes (6). CDCrel-1 also has been referred to as PNUT-like based on its similarity to the well characterized Drosophila septin (7, 8).
Recent reports have provided insights into a possible function for CDCrel-1 in neurons. Scheller and coworkers (9) purified a protein(s) with the CDCrel-1 sequence as part of the brain sec6–sec8 complex thought to establish polarity in neurons and determine where synapses form. Shortly thereafter, Caltagarone et al. (10) cofractionated CDCrel-1 with SNAP-25-labeled membranes and synaptophysin-marked synaptic vesicles from neurons. The linkage to neurotransmitter release became stronger when Beites et al. (11) characterized dominant-negative mutations in CDCrel-1 that inhibited exocytosis in transfected cell lines. However, a CDCrel-1Null mouse has been generated, and a neuron defect has been difficult to document presumably because of the presence of multiple brain septins and the potential for functional redundancy (12).
There is a growing appreciation for molecular similarities between platelet secretion and the mechanisms that control neurotransmitter release (13, 14). Although the mechanisms controlling neurotransmitter release have been studied intensively, the molecular basis of platelet secretion remains poorly characterized (15, 16). In this report, we characterize platelet CDCrel-1 and demonstrate CDCrel-1 is a substrate for phosphorylation in response to platelet agonists. Immunopurification of platelet CDCrel-1 results in copurification of syntaxin 4, which is consistent with a role for CDCrel-1 in exocytosis and with our localization of CDCrel-1 to the area surrounding platelet-storage granules. Moreover, we present data using platelets from CDCrel-1Null mice demonstrating a role for CDCrel-1 in platelet secretion and aggregation.
Material and Methods
Western Blotting and Immunological Reagents.
Platelets were isolated from human blood by using centrifugation to generate platelet-rich plasma (PRP). Briefly, the blood was centrifuged at 700 × g for 5 min, and PRP was removed from the supernatant. The platelets then were pelleted by centrifugation at 1,800 × g and lysed at 4°C in a 50% modified Hepes Tyrode's buffer (0.1 M Hepes/1.4 M NaCl/27 mM KCl/4 mM NaH2PO4/0.1 M NaHCO3/dextrose, pH 7.4) and 50% solubilization buffer at pH 7.4 [2% Triton X-100/0.1 M Tris base/0.01 M EGTA/0.15 M NaCl/leupeptin (20 μM)/pepstatin (10 μM)/Pefabloc (2 mM)] (17). SDS/PAGE was performed after reduction and denaturation of samples. After electrophoresis the samples were transferred to nitrocellulose. The anti-CDCrel-1 monoclonal antibody LJ-33 has been described previously (8). The anti-syntaxin 4 monoclonal antibody (catalog no. S40220) was purchased from Transduction Laboratories (Lexington, KY). Protein molecular weight standards were MultiMark multicolored proteins from Invitrogen.
Phosphorylation of CDCrel-1.
Phosphorylation in permeabilized platelets was performed essentially as described by Reed et al. (18). Blood from healthy volunteers was drawn into acid–citrate–dextrose supplemented with 10 μM prostaglandin E1 (Sigma) and centrifuged at 700 × g for 5 min. Platelets were washed three times in 140 mM NaCl/2.7 mM KCl/0.47 mM NaH2PO4/20 mM Hepes/2.77 mM dextrose (pH 6.5) and resuspended at 1.4 × 109 cells per ml in 20 mM Pipes [piperazine N,N′-bis(ethanesulfonic acid)]/150 mM glutamate/7 mM magnesium diacetate/5 mM dextrose/5 mM EGTA (pH 7.4). Platelets then were incubated with or without 1 μM of the protein kinase inhibitor Ro-31-8220 (3-{1-[3-(amidinothio)propyl]-1H-indol-3-yl}-3-(1-methyl-1H-indol-3-yl)maleimide methanesulfonate (Calbiochem)). The platelet suspension (100 μl) was mixed with 1 μl of Na2ATP (500 mM, Sigma), 3 μl of [γ-32P]ATP [6,000 Ci/mmol (1 Ci = 37 GBq), Perkin–Elmer], 2 μl of 20 mM CaCl2, and 29 μl of 100 μg/ml saponin (Eastman Kodak) prewarmed at 30°C. Platelets (final volume of 140 μl) then were incubated with 5 μl of one of the following agonists: 28 units/ml thrombin (Sigma), 28 μM phorbol 12-myristate 13-acetate (Sigma), or 840 μg/ml bovine fibrillar collagen (type I) prepared as described (19). The no-agonist controls received 5 μl of 280 μM prostaglandin E1. Platelets suspensions were incubated in the presence of agonists for 5 min at 30°C. The reaction was stopped by adding 1 volume of lysis buffer (1% SDS/5 mM NaF/50 mM sodium orthovanadate/2 mM EDTA/1 mM Pefabloc/2 mM DTT/0.2 mg/ml leupeptin/100 units/ml aprotinin). Samples were boiled for 3 min and cooled on ice. Two milliliters of radioimmunoprecipitation buffer [1.25% (wt/wt) Nonidet P-40/1.25% sodium deoxycholate (wt/vol)/2 mM EDTA/12.5 mM NaPO4/0.2 mM sodium orthovanadate/50 mM NaF/100 units/ml aprotinin (pH 7.2)] was added to the mixture, and samples were centrifuged at 10,000 × g for 15 min. Denatured supernatants were incubated with LJ-33 coupled to agarose-immobilized rProtein A (Repligen) (2 h at 22°C). Immobilized protein A was washed extensively with radioimmunoprecipitation buffer, mixed with SDS/PAGE loading buffer containing β-mercaptoethanol, boiled for 5 min, and centrifuged for 2 min at 10,000 × g. Phosphorylated proteins were identified after electrophoresis in SDS/PAGE (10%) and autoradiography.
Immunopurification of CDCrel-1.
A rabbit polyclonal antibody was prepared from injections of a bacterially expressed amino-terminal fragment of human CDCrel-1 (8). The antibody was purified with three 50% saturated ammonium sulfate cuts, dialyzed against Hepes-buffered saline (pH 7.4), and purified with protein A-Sepharose. The purified antibody (1.6 mg/ml) was coupled to AminoLink Plus beads (pH 10.0, Pierce) by using 4 ml of purified antibody.
PRP was prepared from 60 ml of blood. The platelets were washed in modified Hepes/Tyrode's buffer (pH 7.4) in the presence of prostaglandin E1 as described above. Platelets were lysed in solubilization buffer and applied to the anti-CDCrel-1 immunoaffinity column in the presence of PBS. The column was washed, and the antigen was eluted in 0.1 M glycine (pH 2.5). The sample was neutralized immediately with 1 M Tris (pH 9.5) according to the protocol supplied with AminoLink Plus beads. After elution the proteins were concentrated by using Aquacide (Calbiochem) and dialyzed against Hepes-buffered saline (pH 7.4). The samples then were subjected to electrophoresis, Coomassie staining, or Western transfer.
Electron Microscopy and Immunogold Labeling.
The preparation of washed platelets and the basic electron microscopy procedure are similar to those described (20). For immunogold labeling, washed human platelet samples were resuspended in Tyrode's buffer postfixed in 1.25% (vol/vol) glutaraldehyde for 1 h at room temperature and, after washing, infused with 2.3 M sucrose before being frozen in propane and then in liquid nitrogen. Ultrathin sections of ≈70 nm were cut at −120°C and placed on collodion-coated nickel grids. The grids were incubated for 10 min on drops of washing buffer consisting of PBS supplemented with 0.5% albumin and then for 15 min on drops of PBS supplemented with FCS. A 1-h incubation with the purified anti-CDCrel-1 polyclonal antibody (10 μg/ml) was followed by two similar incubations to amplify the immunoreactive signal. The samples were treated with FITC-conjugated F(ab′)2 fragments of sheep anti-rabbit IgG and then a rabbit anti-FITC-purified Ig (Dako). Finally, the grids were incubated with gold-labeled anti-rabbit IgG (AuroProbe EM, Amersham Pharmacia) and rinsed abundantly. The cryosections were stained by uranyl acetate. After washing, the sections were embedded in a thin film of methylcellulose before observation with a Jeol JEM-1010 transmission electron microscope at 80 kV.
CDCrel-1-Deficient Mice.
The generation and characterization of CDCrel-1Null mice have been described (12). Offspring from heterozygous crosses were genotyped by Southern blotting, and littermates were matched before blood drawing for each assay.
Platelet Aggregation Assays.
Blood was drawn from wild-type, CDCrel-1Het, and CDCrel-1Null littermates from a retroorbital sinus by using heparin-coated capillaries (Drummond Scientific, Broomall, PA) cut to 1 cm in length. Up to 1 ml of blood was collected in Eppendorf tubes containing 200 units of heparin. Blood from 4–5-week-old littermates with the same genotype was pooled. The volumes and platelet counts were measured, and an equal volume of 20 mM Hepes/0.15 M NaCl (pH 7.4) was added to normalize platelet counts. Mouse PRP (mPRP) was removed after centrifugation at 450 × g for 2 min. Platelets were counted in a Baker cell counter. Mouse platelet-poor plasma was prepared by spinning the remaining red cells and diluted plasma at 1,800× g for 5 min. Stirred aggregation profiles were generated in a Chrono-Log (Havertown, PA) aggregometer using 3.5 × 105 platelets per μl. The thromboxane agonist, U46619 (Sigma), was dried under a stream of nitrogen and resuspended to 10 mM in 10% ethanol/90% sodium carbonate (0.2 mg/ml). Before use in platelet studies the U46619 was diluted 10-fold in 0.15 M NaCl and added to the PRP to give the indicated final concentration.
[14C]Serotonin Release Assays.
mPRP was incubated with [14C]serotonin (5-hydroxy[side chain-2-14C]tryptamine with creatinine sulfate; Amersham Pharmacia). [14C]Serotonin (2 μl) was added per ml of PRP. After a 30-min incubation at 37°C, uptake was blocked by adding 1 μl of 1 mM imipramine (Sigma)/ml of PRP. Pilot studies demonstrated an 88% uptake of labeled serotonin for wild-type, CDCrel-1Het, and CDCrel-1Null mice. mPRP (350 μl) was prewarmed at 37°C, stirred at 1,000 rpm in an aggregometer (Chrono-Log), and mixed with 50 μl of agonist, and the aggregation tracing was recorded for 3 min. Then, 1 μl of indomethacin (10 mM in ethanol) was added, and 150 μl of the mPRP sample was fixed with an equal volume of 2.5% formaldehyde. The sample then was spun at 10,000 × g for 2 min, and a 50-μl aliquot of the supernatant was transferred to a scintillation vial for counting. The results of Table 1 are presented as a percent release of the total serotonin uptake calculated according to the following equation: cpm sample − cpm mouse platelet-poor plasma/cpm mPRP − cpm mouse platelet-poor plasma × 100 = percentage of [14C]serotonin release
Table 1.
Percentage of [14C]serotonin release during agonist stimulation
| Collagen, μg/ml
|
U46619,
μM
|
ADP, μM
|
||||||
|---|---|---|---|---|---|---|---|---|
| 1.25 | 2.5 | 5 | 2.5 | 7.5 | 15 | 1 | 10 | |
| Wild type | 2.8 | 11.7 | 27.7 | 1.9 | 11.7 | 14.9 | 2.9 | 2.0 |
| (0.8) | (2.4) | (3.8) | (0.8) | (1.1) | (1.4) | (2.2) | (0.9) | |
| CDCrel-1Null | 8.3 | 23.2 | 37.3 | 5.1 | 17.6 | 26.3 | 1.2 | 2.0 |
| (1.9) | (1.4) | (0.7) | (0.5) | (1.5) | (2.4) | (0.7) | (1.1) | |
Standard error of the mean is given in parentheses, n = 5.
Results
Platelet CDCrel-1.
The generation and characterization of anti-CDCrel-1 monoclonal antibody LJ-33 has been described (8). In human platelet lysates, this antibody identifies a 45-kDa immunoreactive species (Fig. 1). However, LJ-33 does not recognize native or nondenatured CDCrel-1, presumably reflecting the use of a bacterially expressed fragment for immunization (data not shown). Therefore, rabbit antiserum also was generated by using the same recombinant fragment. This serum contains an IgG capable of immunopurifying CDCrel-1 from a lysate of purified human platelets. Shown in Fig. 2 Left is the Coomassie-stained gel of immunopurified platelet CDCrel-1. Again, a 45-kDa species is the most prevalent protein with several minor bands of both slower and faster mobility. The identification of the major Coomassie-staining band as CDCrel-1 was confirmed by probing the purified samples with anti-CDCrel-1 monoclonal antibody LJ-33 (Fig. 2 Middle).
Figure 1.

(A) Mammalian septins range from 350–500 aa in length and contain a conserved central core domain of ≈300 residues (3, 27, 34). Within the core domain is a polybasic sequence proposed to interact with phosphatidylinositol 4,5-bisphosphate and sequences comprising a P-loop motif characteristic of GTP/ATP-binding proteins (26, 27). (B) A local sequence alignment profile is shown for a number of septin proteins within their polybasic motif. A serine residue (asterisk) flanked by residues fitting the consensus sequence for phosphorylation by protein kinase C is highlighted (35). Phosphorylation by protein kinase C has been documented in the septin Nedd5 but is lacking in G-septin (21). The G-septin sequence diverges from the protein kinase C substrate recognition motif. (C) Shown is the autoradiograph from a Western blot of human platelet lysate treated with monoclonal antibody LJ-33 prepared against a recombinant fragment of CDCrel-1. LJ-33 recognizes a single species with a mobility of 45 kDa.
Figure 2.
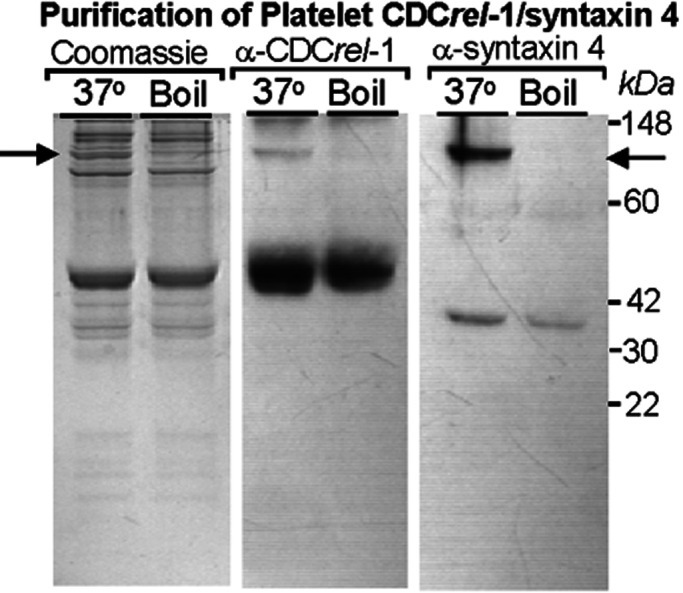
An immunoaffinity column was prepared by using a purified rabbit serum generated against CDCrel-1 antigen. Approximately 0.5 mg of protein was purified from 1 × 1010 platelets. The immunopurified material (80 μg) was electrophoresed through a 12% SDS/PAGE and stained with Coomassie blue (Left). The predominant species migrates at 45 kDa and is recognized by monoclonal antibody LJ-33 (Middle). A number of fainter staining bands migrating in the 30–40-kDa range were obtained also along with a number of higher molecular weight species ranging from ≈70 to 100 kDa. A previous study suggested an interaction between CDCrel-1 and syntaxins (11). Thus, we investigated whether any of the copurified proteins could be platelet syntaxin 4. Indeed, Western analysis confirmed the presence of syntaxin 4 (35 kDa) and demonstrated its presence in a higher molecular weight SDS-resistant, heat-sensitive complex (see arrow). The anti-CDCrel-1 monoclonal antibody also recognizes a heat-sensitive complex with a similar migration to the complex containing syntaxin 4.
A number of copurifying protein species were visible in the Coomassie-stained gel of immunopurified CDCrel-1 (Fig. 2 Left). We also observed that one of the slower migrating species reacted with LJ-33 and was present in an SDS-resistant, heat-sensitive complex, because boiling of the samples abolished the slower migrating immunoreactive species (Fig. 2 Middle). A previous report had established that brain CDCrel-1 interacts with syntaxins (11), a critical protein of the SNAP-receptor complex. Moreover, Flaumenhaft et al. (13) reported that platelet syntaxin 4 exists in an SDS-resistant, heat-sensitive macromolecular complex similar to that immunopurified by LJ-33 in Fig. 2 Middle. Thus, we probed our immunopurified preparation of CDCrel-1 for the presence of syntaxin 4. Indeed, as shown in Fig. 2 Right, immunoreactive syntaxin 4 is seen migrating at ≈35 kDa and also is present in an SDS-resistant, heat-sensitive complex with a mobility that is indistinguishable from the macromolecular complex containing CDCrel-1. Thus, syntaxin 4 was copurified by using the anti-CDCrel-1 antiserum, and both syntaxin 4 and CDCrel-1 exist in a heat-sensitive macromolecular complex with a similar mobility when analyzed by SDS/PAGE.
In Situ Localization of Platelet CDCrel-1.
The anti-human CDCrel-1 polyclonal serum was used to investigate the localization of CDCrel-1 within the ultrastructure of the platelet. As shown in Fig. 3, immunogold labeling of CDCrel-1 displayed a marked preference for areas surrounding α-granules. Whether this localization included platelet-dense granules could not be determined, because the method for platelet analysis did not allow their morphological characterization. Controls were performed in the absence of primary antibody and showed no labeling (data not shown).
Figure 3.
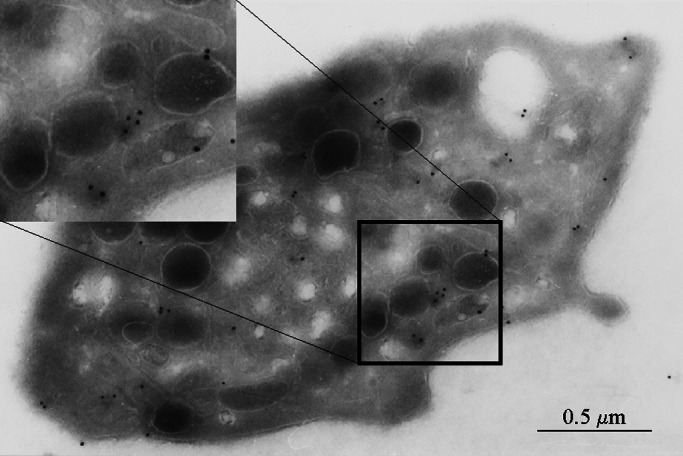
Shown is a photograph obtained from transmission electron microscopy of an unstimulated human platelet. The grids were reacted with an anti-CDCrel-1 polyclonal antibody, and the in situ localization of CDCrel-1 was identified by gold particles after an amplification of the primary signal (see Material and Methods).
Phosphorylation of Platelet CDCrel-1 in Response to Agonists.
We identified a conserved protein kinase C recognition site within the linear CDCrel-1 sequence (Fig. 1). A similar sequence is present in the human septin Nedd5, and this protein can undergo phosphorylation (21). The ability of CDCrel-1 to become phosphorylated in the presence of platelet agonists was investigated by incubating permeabilized platelets with [γ-32P]ATP. After permeabilization, stimulation, and labeling of platelets, the CDCrel-1 antigen was denatured slightly and immunopurified using LJ-33. The immunopurified products were analyzed by SDS/PAGE and autoradiography. Phosphorylation was observed in platelets stimulated with thrombin, phorbol 12-myristate 13-acetate, and collagen (Fig. 4). Moreover, phosphorylation of CDCrel-1 was blocked in the presence of the protein kinase inhibitor Ro-31-8220 (Fig. 4B). These results demonstrate the potential for functional regulation of CDCrel-1 in the presence of typical platelet agonists.
Figure 4.

Washed human platelets were incubated with saponin and [γ-32P]ATP to determine whether CDCrel-1 is phosphorylated in the presence of agonists. After treatment with agonists, platelets were solubilized, immunoprecipitated, and subjected to SDS/PAGE. Autoradiographic images are shown with the position of CDCrel-1 highlighted by the arrow. (A) Platelet samples are shown treated with 10 μM prostaglandin E1 (no agonist), 1 unit/ml thrombin, and 1 μM phorbol 12-myristate 13-acetate (PMA). (B) Phosphorylation of CDCrel-1 by a protein kinase was confirmed by blocking the labeling of CDCrel-1 with the protein kinase C inhibitor Ro-31-8220. Results using the agonists phorbol 12-myristate 13-acetate (1 μM) and collagen (30 μg/ml) are shown in the presence (+) and absence (−) of Ro-31-8220.
Characterization of Platelets from CDCrel-1Null Mice.
The generation of a CDCrel-1Null mouse has been described in a separate report (12). Briefly, the mouse lacks the ability to express CDCrel-1 protein because of the replacement of exons 4, 5, and 6 with a neomycin-resistance cassette. The resultant homozygous-deficient mice are viable with no gross abnormalities or apparent problems with viability. Studies to date have been unable to document any altered neurotransmitter release (12). However, given the expanding nature of the septin protein family, the apparent lack of an obvious neural phenotype may reflect functional redundancy among the family of brain-expressed septins (12). Indeed, several septins change their expression level in the absence of murine CDCrel-1 (12).
Platelet counts from CDCrel-1Null mice are in the normal range. There are no changes in platelet size as viewed by the forward-scatter profile produced by fluorescence-activated cell sorter analysis, nor were there any obvious changes to the platelet ultrastructure viewed by transmission electron microscopy (data not shown). Mice containing heterozygous CDCrel-1 alleles (CDCrel-1Het) were crossed and produced the expected three CDCrel-1 genotypes, wild-type (CDCrel-1WT), heterozygous (CDCrel-1Het), and homozygous-deficient (CDCrel-1Null). After the genotypic identification of pups produced from heterozygous crosses, we examined platelet aggregation in PRP prepared from littermates with a CDCrel-1WT or CDCrel-1Null genotype (Fig. 5). By using bovine insoluble fibrillar collagen (type I) as an agonist, we observed platelet aggregation with subthreshold levels of collagen in platelet preparations from the CDCrel-1Null mouse. Using agonists such as ADP that directly aggregate platelets and do not induce secretion, no difference was observed between CDCrel-1Null platelets and platelets from their wild-type littermates (Fig. 5). Moreover, the same results were obtained by comparing platelets from a CDCrel-1Null mouse to a panel of platelets obtained from generic wild-type mice including Balb/C, 129Sv/J, Black 6, and C57/SBJ (data not shown). Thus, the CDCrel-1Null platelets respond to subthreshold levels of collagen and support an enhanced aggregation with an agonist requiring a platelet secretory response.
Figure 5.

Blood was drawn from wild-type (WT) and CDCrel-1Null (KO) littermates. Blood from mice with the same genotype was pooled. Shown are platelet-aggregation profiles using insoluble fibrillar collagen or ADP at the indicated concentrations. These results are representative of aggregation experiments performed with mPRP obtained from pooled blood isolated from littermates of the same genotype (see Material and Methods).
To confirm these findings, we investigated the release of stored serotonin from mice containing wild-type and CDCrel-1Null alleles (Table 1). Here, the results mirrored those observed with platelet aggregation. The CDCrel-1Null platelet releases more stored serotonin by using subthreshold levels of collagen. Serotonin release from ADP-stimulated platelets was indistinguishable from background levels, reflecting the lack of ADP to stimulate platelet secretion although it can support platelet aggregation (Fig. 5). Additional serotonin-release assays were performed using the thromboxane A2 analogue U46619. In the presence of U46619, CDCrel-1Null platelets released more serotonin than platelets from CDCrel-1WT animals (Table 1), confirming that the CDCrel-1Null platelet has a hyperresponsive degranulation phenotype.
Discussion
Central to normal platelet function is the ability of platelets to secrete effector molecules from storage granules (14). The coordinated release of platelet-storage granule components is critical for the role of platelets in hemostasis and tissue repair at the site of vascular injury. Specifically, platelets contain three different types of storage granules, the unique α-granules, dense (or δ) granules, and lysosomal granules. The α-granules secrete soluble adhesion molecules involved in hemostasis, protease inhibitors to ensure growth of a fibrin clot, and growth factors to stimulate tissue repair. Dense granules secrete small molecule agonists such as ADP, calcium, and magnesium, each critical to the platelet activation response. The granular types and composition are similar for human and mouse platelets. One striking difference that is relevant to the present study is the ability of collagen to induce the release reaction in both species, whereas ADP causes a release in only human platelets (22). The physiologic relevance of platelet release is exemplified best in humans with congenital defects in release and the resulting bleeding phenotype (23). However, the molecular basis of most of these phenotypes is poorly understood, reflecting the lack of information on the multitude of events occurring during the platelet release.
Analogous to neurotransmitter release, platelet SNAP-receptor proteins have been described as bridging the granule membrane to the plasma membrane (14). Kinoshita et al. (24), using immunogold labeling of brain CDCrel-1, localized the protein to presynaptic vesicles in nerve terminals. Our localization of platelet CDCrel-1 to areas surrounding storage granules further supports some of the known functional redundancies between neurotransmitter release and the platelet response during hemostasis and thrombosis. Other proteins such as SNAP-23, syntaxin 2, and syntaxin 4 also have been associated with both neurotransmitter release and the platelet secretory response (13, 25). Although key proteins have been identified, the regulatory elements/factors that facilitate docking of the vesicle after platelet stimulation are the key mechanisms controlling platelet function and need to be defined (14). The current study suggests that platelet CDCrel-1 can regulate the platelet secretion response. This hypothesis is based on (i) the identity of platelet CDCrel-1 (Fig. 1), (ii) the demonstration of an interaction between CDCrel-1 and syntaxin 4 (Fig. 2), (iii) immunogold localization of CDCrel-1 to membranous areas surrounding α-granules (Fig. 3), (iv) phosphorylation of CDCrel-1 in response to platelets agonists (Fig. 4), and (v) characterization of a CDCrel-1 knockout mouse with an enhanced platelet secretion response (Fig. 5 and Table 1). Together these results support a hypothesis in which CDCrel-1 has an inhibitory role in the platelet release reaction.
An inhibitory role for CDCrel-1 in secretion was proposed originally by Beites et al. (11) based on their characterization of CDCrel-1 transfected into heterologous cells. In their study, transfection of HIT-T15 cells with wild-type CDCrel-1 inhibited secretion, whereas GTPase dominant-negative mutants enhanced secretion. They proposed that CDCrel-1 might regulate neurotransmitter release through interactions with syntaxin, because they were able to document a direct interaction between CDCrel-1 and several different syntaxin isoforms. However, unlike the platelet CDCrel-1Null phenotype, a brain phenotype in CDCrel-1Null mice has been difficult to document (12). Most likely, this result reflects some degree of functional redundancy among individual septins. Indeed, in the absence of CDCrel-1, the neural cells in the CDCrel-1Null mouse alter their expression of other septins (12). It is intriguing to speculate that the lack of brain phenotype with the presence of a platelet phenotype is caused in part by the inability of platelets to compensate or change their gene expression pattern. Thus, neurons are able to overcome the lack of CDCrel-1 because of their ability to alter levels of other brain septins and some potential functional redundancies. In addition, for all the known similarities between neurotransmitter release and platelet release, the presence of platelet phenotype may reflect an uncharacterized mechanistic difference between platelet secretion and neurotransmitter release (14).
The recognition of CDCrel-1 as a negative regulator of the platelet response is based on the interpretation of data obtained with a complete absence of CDCrel-1 in the null animal. It is conceivable this hypothesis is misleading, because it excludes the possibility for regulation or timing of vesicle release via a mechanism involving CDCrel-1. For example, CDCrel-1 could be both inhibitory and prosecretory depending on the functional state of the septin. Our observation that CDCrel-1 is phosphorylated in the presence of platelet agonists is supportive of a hypothesis that the functional state of CDCrel-1 can be regulated. In addition, a hallmark feature of septins is the presence of GTP and phosphatidylinositol 4,5-bisphosphate binding motifs in the central core domain (refs. 26 and 27; Fig. 1). Structure/function studies of these motifs may provide some novel insights into the mechanisms regulating the platelet exocytic machinery, and these will become the focus of future studies.
The generation of additional homozygous-deficient animals lacking other septin genes presents an excellent opportunity to determine the function of murine septins and address the potential for functional redundancy within this protein family. This is particularly important when considering organs such as brain, in which there seems to be a large repertoire of septin proteins expressed. To date, 10 different human septin genes have been identified with a common theme of alternative 5′ mRNA processing. Thus, the possibility for a large number of septin proteins and/or isoforms makes their characterization a somewhat daunting task. In preliminary studies we have observed expression of multiple platelet septins, but the more compelling question is whether each septin contributes to a unique molecular event. Yeast septins participate in aspects of cytoplasmic partitioning during cytokinesis or yeast sporulation (28–33). Both processes require some degree of cytoplasmic reorganization. Indeed, cytoplasmic reorganization is likely to be key to a number of events unique to megakaryocytes and platelets. The potential for septins to be involved in aspects of megakaryocyte biology where dynamic membrane movement occurs opens up the possibility for involvement in megakaryocyte maturation, platelet formation and release, or as shown in this study, the platelet secretion response in hemostasis. Thus, the characterization of CDCrel-1 as a prototypic platelet septin may represent a beginning for the unraveling of a number of key events in megakaryocyte and platelet biology.
Acknowledgments
We are grateful for the technical support of Mr. Hon Tran and Jim Roberts. This work was performed using funds from National Heart, Lung, and Blood Institute Grant HL50545 (to J.W.), the Canadian Institutes of Health Research (to W.S.T.), and the Associazione per la Lotta alla Trombosi (to M.C.). We acknowledge the Sam and Rose Stein Charitable Trust supporting the DNA core facility within the Department of Molecular and Experimental Medicine at The Scripps Research Institute.
Abbreviations
- CDCrel-1
cell division control-related-1
- PRP
platelet-rich plasma
- mPRP
mouse PRP
References
- 1.Hartwell L H, Culotti J, Pringle J R, Reid B J. Science. 1974;183:46–51. doi: 10.1126/science.183.4120.46. [DOI] [PubMed] [Google Scholar]
- 2.Chant J. Cell. 1996;84:187–190. doi: 10.1016/s0092-8674(00)80972-1. [DOI] [PubMed] [Google Scholar]
- 3.Kartmann B, Roth D. J Cell Sci. 2001;114:839–844. doi: 10.1242/jcs.114.5.839. [DOI] [PubMed] [Google Scholar]
- 4.Zieger B, Hashimoto Y, Ware J. J Clin Invest. 1997;99:520–525. doi: 10.1172/JCI119188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neufeld T P, Rubin G M. Cell. 1994;77:371–379. doi: 10.1016/0092-8674(94)90152-x. [DOI] [PubMed] [Google Scholar]
- 6.Yagi M, Zieger B, Roth G J, Ware J. Gene. 1998;212:229–236. doi: 10.1016/s0378-1119(98)00146-2. [DOI] [PubMed] [Google Scholar]
- 7.McKie J M, Sutherland H F, Harvey E, Kim U J, Scambler P J. Hum Genet. 1997;101:6–12. doi: 10.1007/s004390050576. [DOI] [PubMed] [Google Scholar]
- 8.Zieger B, Tran H, Hainmann I, Wunderle D, Zgaga-Griesz A, Blaser S, Ware J. Gene. 2000;261:197–203. doi: 10.1016/s0378-1119(00)00527-8. [DOI] [PubMed] [Google Scholar]
- 9.Hsu S C, Hazuka C D, Roth R, Foletti D L, Heuser J, Scheller R H. Neuron. 1998;20:1111–1122. doi: 10.1016/s0896-6273(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 10.Caltagarone J, Rhodes J, Honer W G, Bowser R. NeuroReport. 1998;9:2907–2912. doi: 10.1097/00001756-199808240-00042. [DOI] [PubMed] [Google Scholar]
- 11.Beites C L, Xie H, Bowser R, Trimble W S. Nat Neurosci. 1999;2:434–439. doi: 10.1038/8100. [DOI] [PubMed] [Google Scholar]
- 12.Peng X-R, Jia Z, Ware J, Trimble W S. Mol Cell Biol. 2002;22:378–387. doi: 10.1128/MCB.22.1.378-387.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flaumenhaft R, Croce K, Chen E, Furie B, Furie B C. J Biol Chem. 1999;274:2492–2501. doi: 10.1074/jbc.274.4.2492. [DOI] [PubMed] [Google Scholar]
- 14.Reed G L, Fitzgerald M L, Polgár J. Blood. 2000;96:3334–3342. [PubMed] [Google Scholar]
- 15.Sudhof T C. Nature (London) 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 16.Jahn R, Sudhof T C. Annu Rev Biochem. 1999;68:863–911. doi: 10.1146/annurev.biochem.68.1.863. [DOI] [PubMed] [Google Scholar]
- 17.Gu M, Du X. J Biol Chem. 1998;273:33465–33471. doi: 10.1074/jbc.273.50.33465. [DOI] [PubMed] [Google Scholar]
- 18.Reed G L, Houng A K, Fitzgerald M L. Blood. 1999;93:2617–2626. [PubMed] [Google Scholar]
- 19.Folie B J, McIntire L V, Lasslo A. Blood. 1988;72:1393–1400. [PubMed] [Google Scholar]
- 20.Nurden P, Poujol C, Durrieu-Jais C, Winckler J, Combrie R, Macchi L, Bihour C, Wagner C, Jordan R, Nurden A T. Blood. 1999;93:1622–1633. [PubMed] [Google Scholar]
- 21.Xue J, Wang X, Malladi C S, Kinoshita M, Milburn P J, Lengyel I, Rostas J A P, Robinson P J. J Biol Chem. 2000;275:10047–10056. doi: 10.1074/jbc.275.14.10047. [DOI] [PubMed] [Google Scholar]
- 22.Tsakiris D A, Scudder L, Hodivala-Dilke K, Hynes R O, Coller B S. Thromb Haemostasis. 1999;81:177–188. [PubMed] [Google Scholar]
- 23.Rao A K. Am J Med Sci. 1998;316:69–76. doi: 10.1097/00000441-199808000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Kinoshita A, Noda M, Kinoshita M. J Comp Neurol. 2000;428:223–239. doi: 10.1002/1096-9861(20001211)428:2<223::aid-cne3>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 25.Chen D, Bernstein A M, Lemons P P, Whiteheart S W. Blood. 2000;95:921–929. [PubMed] [Google Scholar]
- 26.Zhang J, Kong C, Xie H, McPherson P S, Grinstein S, Trimble W S. Curr Biol. 1999;9:1458–1467. doi: 10.1016/s0960-9822(00)80115-3. [DOI] [PubMed] [Google Scholar]
- 27.Trimble W S. J Membr Biol. 1999;169:75–81. doi: 10.1007/s002329900519. [DOI] [PubMed] [Google Scholar]
- 28.Frazier J A, Wong M L, Longtin M S, Pringle J R, Mann M, Mitchison T J, Field C. J Cell Biol. 1998;143:737–749. doi: 10.1083/jcb.143.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooper J A, Kiehart D P. J Cell Biol. 1996;134:1345–1348. doi: 10.1083/jcb.134.6.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barral Y, Mermall V, Mooseker M S, Snyder M. Mol Cell. 2000;5:841–851. doi: 10.1016/s1097-2765(00)80324-x. [DOI] [PubMed] [Google Scholar]
- 31.Takizawa P A, DeRisi J L, Wilhelm J E, Vale R D. Science. 2000;290:341–344. doi: 10.1126/science.290.5490.341. [DOI] [PubMed] [Google Scholar]
- 32.Ozsarac N, Bhattacharyya M, Dawes I W, Clancy M J. Gene. 1995;164:157–162. doi: 10.1016/0378-1119(95)00438-c. [DOI] [PubMed] [Google Scholar]
- 33.De Virgilio C, DeMarini D J, Pringle J R. Microbiology. 1996;142:2897–2905. doi: 10.1099/13500872-142-10-2897. [DOI] [PubMed] [Google Scholar]
- 34.Longtine M S, DeMarini D J, Valencik M L, Al-Awar O S, Fares H, De Virgilio C, Pringle J R. Curr Opin Cell Biol. 1996;8:106–119. doi: 10.1016/s0955-0674(96)80054-8. [DOI] [PubMed] [Google Scholar]
- 35.Pinna L A, Ruzzene M. Biochim Biophys Acta. 1996;1314:191–225. doi: 10.1016/s0167-4889(96)00083-3. [DOI] [PubMed] [Google Scholar]