

Abstract
Background
Neutrophil apoptosis may play a critical role in the resolution of inflammation by stimulating anti-inflammatory cytokine generation from monocytes. In this study, we investigated the effect of apoptotic neutrophils on interleukin (IL)-10 and IL-1β production from monocytes in response to Porphyromonas gingivalis lipopolysaccharide.
Methods
Peripheral blood neutrophils from healthy individuals were isolated by sodium diatrizoate density gradient centrifugation. In order to induce apoptosis, neutrophils were cultured for 24 hours in modified Dulbecco’s medium supplemented with 10% autologous serum. Cell apoptosis was quantified by Annexin V positivity and loss of CD16 expression on the cell surface. Peripheral blood mononuclear cells were isolated from the same subjects; monocytes were purified by magnetic cell sorting and cultured with or without apoptotic or fresh neutrophils. Lipopolysaccharide from Porphyromonas gingivalis was used for cell stimulation. IL-1β and IL-10 levels in supernatants were determined by enzyme-linked immunosorbent assay (ELISA).
Results
IL-10 generation was significantly increased in monocytes cultured with apoptotic neutrophils compared to monocytes alone or cocultured with fresh neutrophils (P <0.05). IL-1β was suppressed both in resting and lipopolysaccharide-stimulated monocytes in the presence of apoptotic neutrophils compared to monocytes alone or monocytes cultured with fresh neutrophils at all time points (P <0.05).
Conclusion
Neutrophil apoptosis provides a signal to monocytes, changing the phenotype of the monocyte resulting in the production of anti-inflammatory cytokines and suppression of proinflammatory cytokines in response to lipopolysaccharide.
Keywords: Apoptosis, cytokines, interleukin-1, interleukin-10, lipopolysaccharides, monocytes, neutrophils, Porphyromonas gingivalis
The inflammatory response is a highly dynamic and complex process that is central to the development and progression of periodontitis. Bacterial products interact with gingival epithelium to induce the expression of adhesion molecules that leads to the production of proinflammatory cytokines and chemokines by neutrophils and monocytes/macrophages.1–5 However, the kinetics and interactions of neutrophils and other specialized effector cells, such as monocytes or macrophages, are poorly understood in relation to periodontal disease pathogenesis. Furthermore, little is known about how anti-inflammatory signals are generated from the endogenous reactions among these cells.3,6,7
Monocytes are activated and attracted to sites of inflammation by microbial products.8–10 Generation of tumor necrosis factor-alpha (TNF-α) followed by other proinflammatory cytokines, such as interleukin-1 beta (IL-1β), marks an important stage in the monocyte/macrophage-mediated inflammatory response.11 In addition to this proinflammatory role, monocytes/macrophages are also involved in the resolution of inflammation through the production of anti-inflammatory cytokines such as IL-10. IL-10 reduces the production of proinflammatory cytokines and regulates lipopolysaccharide (LPS) tolerance and phagocytic clearance of opsonized apoptotic cells.12–15 Macrophages also contribute to the resolution of inflammation by inducing neutrophil apoptosis.16 Thus, monocyte function and the balance between the pro-and anti-inflammatory cytokines are central to the effectiveness of the inflammatory response.5,8,9,17
Apoptosis, or programmed cell death, is a form of physiological cell death.18–20 It is increased in the presence of infection, inflammation, or tissue remodeling. Local specialized or non-professional cells rapidly remove apoptotic cells generated in these physiological or pathological conditions without provoking the release of proinflammatory cytokines.21,22 Recent reports indicate that these mechanisms have significant regulatory effects on inflammatory responses.23,24 It has been shown that apoptotic cells may play an important role in the resolution of inflammation by reducing the production of proinflammatory cytokines such as IL-1β and TNF-α and stimulating the generation of anti-inflammatory cytokines such as transforming growth factor-beta (TGF-β) and IL-10.22,25 Apoptotic leukocytes may alter cytokine production from LPS-activated monocytes increasing secretion of IL-10.26 The proinflammatory capacity of neutrophils is downregulated at the gene expression level during the induction of apoptosis.27 In chronic inflammatory diseases such as rheumatoid arthritis, the microenviroment of synovial fluid proinflammatory milieu is responsible for the local persistence of activated long-surviving neutrophils by delaying apoptosis.28
Previous studies have suggested that neutrophil apoptosis is involved in the pathogenesis of chronic periodontal diseases.29–33 The mechanism of neutrophil death and the control of the functional lifespan of neutrophils in the gingival crevice and periodontal pocket are important to understanding the pathogenesis of periodontal diseases.30 It has been shown that granulocyte-monocyte colony stimulating factor (GM-CSF) secreted during the inflammatory response into the gingival crevicular fluid (GCF) reduced apoptosis of neutrophils.34 Neutrophil apoptosis was delayed when neutrophils were incubated with LPS from Porphyromonas gingivalis (P. gingivalis),35,36 suggesting that the retardation of neutrophil apoptosis by P. gingivalis LPS may modulate the transition from acute to chronic inflammation in the periodontal tissues. Taken together, these data imply a principal mechanism through delayed apoptosis by which neutrophils can accumulate in periodontitis lesions, leading to both the acceleration of neutrophil-mediated tissue damage and the insufficiency of anti-inflammatory signals from monocytes. Therefore, the goal of this study was to investigate the effect of apoptotic neutrophils on the production of pro-and anti-inflammatory cytokines (IL-1β and IL-10) from monocytes in response to P. gingivalis LPS.
MATERIALS AND METHODS
Isolation of Human Neutrophils and Induction of Apoptosis
Peripheral blood samples were obtained from four healthy adult volunteers with no clinical or radiographical signs of periodontal disease (mean age: 31.25 ± 6.25 years). All samples were obtained after written consent and approval of the Institutional Review Board at Boston University Medical Center. None of the subjects were smokers. Neutrophils were isolated by discontinuous density gradient centrifugation using polysucrose and sodium diatrizoate.‡ 37 Briefly, peripheral blood (4.5 ml) was layered on the separating medium, and the tubes were centrifuged at 500 × g for 30 minutes. The neutrophil-rich layer was collected, and contaminating erythrocytes were lysed with 0.156 M hypotonic buffer containing ammonium chloride (NH4Cl). The cells were washed twice with phosphate-buffered saline (PBS) (1M PBS, pH = 7.4), resuspended in 50 ml PBS, and counted with a hemocytometer. Cell viability was >98% as assessed by trypan blue exclusion and cell preparations were >99% neutrophils. In order to induce apoptosis, fresh neutrophils (2 × 106/ml) were suspended in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% autologous serum and incubated for 24 hours at 37°C in 5% CO2. Induction of apoptosis was performed as previously described.25
Assessment of Apoptosis
Apoptosis was determined by both Annexin V positivity and loss of CD16 expression on the cell surface.9,38 First, positive selection of the apoptotic cells was based on their ability to bind Annexin V, a 35 kDa phospholipid-binding protein with high affinity to phosphatidylserine.38–45 A commercially available cell isolation kit§ was used for the isolation of apoptotic cells with exposed phosphatidylserine38,40,41 and the purification of apoptotic and non-apoptotic neutrophils.42 Briefly, cells were enriched by labeling with Annexin V microbeads and separated on a magnetic cell sorter (MACS) column in the magnetic field of a separator. The extremely small size of the microbeads prevents mechanical stress, unwanted activation, and functional stimulation of the cells. Cells retain their viability and physiological functions.46,47 The magnetically labeled phosphatidylserine-exposed cells were retained in the column while the unlabeled cells ran through. After removal of the column from the magnetic field, the magnetically retained phosphatidylserine-exposed cells were eluted as the positively selected cell fraction (Fig. 1A). Binding of fluorescein isothiocynate (FITC)-labeled Annexin V was further confirmed by immunofluorescence in a flow cytometer.||
Figure 1.
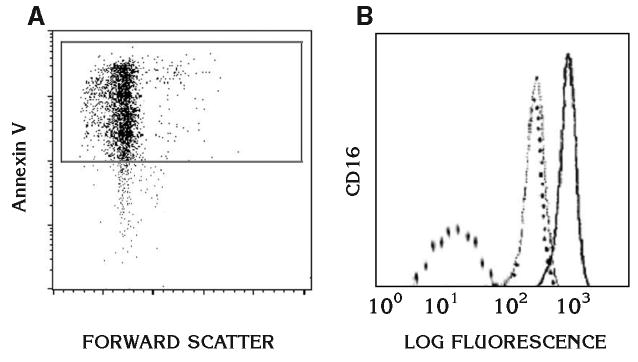
Assessment of apoptosis. Apoptosis was determined by both Annexin V positivity and loss of CD16 expression on the cell surface as described in detail in Materials and Methods. A) The Annexin V positivity by binding of FITC-labeled Annexin V in a flow cytometer. B) Further assessment of apoptosis by the loss of CD16 on the cell surface by FACS analysis. Dashed lines represent negative controls where buffer or control IgG was tested. Solid line represents fresh neutrophils while the thin line shows the shift of CD16 expression on apoptotic neutrophils.
Apoptosis was further assessed by the depletion of CD16 on the cell surface.39 Cultured neutrophils were washed and resuspended in PBS containing 1% bovine serum albumin (BSA) at a concentration of 107 cells/ml. This suspension was mixed with an equal volume of FITC-labeled CD16 monoclonal antibody¶ and incubated for 30 minutes on ice. The cells were then washed and resuspended in PBS/BSA buffer. For control experiments, cells were either incubated with an isotype-matched control primary antibody or only FITC-conjugated secondary antibody. Data were acquired and analyzed using special software# (Fig. 1B). About 80% of neutrophils were found to be apoptotic.
Preparation of LPS From P. gingivalis
P. gingivalis strain A7436 was used in these studies and was maintained on anaerobic blood agar plates.** P. gingivalis cultures were incubated anaerobically at 37°C. P. gingivalis LPS was extracted using a hot phenol-water technique.48,49 The LPS preparation was analyzed for protein contamination by electrophoresis overloading a sodium dodecyl sulfate-12.5% polyacrylamide gel and staining with Coomassie blue and silver nitrate. LPS samples were also examined on commercially prepared 10% to 20% gradient gels. LPS was further analyzed for protein contamination using a bicinchoninic acid protein assay kit.††
Monocyte Isolation and Coculture of Monocytes With Apoptotic Neutrophils
Peripheral blood mononuclear cells were isolated from the same subjects using a discontinuous gradient system (0.25 × 106/ml). Contaminating non-monocytic cells were removed and CD14 + monocyte populations were purified by MACS.‡‡ 50,51 Pure monocytes were resuspended in IMDM (0.25 × 106/ml) with 10% human AB serum in the presence of apoptotic or fresh neutrophils (2.5 × 106/ml). The ratio of monocytes to neutrophils was 1:10. They were then stimulated with P. gingivalis LPS (100 ng/ml; strain A7436) and incubated for 72 hours at 37°C in a 5% CO2 atmosphere in 24-well culture plates. Cell culture supernatants were collected at 24, 48, and 72 hours; centrifuged at 500 × g for 10 minutes at 4°C to remove the particulate debris; and aliquots were stored at −80°C.
Enzyme-Linked Immunosorbent Assay (ELISA)
The release of IL-10 and IL-1β in supernatants was measured by commercially available ELISA kits per the manufacturer’s instructions.§§ Briefly, 96-well microplates were coated with mouse anti-human capture antibody and incubated overnight at room temperature. The plates were washed with PBS containing 0.05% Tween 20, and then blocked by PBS with 1% bovine serum albumin and 5% sucrose. After the addition of diluted samples and standard antigen dilutions, plates were incubated for 2 hours at room temperature. Biotinylated goat anti-human antibodies were used as the detection antibody and streptavidin-horseradish peroxidase (HRP) was added as the conjugate to each well. Equal proportions of hydrogen peroxide and tetramethylbenzidine were used as the substrate solution and sulfuric acid was used to stop the reaction. All samples and standards were run in duplicate and optical density was determined with a microplate reader|| || at 450 nm. Samples above the standard determination range for optical density readings were assayed again and read at an appropriate dilution.
Statistical Analysis
Each experiment was repeated in triplicate and data were expressed as the mean of three separate values for each of four individuals (± standard deviation). Data analysis was performed by Mann-Whitney U test and significance was set at P <0.05.
RESULTS
IL-1β Production by Apoptotic Neutrophil-Treated Human Monocytes in Response to LPS
IL-1β was significantly suppressed in resting monocytes in the presence of apoptotic neutrophils (P <0.01) (Fig. 2). This effect was observed as early as 24 hours and was stable over 72 hours of incubation. When fresh neutrophils were cocultured with monocytes, the suppression of IL-1β generation, although less profound compared to the effect of apoptotic neutrophils, was also statistically different from monocytes alone (P <0.05). In addition, apoptotic neutrophils led to significant reduction in IL-1β secretion from monocytes compared to those cocultured in the presence of fresh neutrophils in the absence of LPS treatment (P <0.05). Overall, unstimulated monocytes did not show any increased levels of IL-1β over time.
Figure 2.
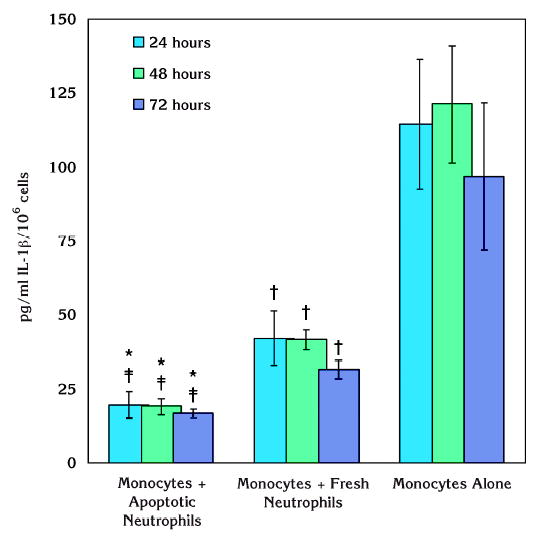
Effect of apoptotic neutrophils on IL-1β production by resting human monocytes. Coculture of unstimulated monocytes with apoptotic neutrophils led to significant suppression of IL-1β generation over 72 hours of incubation (*P <0.01). In the presence of fresh neutrophils, there was also a reduction in IL-1β and the effect, although less obvious than apoptotic neutrophils, was statistically significant compared to monocytes alone (†P <0.05). Apoptotic neutrophils led to a significant reduction in IL-1β secretion from monocytes compared to those cocultured with fresh neutrophils even in the absence of LPS stimulation (‡P <0.05).
Treatment of monocytes alone or in the presence of neutrophils with LPS from P. gingivalis is presented in Figure 3. Apoptotic neutrophils led to significant suppression of IL-1β production of monocytes in response to P. gingivalis LPS. This effect was statistically different compared to either monocytes or monocytes cocultured with fresh neutrophils at all time points (P <0.05). There was no difference between the tested time points over an incubation period of 72 hours although the peak IL-1β levels were observed at 24 hours in pure monocyte or monocyte and fresh neutrophil cultures. Fresh neutrophils led to significant suppression of P. gingivalis LPS-induced IL-1βgeneration from monocytes only at 72 hours compared to monocytes without any neutrophil coculture (P <0.05).
Figure 3.
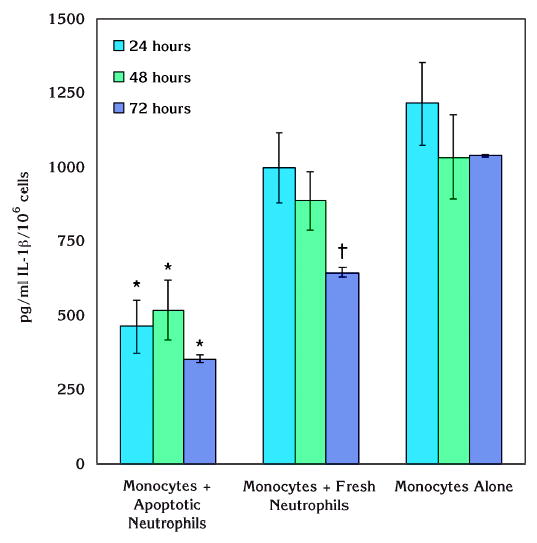
Effect of apoptotic neutrophils on monocytic IL-1β production in response to P. gingivalis LPS. IL-1β production of monocytes in response to P. gingivalis LPS was significantly suppressed when the cells were cultured with apoptotic neutrophils (*P <0.05). This effect was also statistically different compared to monocytes cocultured with fresh neutrophils and stable over 72 hours of incubation (*P <0.05). Fresh neutrophils led to significant reduction in monocytic IL-1βgeneration only at the end of 72 hours (†P <0.05).
IL-10 Production by Apoptotic Neutrophil-Treated Human Monocytes in Response to LPS
In contrast with IL-1β, IL-10 generation by unstimulated monocytes was increased in the presence of apoptotic neutrophils over time (Fig. 4). The increase was statistically significant compared to monocytes alone and monocytes cocultured with fresh neutrophils at 24 and 48 hours (P <0.05). After 72 hours of culture, IL-10 generation was not changed in monocytes that were not stimulated, while fresh neutrophils led to a gradual increase in monocytic IL-10 generation. However, this change was not significant compared to 24 hours or 48 hours. At the end of 72 hours, apoptotic neutrophils and fresh neutrophils did not result in significantly different increases in IL-10 generation by monocytes.
Figure 4.
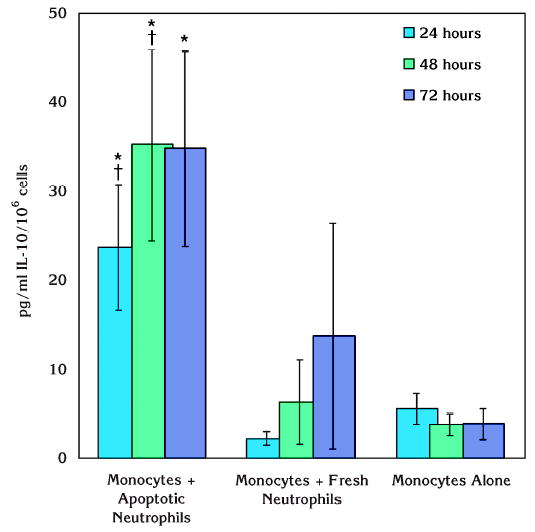
Effect of apoptotic neutrophils on IL-10 production by resting human monocytes. IL-10 generation by monocytes was increased in the presence of apoptotic neutrophils. The difference was statistically significant compared to monocytes alone at all observation time points (*P <0.05). At 24 and 48 hours, this increase was also significantly higher compared to fresh neutrophils cocultured with monocytes (†P <0.05). However, fresh neutrophils did not lead to any significant increase in monocyte IL-10 generation.
We also tested IL-10 production from monocytes in response to P. gingivalis LPS in the presence of apoptotic or fresh neutrophils (Fig. 5). A similar trend was observed in LPS-stimulated cultures as in resting cells. While monocytes did not generate significant amounts of IL-10 in the absence of neutrophils in response to LPS, fresh neutrophils led a slight increase at 24 and 48 hours. However, these changes were not statistically significant. Only at the end of 72 hours, there was a significant increase compared to monocytes alone (P <0.05). When the monocytes were cocultured with apoptotic neutrophils, IL-10 generation was significantly increased in response to P. gingivalis LPS stimulation compared to monocytes alone or cultured with fresh neutrophils at 48 and 72 hours (P <0.05). Overall, monocytes were found not to be a major source of IL-10 over 72 hours of culturing, while the enhancing effect of apoptotic neutrophils on IL-10 generation from monocytes reached to a peak at 48 hours and stayed at this elevated level over 72 hours.
Figure 5.
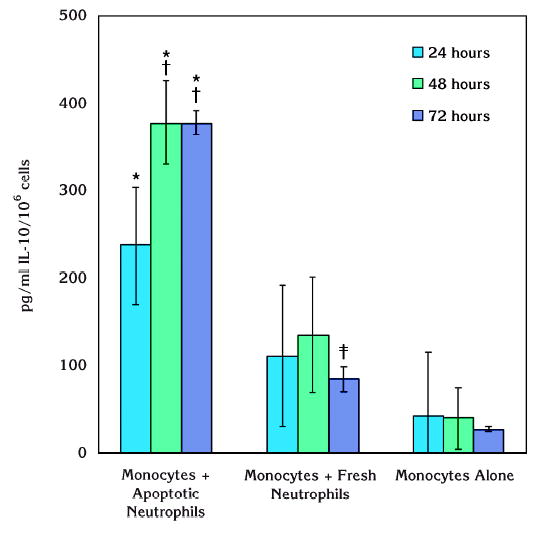
Effect of apoptotic neutrophils on monocytic IL-10 production in response to P. gingivalis LPS. In response to P. gingivalis LPS, IL-10 production from monocytes was significantly increased when cocultured with apoptotic neutrophils (*P <0.05). This effect was also significant compared to monocytes cultured with fresh neutrophils at 48 and 72 hours while there was no difference at 24 hours (†P <0.05). Fresh neutrophils led to a significant increase in IL-10 generation from monocytes only at the end of 72 hours ( ‡P <0.05).
DISCUSSION
Periodontitis is an infection caused by pathogenic Gram-negative bacteria destroying the periodontal connective tissue and alveolar bone.52 Although the presence of bacterial pathogens is necessary for the initiation of periodontal diseases, complex inflammatory and immune responses play a critical role in the progression of disease. Indeed, it has been shown that persistent bacterial stimulation disrupts homeostatic mechanisms and results in release of mediators (IL-1, IL-6, TNF, prostaglandins), which contribute to the pathogenesis of periodontal disease.1,53 These proinflammatory mediators act in various stages of the disease, are essential for the initiation of an inflammatory response and, ultimately, tissue destruction.54 Thus, proinflammatory cytokine activity must be regulated in order to prevent an excessive inflammatory response and abnormal tissue destruction.1,4,53,55 Apoptotic neutrophils, monocytes, soluble mediators, or cell-to-cell contact can play critical roles in the regulation or resolution of periodontal inflammation. In this study, we show that neutrophil apoptosis is an essential process to limit the prolonged effects of proinflammatory activity generated by monocytic phagocytes. We also demonstrate that monocytes can generate significant amounts of IL-10 in response to LPS challenge in the presence of apoptotic neutrophils.
A dynamic balance exists between proinflammatory cytokines and anti-inflammatory components of the immune system. The regulation of inflammation by anti-inflammatory cytokines is complicated by the fact that the immune system has several pathways with multiple elements having similar physiologic effects. The net effect of any cytokine is dependent on the timing of cytokine release, local milieu in which it acts, the presence of competing or synergistic elements, cytokine receptor density, and tissue responsiveness to each cytokine.17,56,57 The pathogenesis of many major diseases is determined by the production of proinflammatory cytokines by monocytes and macrophages. Monocytes and macrophages play an important role among the infiltrating cells in periodontal inflammation.4,53,55 They are known to be the primary targets for LPS and produce proinflammatory cytokines such as IL-1β, TNF-α, IL-6, or IL-8.1,4 A major inhibitor of stimulated, cytokine-producing monocytes and macrophages is the anti-inflammatory cytokine IL-10 secreted largely by helper T cells.58–60 Helper T cell-mediated IL-4 also induces macrophage apoptosis, which reduces the number of infiltrating inflammatory macrophages.61 Macrophage accumulation was found to be continuous in periodontitis due to reduced IL-4 in disease sites.62 Both IL-10 and IL-4 target monocytes and inhibit the release of IL-1β, TNF-α, and oxygen intermediates such as nitric oxide.4,5 However, anti-inflammatory cytokines in addition to IL-10, such as IL-4, and IL-1 receptor antagonist were also found to be upregulated after LPS stimulation, suggesting that they could be important for resolution of inflammation.56,59 The anti-inflammatory effects of monocytes and macrophages can also be balanced by the actions of other cell types in addition to the monocytic cells themselves.9 In this study, we show that monocytes cocultured with apoptotic neutrophils released IL-10 when activated by P. gingivalis LPS. At the same time, IL-1β production in response to LPS was suppressed. Therefore, we propose that interactions of apoptotic neutrophils with LPS-stimulated monocytes may play an important role in early events in periodontal inflammation triggered by innate immune stimuli derived from invading periodontal bacteria, or tissue damage, and provide new insight into the molecular events leading to resolution of inflammation.
Neutrophils are the first-line defense against invading microorganisms and eliminate pathogens by phagocytosis. They play an important role in both the early stage and the termination of inflammation. It has been shown that proinflammatory capacity is down-regulated during the induction of neutrophil apoptosis and is critical for resolution of inflammation in humans.27 Indeed, there is growing evidence that apoptotic neutrophils play an active role in regulation and resolution of inflammation.23,25,27 However, there is little information about the molecular basis for termination of inflammation and endogenous anti-inflammatory signals in periodontal lesions. In the present study, we demonstrated that following interaction with apoptotic neutrophils, monocytes act as suppressors of inflammation in response to LPS challenge. This finding is parallel to the reported data where apoptotic cells enhanced IL-10 production by macrophages in response to E. coli LPS.25 Together, these data show that the induction of anti-inflammatory response by monocytes coupled with the reduction of proinflammatory responses are only specific to apoptotic neutrophils but not fresh neutrophils. Indeed, it has been reported that apoptotic neutrophils can generate the anti-inflammatory signals by the help of phosphatidylserine while fresh neutrophils do not express that molecule on their membrane surface.22 Therefore, cell signaling events mediated by lipid mediators might play a role in this phenomenon.
In conclusion, our findings demonstrate that monocytes increase their anti-inflammatory capacity by the accelerated release of IL-10 in response to P. gingivalis LPS, after they are cocultured with apoptotic neutrophils. They also decrease their proinflammatory capacity during this period. Neutrophil apoptosis provides a signal to monocytes that changes the phenotype and cytokine kinetics of monocytes, resulting in production of anti-inflammatory cytokines in response to P. gingivalis LPS challenges. These data suggest that the character of the neutrophil response in early events in inflammation have a direct effect on the establishment of chronic lesions in periodontitis.
Acknowledgments
This research was supported by a General Clinical Research Center (GCRC) grant (MO1 RR00533) from Boston University, Boston, Massachusetts.
Footnotes
Histopaque 1119 and 1077, Sigma Chemical Co., St. Louis, MO.
Annexin V Micro Bead kit, Miltenyi Biotech, Auburn, CA.
FACScan, BD Bioscience, Franklin Lakes, NJ.
Becton Dickinson Immunocytometry Systems, San Jose, CA.
BD Bioscience.
Pierce, Rockford, IL.
Fisher Scientific Co., Springfield, NJ.
Miltenyi Biotech.
R&D Systems, Minneapolis, MN.
Vmax, Molecular Devices, Sunnyvale, CA.
References
- 1.Zadeh HH, Nichols FC, Miyasaki KT. The role of the cell-mediated immune response to Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in periodontitis. Periodontol 2000. 1999;20:239–288. doi: 10.1111/j.1600-0757.1999.tb00163.x. [DOI] [PubMed] [Google Scholar]
- 2.Takeichi O, Haber J, Kawai T, Smith DJ, Moro I, Taubman MA. Cytokine profiles of T-lymphocytes from gingival tissues with pathological pocketing. J Dent Res. 2000;79:1548–1555. doi: 10.1177/00220345000790080401. [DOI] [PubMed] [Google Scholar]
- 3.Van Dyke TE, Serhan CN. Resolution of inflammation: A new paradigm for the pathogenesis of periodontal diseases. J Dent Res. 2003;82:82–90. doi: 10.1177/154405910308200202. [DOI] [PubMed] [Google Scholar]
- 4.Gemmell E, Yamazaki K, Seymour GJ. Destructive periodontitis lesions are determined by the nature of the lymphocytic response. Crit Rev Oral Biol Med. 2002;13:17–34. doi: 10.1177/154411130201300104. [DOI] [PubMed] [Google Scholar]
- 5.Okada H, Murakami S. Cytokine expression in periodontal health and disease. Crit Rev Oral Biol Med. 1998;9:248–266. doi: 10.1177/10454411980090030101. [DOI] [PubMed] [Google Scholar]
- 6.Pouliot M, Clish CB, Petasis NA, Van Dyke TE, Serhan CN. Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: A role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry. 2000;39:4761–4768. doi: 10.1021/bi992551b. [DOI] [PubMed] [Google Scholar]
- 7.Kinane DF, Lappin DF. Clinical, pathological and immunological aspects of periodontal disease. Acta Odontol Scand. 2001;59:154–160. doi: 10.1080/000163501750266747. [DOI] [PubMed] [Google Scholar]
- 8.Beutler B, Cerami A. Tumor necrosis, cachexia, shock, and inflammation: A common mediator. Annu Rev Biochem. 1988;57:505–518. doi: 10.1146/annurev.bi.57.070188.002445. [DOI] [PubMed] [Google Scholar]
- 9.Vignola AM, Gjomarkaj M, Arnoux B, Bousquet J. Monocytes. J Allergy Clin Immunol. 1998;101:149–152. doi: 10.1016/S0091-6749(98)70378-1. [DOI] [PubMed] [Google Scholar]
- 10.Moss ST, Hamilton JA. Proliferation of a subpopulation of human peripheral blood monocytes in the presence of colony stimulating factors may contribute to the inflammatory process in diseases such as rheumatoid arthritis. Immunobiology. 2000;202:18–25. doi: 10.1016/S0171-2985(00)80048-0. [DOI] [PubMed] [Google Scholar]
- 11.Glauser MP, Zanetti G, Baumgartner JD, Cohen J. Septic shock: Pathogenesis. Lancet. 1991;338:732–736. doi: 10.1016/0140-6736(91)91452-z. [DOI] [PubMed] [Google Scholar]
- 12.Brandtzaeg P, Osnes L, Ovstebo R, Joo GB, Westvik AB, Kierulf P. Net inflammatory capacity of human septic shock plasma evaluated by a monocyte-based target cell assay: Identification of interleukin-10 as a major functional deactivator of human monocytes. J Exp Med. 1996;184:51–60. doi: 10.1084/jem.184.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10 (IL-10) inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Randow F, Syrbe U, Meisel C, Krausch D, Zuckermann H, Platzer C, Volk HD. Mechanism of endotoxin desensitization: Involvement of interleukin 10 and transforming growth factor beta. J Exp Med. 1995;181:1887–1892. doi: 10.1084/jem.181.5.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hart SP, Alexander KM, Dransfield I. Immune complexes bind preferentially to FcgammaRIIA (CD32) on apoptotic neutrophils, leading to augmented phagocytosis by macrophages and release of proinflammatory cytokines. J Immunol. 2004;172:1882–1887. doi: 10.4049/jimmunol.172.3.1882. [DOI] [PubMed] [Google Scholar]
- 16.Meszaros AJ, Reichner JS, Albina JE. Macrophage-induced neutrophil apoptosis. J Immunol. 2000;165:435–441. doi: 10.4049/jimmunol.165.1.435. [DOI] [PubMed] [Google Scholar]
- 17.Kox WJ, Volk T, Kox SN, Volk HD. Immunomodulatory therapies in sepsis. Intensive Care Med. 2000;26 (Suppl 1):S124–128. doi: 10.1007/s001340051129. [DOI] [PubMed] [Google Scholar]
- 18.Fadok VA, Bratton DL, Henson PM. Phagocyte receptors for apoptotic cells: Recognition, uptake, and consequences. J Clin Invest. 2001;108:957–962. doi: 10.1172/JCI14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kerr JF, Wyllie AH, Currie AR. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wyllie AH, Kerr JF, Currie AR. Cell death: The significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 21.Meagher LC, Savill JS, Baker A, Fuller RW, Haslett C. Phagocytosis of apoptotic neutrophils does not induce macrophage release of thromboxane B2. J Leukoc Biol. 1992;52:269–273. [PubMed] [Google Scholar]
- 22.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucas M, Stuart LM, Savill J, Lacy-Hulbert A. Apoptotic cells and innate immune stimuli combine to regulate macrophage cytokine secretion. J Immunol. 2003;171:2610–2615. doi: 10.4049/jimmunol.171.5.2610. [DOI] [PubMed] [Google Scholar]
- 24.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: Clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 25.Byrne A, Reen DJ. Lipopolysaccharide induces rapid production of IL-10 by monocytes in the presence of apoptotic neutrophils. J Immunol. 2002;168:1968–1977. doi: 10.4049/jimmunol.168.4.1968. [DOI] [PubMed] [Google Scholar]
- 26.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 27.Kobayashi SD, Voyich JM, Braughton KR, DeLeo FR. Down-regulation of proinflammatory capacity during apoptosis in human polymorphonuclear leukocytes. J Immunol. 2003;170:3357–3368. doi: 10.4049/jimmunol.170.6.3357. [DOI] [PubMed] [Google Scholar]
- 28.Ottonello L, Frumento G, Arduino N, et al. Delayed neutrophil apoptosis induced by synovial fluid in rheumatoid arthritis: Role of cytokines, estrogens, and adenosine. Ann N Y Acad Sci. 2002;966:226–231. doi: 10.1111/j.1749-6632.2002.tb04219.x. [DOI] [PubMed] [Google Scholar]
- 29.Niederman R, Zhang J, Kashket S. Short-chain carboxylic-acid-stimulated, PMN-mediated gingival inflammation. Crit Rev Oral Biol Med. 1997;8:269–290. doi: 10.1177/10454411970080030301. [DOI] [PubMed] [Google Scholar]
- 30.Crawford JM, Wilton JM, Richardson P. Neutrophils die in the gingival crevice, periodontal pocket, and oral cavity by necrosis and not apoptosis. J Periodontol. 2000;71:1121–1129. doi: 10.1902/jop.2000.71.7.1121. [DOI] [PubMed] [Google Scholar]
- 31.Gamonal J, Sanz M, O’Connor A, et al. Delayed neutrophil apoptosis in chronic periodontitis patients. J Clin Periodontol. 2003;30:616–623. doi: 10.1034/j.1600-051x.2003.00350.x. [DOI] [PubMed] [Google Scholar]
- 32.Ratasirayakorn W, Leone P, Leblebicioglu B, Walters JD. Polyamines found in the inflamed periodontium inhibit priming and apoptosis in human polymorphonuclear leukocytes. J Periodontol. 1999;70:179–184. doi: 10.1902/jop.1999.70.2.179. [DOI] [PubMed] [Google Scholar]
- 33.Stehle HW, Leblebicioglu B, Walters JD. Short-chain carboxylic acids produced by Gram-negative anaerobic bacteria can accelerate or delay polymorphonuclear leukocyte apoptosis in vitro. J Periodontol. 2001;72:1059–1063. doi: 10.1902/jop.2001.72.8.1059. [DOI] [PubMed] [Google Scholar]
- 34.Gamonal J, Bascones A, Acevedo A, Blanco E, Silva A. Apoptosis in chronic adult periodontitis analyzed by in situ DNA breaks, electron microscopy, and immunohistochemistry. J Periodontol. 2001;72:517–525. doi: 10.1902/jop.2001.72.4.517. [DOI] [PubMed] [Google Scholar]
- 35.Preshaw PM, Schifferle RE, Walters JD. Porphyromonas gingivalis lipopolysaccharide delays human polymorphonuclear leukocyte apoptosis in vitro. J Periodont Res. 1999;34:197–202. doi: 10.1111/j.1600-0765.1999.tb02242.x. [DOI] [PubMed] [Google Scholar]
- 36.Hiroi M, Shimojima T, Kashimata M, et al. Inhibition by Porphyromonas gingivalis LPS of apoptosis induction in human peripheral blood polymorphonuclear leukocytes. Anticancer Res. 1998;18:3475–3479. [PubMed] [Google Scholar]
- 37.Kalmar JR, Arnold RR, Warbington ML, Gardner MK. Superior leukocyte separation with a discontinuous onestep Ficoll-Hypaque gradient for the isolation of human neutrophils. J Immunol Methods. 1988;110:275–281. doi: 10.1016/0022-1759(88)90115-9. [DOI] [PubMed] [Google Scholar]
- 38.Dressel R, Elsner L, Quentin T, Walter L, Gunther E. Heat shock protein 70 is able to prevent heat shock-induced resistance of target cells to CTL. J Immunol. 2000;164:2362–2371. doi: 10.4049/jimmunol.164.5.2362. [DOI] [PubMed] [Google Scholar]
- 39.Homburg CH, de Haas M, von dem Borne AE, Verhoeven AJ, Reutelingsperger CP, Roos D. Human neutrophils lose their surface Fc gamma RIII and acquire Annexin V binding sites during apoptosis in vitro. Blood. 1995;85:532–540. [PubMed] [Google Scholar]
- 40.Rickers A, Brockstedt E, Mapara MY, Otto A, Dorken B, Bommert K. Inhibition of CPP32 blocks surface IgM-mediated apoptosis and D4-GDI cleavage in human BL60 Burkitt lymphoma cells. Eur J Immunol. 1998;28:296–304. doi: 10.1002/(SICI)1521-4141(199801)28:01<296::AID-IMMU296>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 41.Kuypers FA, Lewis RA, Hua M, et al. Detection of altered membrane phospholipid asymmetry in subpopulations of human red blood cells using fluorescently labeled annexin V. Blood. 1996;87:1179–1187. [PubMed] [Google Scholar]
- 42.Khwaja A, Tatton L. Caspase-mediated proteolysis and activation of protein kinase Cδ plays a central role in neutrophil apoptosis. Blood. 1999;94:291–301. [PubMed] [Google Scholar]
- 43.Atsumi G, Murakami M, Kojima K, Hadano A, Tajima M, Kudo I. Distinct roles of two intracellular phospholipase A2s in fatty acid release in the cell death pathway. Proteolytic fragment of type IVA cytosolic phospholipase A2α inhibits stimulus-induced arachidonate release, whereas that of type VI Ca2+-independent phospholipase A2 augments spontaneous fatty acid release. J Biol Chem. 2000;275:18248–18258. doi: 10.1074/jbc.M000271200. [DOI] [PubMed] [Google Scholar]
- 44.Fadok VA, Bratton DL, Frasch SC, Warner ML, Henson PM. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 1998;5:551–562. doi: 10.1038/sj.cdd.4400404. [DOI] [PubMed] [Google Scholar]
- 45.Henson PM, Bratton DL, Fadok VA. The phosphatidylserine receptor: A crucial molecular switch? Nat Rev Mol Cell Biol. 2001;2:627–633. doi: 10.1038/35085094. [DOI] [PubMed] [Google Scholar]
- 46.Manz R, Assenmacher M, Pfluger E, Miltenyi S, Radbruch A. Analysis and sorting of live cells according to secreted molecules, relocated to a cell-surface affinity matrix. Proc Natl Acad Sci (USA) 1995;92:1921–1925. doi: 10.1073/pnas.92.6.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oelke M, Moehrle U, Chen JL, et al. Generation and purification of CD8+ melan-A-specific cytotoxic T lymphocytes for adoptive transfer in tumor immunotherapy. Clin Cancer Res. 2000;6:1997–2005. [PubMed] [Google Scholar]
- 48.Duncan RL, Jr, Hoffman J, Tesh VL, Morrison DC. Immunologic activity of lipopolysaccharides released from macrophages after the uptake of intact E. coli in vitro. J Immunol. 1986;136:2924–2929. [PubMed] [Google Scholar]
- 49.Westphal O, Jann K. Bacterial lipopolysaccharide: Extraction with phenol water and further applications of the procedure. Methods Carbohydr Chem. 1965;5:83–91. [Google Scholar]
- 50.Flo RW, Naess A, Lund-Johansen F, et al. Negative selection of human monocytes using magnetic particles covered by anti-lymphocyte antibodies. J Immunol Methods. 1991;137:89–94. doi: 10.1016/0022-1759(91)90397-x. [DOI] [PubMed] [Google Scholar]
- 51.Pickl WF, Majdic O, Kohl P, et al. Molecular and functional characteristics of dendritic cells generated from highly purified CD14+ peripheral blood monocytes. J Immunol. 1996;157:3850–3859. [PubMed] [Google Scholar]
- 52.Kornman KS, Page RC, Tonetti MS. The host response to the microbial challenge in periodontitis: Assembling the players. Periodontol 2000. 1997;14:33–53. doi: 10.1111/j.1600-0757.1997.tb00191.x. [DOI] [PubMed] [Google Scholar]
- 53.Oringer RJ. Modulation of the host response in periodontal therapy. J Periodontol. 2002;73:460–470. doi: 10.1902/jop.2002.73.4.460. [DOI] [PubMed] [Google Scholar]
- 54.Page RC. The role of inflammatory mediators in the pathogenesis of periodontal disease. J Periodont Res. 1991;26:230–242. doi: 10.1111/j.1600-0765.1991.tb01649.x. [DOI] [PubMed] [Google Scholar]
- 55.Seymour GJ, Gemmell E. Cytokines in periodontal disease: Where to from here? Acta Odontol Scand. 2001;59:167–173. doi: 10.1080/000163501750266765. [DOI] [PubMed] [Google Scholar]
- 56.Dinarello CA. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. Int Rev Immunol. 1998;16:457–499. doi: 10.3109/08830189809043005. [DOI] [PubMed] [Google Scholar]
- 57.Seymour RM, Henderson B. Pro-inflammatory–anti-inflammatory cytokine dynamics mediated by cytokine-receptor dynamics in monocytes. IMA J Math Appl Med Biol. 2001;18:159–192. [PubMed] [Google Scholar]
- 58.Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest. 2000;117:1162–1172. doi: 10.1378/chest.117.4.1162. [DOI] [PubMed] [Google Scholar]
- 59.Keystone E, Wherry J, Grint P. IL-10 as a therapeutic strategy in the treatment of rheumatoid arthritis. Rheum Dis Clin North Am. 1998;24:629–639. doi: 10.1016/s0889-857x(05)70030-2. [DOI] [PubMed] [Google Scholar]
- 60.Allen JB, Wong HL, Costa GL, Bienkowski MJ, Wahl SM. Suppression of monocyte function and differential regulation of IL-1 and IL-1ra by IL-4 contribute to resolution of experimental arthritis. J Immunol. 1993;151:4344–4351. [PubMed] [Google Scholar]
- 61.Ma J, Chen T, Mandelin J, et al. Regulation of macrophage activation. Cell Mol Life Sci. 2003;60:2334–2346. doi: 10.1007/s00018-003-3020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bingisser R, Stey C, Weller M, Groscurth P, Russi E, Frei K. Apoptosis in human alveolar macrophages is induced by endotoxin and is modulated by cytokines. Am J Respir Cell Mol Biol. 1996;15:64–70. doi: 10.1165/ajrcmb.15.1.8679223. [DOI] [PubMed] [Google Scholar]