

Abstract
Recurrent exposure of the developing fetus to cocaine produces persistent alterations in structure and function of the cerebral cortex. Neurons of the cerebral cortex are derived from two sources: projection neurons from the neuroepithelium of the dorsal pallium and interneurons from the ganglionic eminence of the basal telencephalon. The interneurons are GABAergic and reach the cerebral cortex via a tangential migratory pathway. We found that recurrent, transplacental exposure of mouse embryos to cocaine from embryonic day 8 to 15 decreases tangential neuronal migration and results in deficits in GABAergic neuronal populations in the embryonic cerebral wall. GABAergic neurons of the olfactory bulb, which are derived from the ganglionic eminence via the rostral migratory pathway, are not affected by the cocaine exposure suggesting a degree of specificity in the effects of cocaine on neuronal migration. Thus, one mechanism by which prenatal cocaine exposure exerts deleterious effects on cerebral cortical development may be by decreasing GABAergic neuronal migration from the ganglionic eminence to the cerebral wall. The decreased GABA neuron migration may contribute to persistent structural and functional deficits observed in the exposed offspring.
Keywords: cerebral cortex, cocaine, GABA, ganglionic eminence, neurogenesis, neuronal migration
Introduction
Animal models have established that recurrent exposure to cocaine during pregnancy disrupts fetal brain development causing lasting changes in the cellular architecture of the brain. Cocaine penetrates the placental barrier and interferes with monoamine uptake and storage mechanisms in the fetus (Akbari et al., 1992; Meyer et al., 1993; Kosofsky et al., 1994; X.-H. Wang et al., 1995; Shearman et al., 1996; Levitt et al., 1997; Mayes, 1999; Jones et al., 2000). Monoamines are among the earliest neurochemical systems to develop in the embryo and can influence neurogenesis and neuronal and glial cell differentiation (Molliver, 1982; Lauder, 1988; Cases et al., 1995, 1996; Reinoso et al., 1996; Levitt et al., 1997; Bongarzone et al., 1998; Vitalis et al., 1998; Ohtani et al., 2003). Previous reports showing that exposure of the fetal brain to cocaine disrupted the cytoarchitecture of the cerebral cortex in primates, rodents and lagomorphs suggested aminergic bases for the effects of cocaine (Gressens et al., 1992a,b; Lidow, 1995; Mayes, 1999; Jones et al., 2000; Lidow and Song, 2001; Lidow et al., 2001; Morrow et al., 2003).
There are two major classes of neurons in the cerebral cortex: projection neurons and interneurons. The projection neurons arise from the neuroepithelium of the cerebral wall and interneurons from the ganglionic eminence in the basal forebrain (Anderson et al., 1997, 1999; Lavdas et al., 1999; Marin et al., 2000; Wichterle et al., 2001; Nery et al., 2002). Projection neurons migrate radially from their site of origin in the cortical neuroepithelium to the marginal zones of the cerebral wall, guided by radial glial fascicles. Interneurons migrate tangentially from the ganglionic eminence to the cerebral wall, probably guided by signaling molecules (Wu et al., 1999; Marin et al., 2001; Mason et al., 2001; Powell et al., 2001), axons in the nascent internal capsule (Denaxa et al., 2001) and other cues not identified yet. Upon arrival in the cerebral wall, interneurons migrate further in radial or tangential directions (Nadarajah et al., 2001, 2002; Ang et al., 2003) to assume their final positions. The ganglionic eminence can be divided into medial, lateral and caudal divisions (MGE, LGE and CGE, respectively) based on gene expression patterns (Anderson et al., 1999; Marin et al., 2000; Nery et al., 2002, 2003). Although all three subdivisions produce GABAergic neurons that migrate long distances away from their sites of origin, MGE and CGE are the predominant sources of GABAergic neurons of the cerebral wall and LGE is the predominant source of GABAergic neurons of the striatum and the olfactory bulb.
The effects of cocaine on the development of cerebral cortical cytoarchitecture may be due to its effects on the generation and/or migration of projection neurons, interneurons or both. We report that exposure of embryonic mice to cocaine decreases tangential migration of neurons from the ganglionic eminence to the cerebral cortex and produces deficits in the relative numbers of GABA immunoreactive neurons in the embryonic cerebral wall. Although GABAergic neurons of the olfactory bulb also are derived from the ganglionic eminence, their distribution is unaffected by prenatal cocaine exposure. Therefore, cocaine appears to selectively influence the migration of a subset of GABAergic interneurons.
Materials and Methods
Animals
Timed-pregnant Swiss–Webster mice were obtained from Taconic Farms (Germantown, NY). A transplacental cocaine exposure paradigm described previously (Kosofsky et al., 1994; Wilkins et al., 1998) was abbreviated so as to expose mouse embryos to cocaine twice daily from the morning of embryonic day 8 (E8; day of conception = E0) to the evening of E14, inclusive. At the beginning of the experiment, pregnant dams of comparable weight were assigned to the following four groups: cocaine 20 mg/kg/day (COC-20), cocaine 40 mg/kg/day (COC-40), saline pair-fed (SPF-40) and saline control (SAL). The dams were handled for 5 min each morning, beginning on the 6th day of pregnancy (corresponding to E6). The dams were placed on a liquid diet (Bioserve, Frenchtown, NJ) and the amount of food intake and body weight were recorded daily beginning on E7. From E8 onwards, cocaine was injected subcutaneously to the COC-20 and COC-40 groups at a dose of 20 or 40 mg/kg/day, respectively, divided twice daily (7 a.m. and 7 p.m.) through E14. Dams in the SPF-40 and SAL groups received saline injections, twice daily, from E8 to E14 at the same time that the COC-20 and COC-40 dams received their cocaine injections. The daily food intake of each dam in the SPF-40 group was restricted to the amount consumed by the dam in the COC-40 group with which it was paired on the same gestational day. The SPF-40 group served as a nutritional control for the COC-40 group, whose food intake is reduced compared to the SAL and COC-20 groups, as reported previously (Kosofsky et al., 1994; Wilkins et al., 1998). The dams were singly housed in a temperature and humidity controlled environment, on a 12 h light/dark cycle with water available ad libidum. The dams were anesthetized on E15 (Ketamine, 50 mg/kg body weight and Xylazine, 10 mg/kg body weight, i.p.) at 9 a.m. and the embryos were removed by hysterectomy for further analyses. All of the experimental procedures performed on the mice were in full compliance with institutional guidelines and the NIH ‘Guide for the Care and Use of Laboratory Animals’.
Brain Slice Preparation and Culture
Neuronal migration was assayed in slice preparations of the embryonic telencephalon as described previously (Tobet et al., 1994; Anderson et al., 1997). Briefly, E15 embryos were removed one at a time from deeply anesthetized dams, utilizing pups from a minimum of three litters for each of the four prenatal treatment groups. Age of the embryos was confirmed by crown-rump length, (13 –15 mm) and other external features (Theiler, 1972). The embryos were decapitated and the heads were embedded in 8% agarose (Type VII, Sigma Chemical Company, St Louis. MO). Coronal sections of the embryonic head were cut at a thickness of 250 μm on a Vibratome. The sections were cultured individually on membrane filters in polycarbonate transwells (Costar, Corning, NY) and placed in a six-well cell culture plate. Neurobasal medium (GIBCO-BRL) containing 2% B-27 supplement, penicillin, streptomycin and glutamine was added beneath the filter into each well. The slices were maintained in culture for 2 days. The medium was replaced at the end of the first 24 h.
DiI-Labeling of Cells in the Slice Preparations
Glass shards coated with DiI (Molecular Probes, Eugene, OR) were prepared (Bhide and Frost, 1991) and inserted into the ganglionic eminence (~200–400 μm ventrolateral to the pallial-striatal angle; Fig. 1) with the aid of a stereomicroscope immediately after plating each slice. Following 2 days in culture, the slices were fixed with 2% paraformaldehyde in 0.1 M phosphate buffer, pH 7.2 and viewed as whole-mounts in a fluorescence microscope fitted with TRITC filters.
Figure 1.
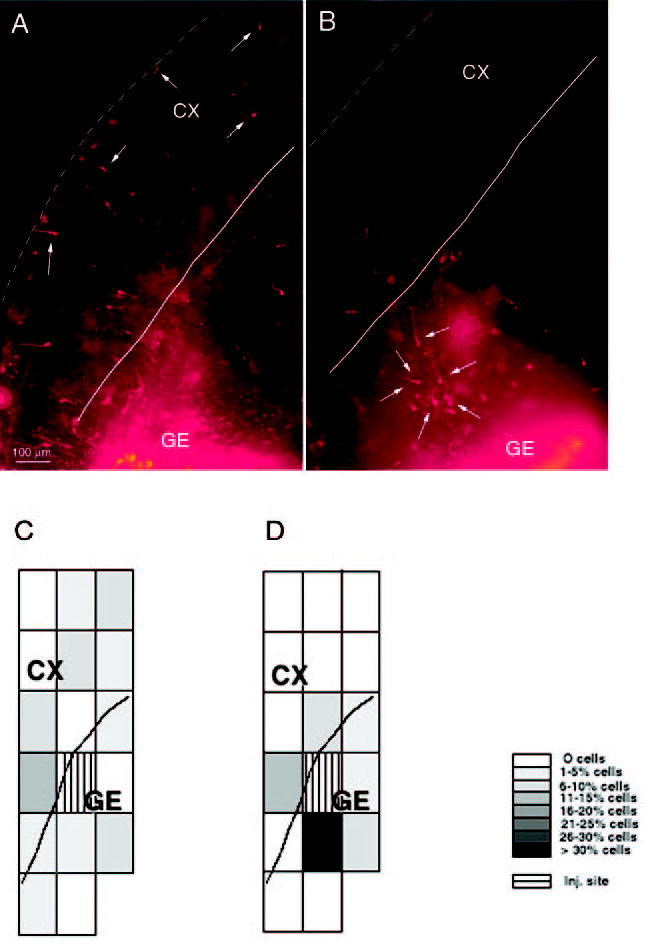
Distribution of DiI-labeled cells in slice preparations from E15 mice maintained for 2 days in vitro (A, B) and a graphical representation of the extent of DiI-labeled cell migration in ‘typical’ SAL and COC-40 slices (C, D). The DiI was deposited in the ganglionic eminence (GE) after plating the slices. There are fewer DiI-labeled cells in the cortex in the slice from the COC-40 group (B) compared to the slice from the SAL group (A), although many labeled cells are present close to the injection site in both slices. To obtain a quantitative measure of the overall extent of DiI-labeled cell migration in the slices and to give a visual indication of the extent of neuronal migration, we overlaid a grid of rectangles or boxes on the microscopic image of the slices (C, D). We calculated the percentage of DiI-labeled cells in each grid box (as a percentage of the total number of labeled cells in the slice). The percentage was coded in 5% increments of gray values (see the gray scale legend to the right of D) and a pictorial representation of the gray scale was prepared. The majority (>30%) of the DiI-labeled cells are in the box closest to the injection site (GE) in the COC-40 case (D). In the slice from the SAL case (C), the DiI-labeled cells are dispersed widely such that no single box contains more than 15% of the cells. C and D are oriented in the same direction as A and B. The dotted lines in A and B indicate the pial surface, and the solid lines in A, B, C and D indicate the boundary between the cerebral wall and the ganglionic eminence or ventricular surface. CX = cerebral cortex.
Measurement of the Size of the DiI Injection Site
The extent of the DiI injection site was delimited for each slice as a solidly intense fluorescent area. A laser printer plot for each case showed the location of each DiI-labeled cell in the entire slice as well as the extent of the DiI injection. The area of the DiI placement was measured from the laser plots using IPLab (v. 3.5) software. A single investigator blinded to the identity of the prenatal treatment group performed the analyses.
Analysis of Cell Migration in the Slice Preparations
The position of DiI-labeled neurons was recorded with respect to the boundaries of the slice using an image analysis system (IBAS Image Analyzer; Zeiss/Kontron) interfaced to the microscope. A single investigator blinded to the identity of the prenatal treatment group performed the analyses. A laser printer plot for each case showed the location of each DiI-labeled cell in the entire slice. An overlay grid of rectangles (each 640 μm × 480 μm) — ‘grid boxes’ — was placed on the image of the slice to measure the overall extent of neuronal migration. The percentage of DiI-labeled cells in each grid box (as a percentage of the total number of labeled cells in the slice) was calculated. The percentage was coded in 5% increments of gray values to give a visual indication of the extent of neuronal migration (Fig. 1C,D). We then calculated the percentage of DiI-labeled cells that entered the cerebral wall by analyzing the percentage of labeled cells reaching the cortex (Figs 1 and 2). For this measurement, any DiI-labeled cell found dorsal and lateral to the caudate–pallial angle was considered to have reached the cortex (Fig. 1). The number of such cells in a slice was expressed as a percentage of the total number of DiI-labeled cells in that slice. The data were obtained from at least six forebrain slices from embryos obtained from at least three litters, for each of the four prenatal treatment groups.
Figure 2.

Mean ± SEM values for the percentage of DiI-labeled cells entering the cerebral wall in slice preparations maintained in vitro. DiI was deposited in the ganglionic eminence. * = value is significantly different (P < 0.05) from the SAL group and + = value is significantly different from the SPF-40 group. Statistical significance between two groups was analyzed using the Tukey–Kramer test. The data were obtained from 6–10 slices for each of the experimental groups.
GABA Immunohistochemistry
E15 mice exposed to cocaine from E8 to E14 were perfused with 2% acrolein in 0.1 M phosphate buffer, pH 7.2, and the brains were fixed overnight at 4°C in the same fixative, cryoprotected in 30% sucrose and sectioned at 50 μm thickness in the coronal plane on a sledge microtome. The sections were processed for immunohistochemistry using antibodies to GABA (rabbit polyclonal, 1:3000, Incstar, Still-water, MN), biotinylated secondary antibody (Jackson Immunolabs, Westgrove, PA), avidin–biotin kit (Vector Labs, Burlingame, CA) and nickel ammonium sulfate (0.02%) enhancement of diaminobenzidine reaction product.
Calculation of GABA-positive Cell Numbers
GABA-positive cells were counted by a single investigator blinded to prenatal treatment group in 50 μm thick sections through the presumptive medial prefrontal cortex, somatosensory cortex and the olfactory bulb. The counts were performed using a 40× objective lens in a sector that extended from the ventricular border to the pial surface of the cerebral wall or the olfactory bulb. The counts were expressed as number of cells per 350 μm2. Subsequently, the number of GABA-positive cells was reanalyzed separately for the different laminae of the cerebral wall: ventricular zone/subventricular zone (VZ/SVZ), intermediate zone (IZ), subplate/cortical plate (SP/CP) and the marginal zone (MZ). In the olfactory bulb, the counts were reanalyzed separately for the granule cell layer (GCL), mitral cell/external plexiform layers (MCL/EPL), periglomerular layer and nerve layers (PGL/NL). We analyzed 5–7 brains of embryos obtained from at least three litters for each of the four prenatal treatment groups, utilizing three sections from each brain.
Measurement of Thickness of the Cerebral Wall and the Olfactory Bulb
We measured the radial dimension (i.e. thickness) of the cerebral wall in the presumptive medial prefrontal cortex and the somatosensory cortex, and of the olfactory bulb. The measurements were performed by a single investigator blinded to prenatal treatment group using a calibrated ocular grid superimposed on the image of the section under a microscope. We analyzed 5–7 brains of embryos obtained from at least three litters for each of the four prenatal treatment groups, utilizing three sections from each brain.
Analysis of Bromodeoxyuridine (BrdU) Labeling Index
For these experiments, each dam received a single injection of BrdU (50 mg/kg, i.p.; Sigma) at 9 a.m. on E15. The dams were anesthetized and killed 2.0 h after the BrdU injection. Thus, the BrdU was administered 14 h after the final cocaine exposure, which occurred around 7 p.m. on E14. The embryos were removed and the heads were immersed in 70% ethanol. The brains were dissected, embedded in paraffin wax and 4 μm thick coronal sections were cut through the entire forebrain. The sections were processed for BrdU immunohistochemistry and stained with basic fuchsin (Bhide, 1996). Examples of the histological material are shown in Figure 9. BrdU labeling index (LI, BrdU labeled cells ÷ all cells) was calculated in the VZ and SVZ of the lateral ganglionic eminence (LGE), medial ganglionic eminence (MGE) and the medial prefrontal cortex. A single investigator performed the cell counts without knowledge of the identity of the treatment group to which a given section belonged. We analyzed two or three embryos obtained from at least two litters from each of the four prenatal treatment groups, utilizing four sections from each brain region studied. Preliminary analyses showed that the sampling procedure produced SEM values that were <10% of the mean value for a given experimental group. Increasing the number of sections from four to six per brain, the number of brains from two to three per litter or increasing the number of litters from two to three per experimental group did not reduce the SEM values further.
Figure 9.
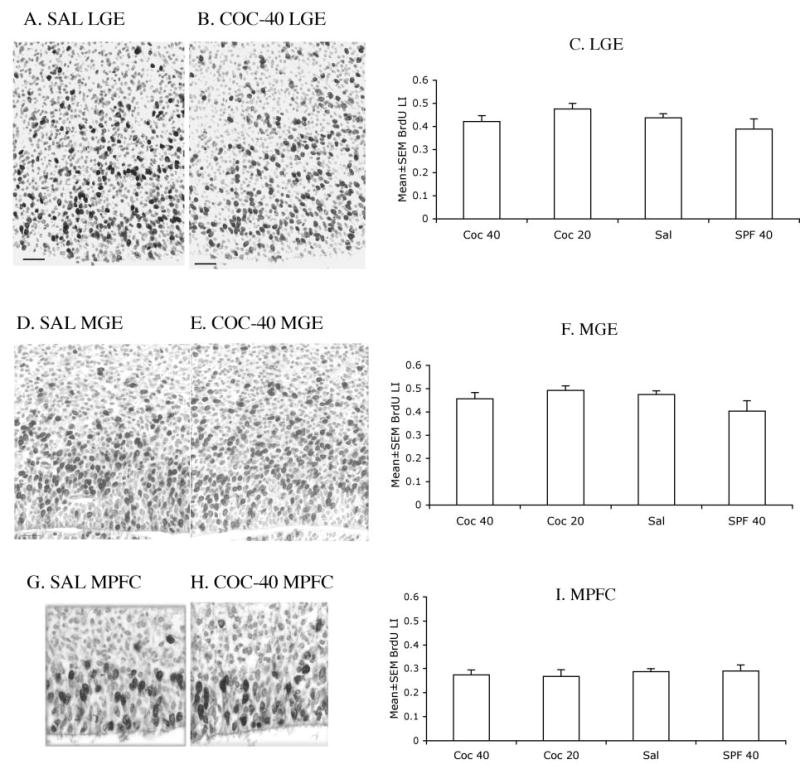
Photomicrographs of 4 μm thick, paraffin embedded coronal sections of the lateral ganglionic eminence (LGE; A, B), medial ganglionic eminence (MGE; D, E) and medial prefrontal cortex (MPFC; G, H) from E15 mice in the SAL (A, D, G) and COC-40 (B, E, H) groups. The sections were processed for BrdU immunohistochemistry using cobalt-nickel intensification of the diaminobenzidine reaction product and stained with basic fuchsin. BrdU-labeled nuclei appear black and the basic fuchsin stained nuclei appear pale gray. The micrographs are oriented such that the ventricular border is at the bottom and the pial surface toward the top. Sections such as these were used to calculate the BrdU labeling indices. Mean ± SEM values for 2.0 hr BrdU labeling index in the LGE (C), MGE (F) and MPFC (I) are shown. The differences among the groups were not statistically significant (P > 0.05). Four sections from each region of each of four or five brains per experimental group were analyzed.
Estimation of Cell Packing Density
We used the basic fuchsin stained 4 μm thick, paraffin-embedded coronal sections of the embryonic brains to calculate the number of nuclei within a 120 × 120 μm2 sector of the medial prefrontal cortex, LGE and MGE. A single investigator performed the cell counts without knowledge of the identity of the treatment group to which a given section belonged. We analyzed two or three embryos obtained from at least two litters from each of the four prenatal treatment groups, utilizing four sections from each brain region studied.
Statistical Analysis
Measurements were compared statistically using two-way analysis of variance (ANOVA). In instances where the two-way ANOVA established main effects of prenatal cocaine or malnutrition to be statistically significant (P < 0.05), pairwise comparisons of the various prenatal treatment groups were performed using the Tukey–Kramer test.
Results
Neuronal Migration
The DiI injection site in the ganglionic eminence/developing striatum could be identified by the presence of a very high density of labeled cells and intense fluorescence (Fig. 1). The size of the injection was not significantly different among the four experimental groups as shown by two-way ANOVA [F(3,31) = 0.118, P = 0.9982]. DiI-labeled cells migrated from the injection site into the striatum as well as the cerebral wall during the 2 day culture period (Fig. 1).
The graphic depictions of the extent of neuronal migration obtained using the grid box overlay method (Fig. 1C,D) indicated a marked reduction in the overall migration of labeled cells in the COC-40 and COC-20 groups compared to the SAL and SPF-40 groups. To quantify the extent of neuronal migration from the ganglionic eminence to the cerebral wall, we calculated the percentage of DiI-labeled cells that had entered the cerebral wall. There was a significant reduction in the percentage of DiI-labeled cells entering the cerebral wall in the COC-40 and COC-20 groups compared to the SAL and SPF-40 groups (Fig. 2). Two-way ANOVA of the data showed a significant main effect of prenatal cocaine exposure on this measurement (F = 63.35, P = 0.0001), but no significant effect of malnutrition (F = 3.65, P > 0.05), nor a significant interaction between prenatal cocaine and malnutrition (F = 0.48, P > 0.05). The Tukey–Kramer test indicated that both the COC-40 and COC-20 groups showed significantly lower values compared to both the SAL and SPF-40 groups (Fig. 2A). Thus, prenatal cocaine exposure, but not malnutrition, significantly reduced the percentage of cells entering the cerebral wall. The difference between the COC-40 and the COC-20 groups was not statistically significant (Tukey–Kramer pairwise comparison, data not shown).
Numerical Density of GABA-Positive Cells
Since most of the tangentially migrating neurons entering the cerebral wall and rostrally migrating neurons entering the olfactory bulb are GABAergic and since prenatal cocaine exposure appeared to reduce tangential neuronal migration from the ganglionic eminence to the cerebral wall, we calculated the numerical density of GABA-positive cells in the presumptive medial prefrontal and somatosensory cortices and the olfactory bulb. Visual inspection of the sections processed for GABA immunohistochemistry revealed a reduction in the numerical density of GABA-positive cells in the medial prefrontal and somatosensory cortices of the COC-40 and COC-20 groups compared to the SAL and SPF-40 groups (Fig. 3C,D). No such changes were evident by visual inspection of the sections of the olfactory bulb (Fig. 3E,F).
Figure 3.
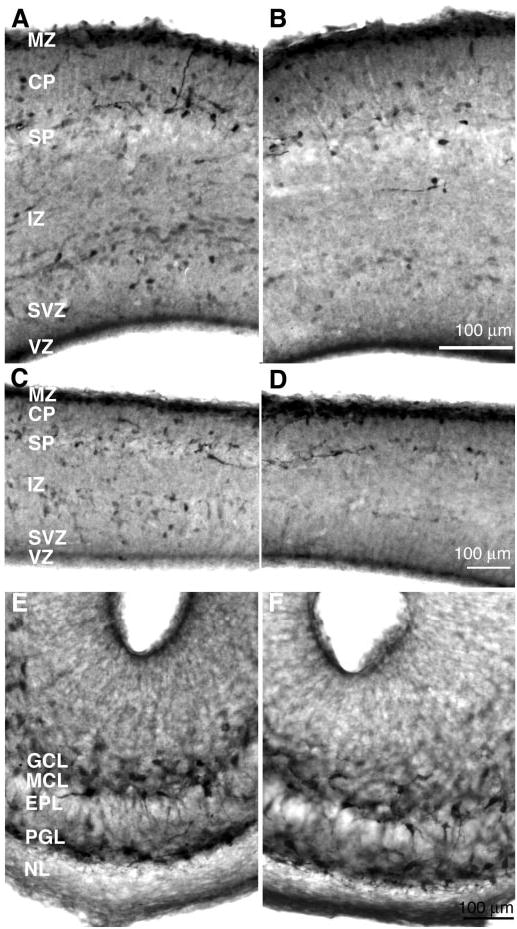
Photomicrographs of coronal sections through the medial prefrontal cortex (A, B), somatosensory cortex (C, D) and olfactory bulb (E, F) of E15 mouse embryos from the SAL (left panel) and COC-40 (right panel) groups, processed for GABA immunohistochemistry. GABA positive cells appear dark and are distributed throughout the cerebral wall and olfactory bulb. There are fewer GABA-positive profiles in the COC-40 group compared to the SAL group in the medial prefrontal and somatosensory regions. The numbers of GABA-positive profiles appear to be similar in the olfactory bulbs of the SAL and COC-40 mice. Abbreviations: VZ = ventricular zone; SVZ = subventricular zone; IZ = intermediate zone; SP = subplate; CP = cortical plate; MZ = marginal zone; GCL = glomerular cell layer; MCL = mitral cell layer; EPL = external plexiform layer; PGL = periglomerular layer; NL = nuclear layer.
Next, we compared the numerical densities of GABA-positive cells in the three regions among the four experimental groups. The data were analyzed by using two-way ANOVA. There was a significant main effect of cocaine treatment on GABA-positive cell numerical density in the medial prefrontal and somatosensory cortices (medial prefrontal cortex: F = 44.38, P = 0.0001; somatosensory cortex: F = 95.98, P = 0.0001), but no significant main effect of malnutrition (medial prefrontal cortex: F = 0.55, P > 0.05; somatosensory cortex: F = 0.39, P > 0.05). Interaction between prenatal cocaine and malnutrition was not significant (medial prefrontal cortex: F = 1.29, P > 0.05; somatosensory cortex: F = 2.4, P > 0.05). In contrast, neither prenatal cocaine exposure nor malnutrition produced significant main effects on GABA-positive cell numerical density in the olfactory bulb (cocaine: F = 0.004, P > 0.05; malnutrition: F = 0.478, P > 0.05).
The Tukey-Kramer test indicated that the GABA-positive cell numerical densities in both the COC-20 and COC-40 groups were significantly lower than those in the SAL and the SPF-40 groups in both the medial prefrontal (Fig. 4A) and somatosensory (Fig. 4B) cortices. The difference between the COC-40 and the COC-20 groups was not statistically significant (Tukey–Kramer pairwise comparison, data not shown). As mentioned earlier, there was no significant difference among the four groups in the olfactory bulb (Fig. 4C). Thus, prenatal cocaine exposure, but not malnutrition, significantly decreased the numerical density of GABA-positive cells in the two cortical regions. Neither cocaine nor malnutrition produced significant effects in the olfactory bulb.
Figure 4.
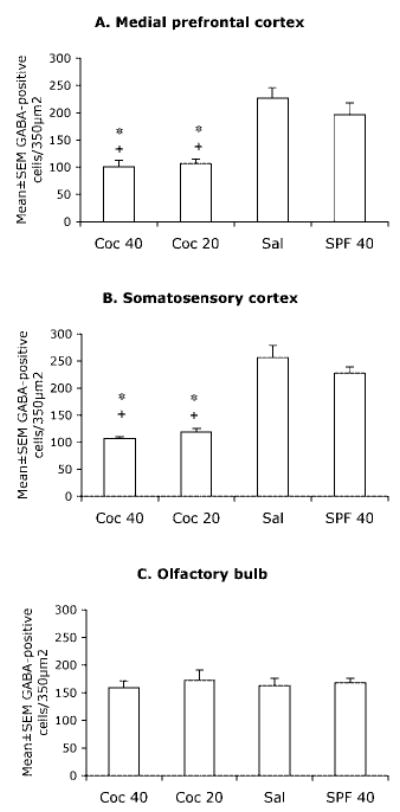
Mean ± SEM values for numerical densities of GABA-positive cells in the presumptive medial prefrontal cortex (A), somatosensory cortex (B) and the olfactory bulb (C) of E15 mice in the four experimental groups. * = value is significantly different (P < 0.05) from the SAL group and + = value is significantly different from the SPF-40 group. Statistical significance between two groups was analyzed using the Tukey–Kramer test. Sections from 5–7 brains were analyzed for each experimental group.
GABA-positive cells are distributed throughout the radial extent of the prefrontal cortex, somatosensory cortex and the olfactory bulb (Fig 3). The radial dimensions of these regions can be divided into laminae based on histological criteria. We analyzed the GABA-positive cell numerical densities separately in the different laminae of the three regions and compared the data by using two-way ANOVA. We found a significant effect of prenatal cocaine treatment in the VZ/SVZ, IZ and SP/CP laminae in both the medial prefrontal and somatosensory cortices (medial prefrontal cortex, VZ/SVZ: F = 25.19, P = 0.0001; IZ: F = 37.98, P = 0.0001; SP/CP: F = 29.67, P = 0.0001; somatosensory cortex, VZ/SVZ: F = 14.4, P = 0.001; IZ: F = 46.16, P = 0.267; SP/CP: F = 50.37, P = 0.001). However, in those regions, there was no significant effect of malnutrition (medial prefrontal cortex, VZ/SVZ: F = 1.30, P > 0.05; IZ: F = 2.06, P > 0.05; SP/CP: F = 0.61, P > 0.05; somatosensory cortex, VZ/SVZ: F = 3.25, P > 0.05; IZ: F = 1.49, P > 0.05; SP/CP: F = 0.452, P > 0.05), or an interaction between cocaine and malnutrition.
Two-way ANOVA showed a significant main effect of prenatal cocaine exposure (F = 18.1, P = 0.004) and malnutrition (F = 7.52, P = 0.012) in the MZ of the somatosensory cortex. Neither prenatal cocaine nor malnutrition showed significant main effects in the MZ of the medial prefrontal cortex (cocaine: F = 3.29, P > 0.05; malnutrition: F = 1.86, P > 0.05), nor was a significant interaction of cocaine and malnutrition evident in the MZ in either cortical region.
Finally, two-way ANOVA of the data showed that neither prenatal cocaine exposure nor malnutrition produced significant main effects in GABA-positive cell numerical density in any lamina of the olfactory bulb.
We performed the Tukey–Kramer test to determine if the differences between any two given experimental groups were statistically significant. This test indicated that the GABA-positive cell numerical density was significantly lower in both the COC-20 and COC-40 groups compared to the SAL group in IZ and SP/CP laminae of the medial prefrontal and somatosensory cortices (Figs 5B,C and 6B,C, respectively).
Figure 5.
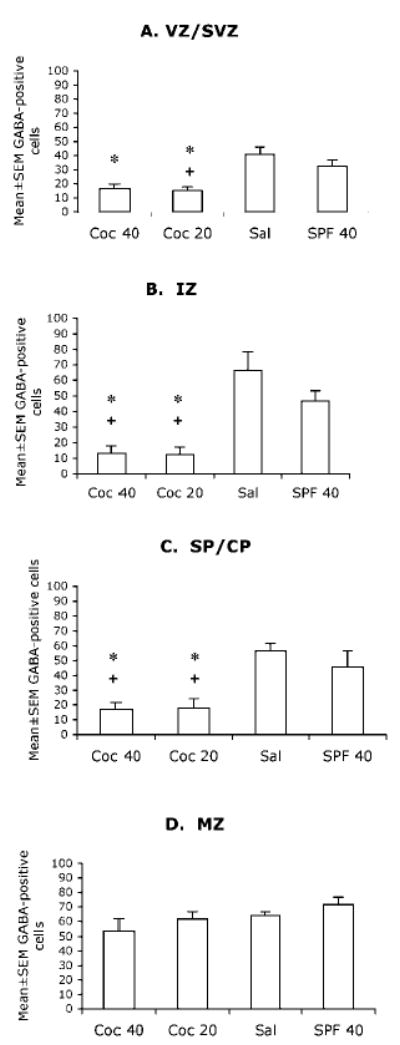
Mean ± SEM values for numerical densities of GABA-positive cells in the different laminae of the presumptive medial prefrontal cortical region of E15 mice in the four experimental groups. VZ/SVZ = ventricular and subventricular zones; IZ = intermediate zone; SP/CP = subplate and cortical plate; MZ = marginal zone. * = value is significantly different (P < 0.05) from the SAL group and + = value is significantly different from the SPF-40 group. Statistical significance between two groups was analyzed using the Tukey–Kramer test. Sections from 5–7 brains were analyzed for each experimental group.
The GABA-positive cell numerical density was lower in the COC-20 and COC-40 groups compared to the SAL group in the VZ of the medial prefrontal (Fig. 5A) and somatosensory cortices (Fig. 6A). The COC-20 group showed significantly lower GABA-cell numerical density compared to the SPF-40 group in the VZ of the medial prefrontal cortex (Fig. 5A) but not the VZ of the somatosensory cortex (Fig. 6A).
Figure 6.

Mean ± SEM values for numerical densities of GABA-positive cells in the different laminae of the presumptive somatosensory cortical region of E15 mice in the four experimental groups. VZ/SVZ = ventricular and subventricular zones; IZ = intermediate zone; SP/CP = subplate and cortical plate; MZ = marginal zone. * = value is significantly different (P < 0.05) from the SAL group and + = value is significantly different from the SPF-40 group. Statistical significance between two groups was analyzed using the Tukey-Kramer test. Sections from 5–7 brains were analyzed for each experimental group.
There were no significant differences in the numerical density of GABA-positive cells in the MZ of the medial prefrontal cortex among the four groups (Fig. 5D). However, the MZ of the somatosensory cortex showed significantly lower GABA-positive cell numerical density in the COC-40 group compared to the SAL and SPF-40 groups (Fig. 6D).
The GABA-positive cell numerical densities were not significantly different among the four groups in any of the laminae of the olfactory bulb (Fig. 7).
Figure 7.
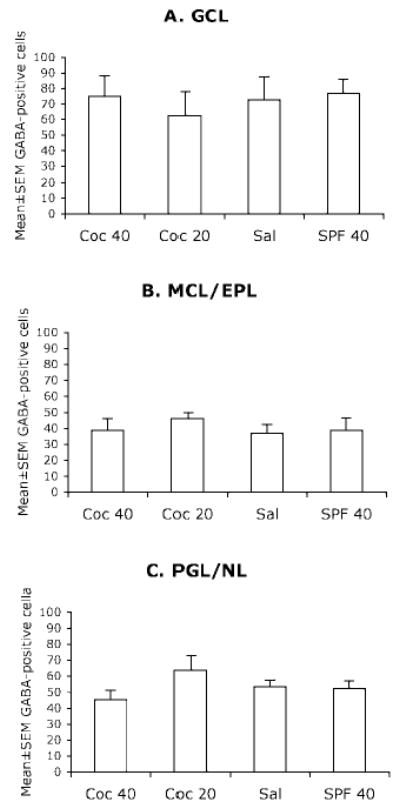
Mean ± SEM values for numerical density of GABA-positive cells in the different laminae of the olfactory bulb of E15 mice in the four experimental groups. GCL = glomerular cell layer; MCL/EPL = mitral cell layer and external plexiform layer; PGL/NL = periglomerular layer and nerve layer. The differences among the 4 groups were not statistically significant (P > 0.05). Sections from 5–7 brains were analyzed for each experimental group.
The differences between SAL and SPF-40 or between COC-20 and COC-40 groups were not statistically significant in any lamina of any of the three regions analyzed (Tukey–Kramer pairwise comparison, data not shown).
The Thickness of the Cerebral Wall and the Olfactory Bulb
Since the numerical density of GABA-positive cells showed significant differences in the medial prefrontal and somatosensory cortices between the four experimental groups, we examined if the radial dimension (i.e. thickness) of those two cortical regions differed among the four prenatal treatment groups. The average thickness of the medial prefrontal and somatosensory cortices was similar in the four prenatal treatment groups when it was analyzed for the entire region (Fig. 8), or separately for each sublamina comprising those regions (data not shown). Two-way ANOVA of the data showed that neither prenatal cocaine treatment nor malnutrition produced significant changes in the thickness of these three regions (medial prefrontal cortex; cocaine: F = 0.24, P > 0.05; malnutrition: F = 1.5, P > 0.05; somatosensory cortex; cocaine: F = 1.8, P > 0.05; malnutrition: F = 0.02, P > 0.05; olfactory bulb; cocaine: F = 0.09, P ≥ 0.05; malnutrition: F = 0.01, P ≥ 0.05).
Figure 8.
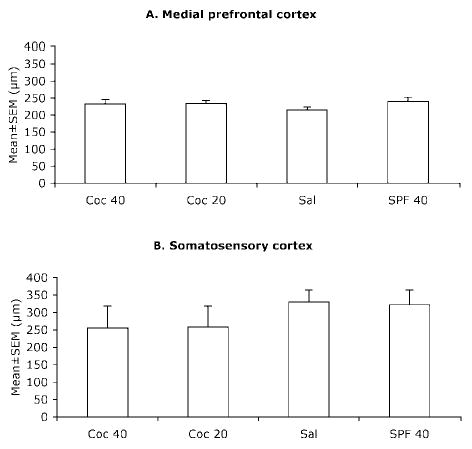
Mean ± SEM values for the radial dimension (thickness) of the presumptive medial prefrontal cortex (A) and the somatosensory cortex (B) of E15 mice in the four experimental groups. The differences were not statistically significant (P > 0.05). Sections from 5–7 brains were analyzed for each experimental group.
BrdU Labeling Index
The 2.0 h BrdU LI was similar in the LGE, MGE and the medial prefrontal cortex in the four prenatal treatment groups (Fig. 9). Two-way ANOVA of the data showed that neither cocaine treatment nor malnutrition produced significant changes in the BrdU LI in any of the three regions analyzed (LGE: cocaine: F =1.67, P > 0.05; malnutrition: F = 0.009, P > 0.05; MGE: cocaine: F = 1.62, P > 0.05; malnutrition: F = 0.41, P > 0.05; MPFC: cocaine: F = 0.873, P > 0.05; malnutrition: F = 0.0003, P > 0.05). There was no significant interaction between prenatal cocaine and malnutrition with regard to changes in BrdU LI in any of the regions analyzed (LGE: F = 3.44, P > 0.05; MGE: F = 3.85, P > 0.05; MPFC: F = 0.01, P > 0.05).
Cell Packing Density
The cell packing density was not significantly different between the four experimental groups in the medial prefrontal cortex, LGE or the MGE (LGE: F = 1.64; P > 0.05; MGE: F = 0.34; P > 0.05; medial prefrontal cortex: F = 0.49; P > 0.05).
Discussion
We show that exposure of the developing mouse brain to cocaine from E8 to E15 reduces neuronal migration from the ganglionic eminence to the cerebral cortex and produces deficits in GABA-positive cell numerical densities in the cerebral wall at E15. The olfactory bulb, a region that receives GABA-ergic neurons from the LGE, does not show significant effects of cocaine exposure on GABA neuron numerical density at E15. Therefore, prenatal cocaine exposure selectively targets tangential neuronal migration to the cerebral wall.
The majority of cells entering the cerebral wall from the ganglionic eminence via the tangential migratory pathway are GABAergic neurons derived from the MGE/CGE (Wichterle et al., 1999, 2001; Parnavelas, 2000; Nery et al., 2002), although at later stages (E14–E16) some LGE-derived GABAergic neurons also enter the cerebral wall (Anderson et al., 2001). The DiI placement in our experiments (Fig. 1) could label the MGE-, LGE- and CGE-derived populations. However, since the majority of neurons entering the cerebral wall are MGE/CGE-derived cells, our data indicate decreased neuronal migration from the MGE/CGE to the cerebral wall.
In addition to the decrease in the percentage of DiI-labeled cells entering the cerebral wall, we found significant reductions in GABA-positive cell numerical densities in both the medial prefrontal and somatosensory regions of the cerebral wall in the cocaine-exposed groups. The early (E12–E14)-versus late (E15)-generated GABA neurons or MGE/CGE- versus LGE-derived GABA-positive neurons may follow different migration trajectories and may populate the different laminae of the cerebral wall (Anderson et al., 1999, 2001). Prenatal cocaine exposure may affect the different populations differently. Therefore, we examined the numerical density of GABA neurons separately in the different laminae of the cerebral wall. We found that the COC-20 and COC-40 groups had significantly lower GABA-positive cells in the IZ and SP/CP laminae compared to the two control groups (SAL and SPF-40) in both the medial prefrontal (Fig. 5B,C) and somatosensory cortices (Fig. 6B,C). The VZ and the MZ laminae showed significant reductions in one cortical area only when the SAL group was compared with the cocaine groups. Thus, the overall effects of cocaine exposure on GABA-positive cell numerical densities appear to be robust in the IZ and SP/CP laminae and less robust in the VZ and MZ laminae. The reasons for the differences in the degree of vulnerabilities of the different laminae and regions (i.e. medial prefrontal versus somatosensory) are unclear. It is possible that GABAergic cell migration within the cerebral wall (Nadarajah et al., 2002) also is affected by the cocaine exposure such that the final laminar destinations of the GABAergic cells are altered. In any event, it appears that GABAergic neurons in all laminae of the cerebral wall are not affected uniformly by the cocaine exposure.
We did not find significant effects of prenatal cocaine exposure on the numerical density of GABA-positive cells in the olfactory bulb indicating that cocaine exposure selectively influenced MGE/CGE-to-cortex tangential neuronal migration and not LGE-to-olfactory bulb rostral neuronal migration. The mechanistic basis of such selectivity remains unknown.
Neuronal migration from the ganglionic eminence to other telencephalic regions is regulated by transcription factors such as Dlx1/2, Nkx 2.1 and Mash-1 (Anderson et al., 1997; Sussel et al., 1999; Corbin et al., 2000; Marin et al., 2000), signaling molecules such as hepatocyte growth factor/scatter factor (Powell et al., 2001, 2003), as well as by extracellular matrix molecules such as Tag-1 (Denaxa et al., 2001). Whether prenatal cocaine exposure interferes with the expression of any of those factors is not known. Another potential mechanism involved with the effects of prenatal cocaine exposure is the activity of neurotransmitter receptors, which are implicated in neuronal migration (Behar et al., 1998, 1999). Cocaine’s effects also may be mediated via its effects on cyclin dependent kinase 5 (Bibb et al., 2001), which is known to influence neuronal migration (Chae et al., 1997; Gilmore et al., 1998; Kwon and Tsai, 1998; Dhavan et al., 2002).
The cocaine-induced decreases in neuronal migration were observed following 2 days in vitro during which time the slices were not exposed to cocaine. In other words, the migration defect persisted for at least 2 days after the final administration of cocaine. Thus, the effects of cocaine exposure on tangential neuronal migration appear to be relatively long lasting. A number of the effects of prenatal cocaine exposure on the CNS, especially changes in neurotransmitter receptors and cortical cytoarchitecture last several weeks to months after the final exposure (Gressens et al., 1992a; H.Y. Wang et al., 1995; Friedman et al., 1996; Friedman and Wang, 1998; Lidow and Song, 2001).
It is possible that prenatal cocaine exposure affected cell proliferation in the LGE and MGE such that fewer cells were generated in those regions, contributing to decreased neuronal migration. Likewise, it is possible that prenatal cocaine exposure affected cell proliferation in the cerebral wall, such that fewer cells were generated in those regions, which could also contribute to alterations in the GABAergic neuron numerical densities in those areas. Several reports show that exposure to cocaine influences cell proliferation in the embryonic brain (Anderson-Brown et al., 1990; Lidow and Song, 2001). Acute and chronic exposures to cocaine can produce different, even opposing, effects on cell proliferation (Anderson-Brown et al., 1990; Lidow and Song, 2001). In the present study BrdU was administered ~14 h after the final exposure to cocaine. The 2.0 h BrdU LI in the LGE, MGE and the cerebral wall did not show significant effects of prenatal cocaine exposure. It is possible that changes in the BrdU LI occurred immediately following one or more cocaine administrations and/or that the recurrent exposure to cocaine from E8 to E14 reduced the response of the proliferating cells by E15. Our cocaine- and BrdU-exposure paradigm would not detect such changes. A detailed analysis of cell cycle kinetics following acute and long-term exposures to cocaine is beyond the scope of the present study and will be required to determine if cocaine exposure alters neurogenesis in our mouse model. We recognize that changes in cell cycle kinetics can occur following cocaine administration and that such changes, even if only transient can alter the number of cells produced by the telencephalic neuroepithelium.
An alternative explanation for the reduced tangential neuronal migration and decreased GABA-positive cell numerical densities observed in the present study may be that the cocaine exposure caused selective death of GABAergic neurons in the LGE/MGE and/or the cerebral wall. Prenatal cocaine exposure is known to increase cell death in the fetal monkey cerebral wall (He et al., 1999). We did not observe any histological evidence of augmented cell death (e.g. higher incidence of pyknotic or swollen nuclei) in the sections of the embryonic brains from either the COC-20 or the COC-40 groups. If cell death had been augmented by the cocaine exposure then the cell packing density should be reduced in those two groups (He et al., 1999). We did not find any evidence of changes in cell packing density in the cerebral wall, LGE or MGE. The thickness of the cerebral wall was also unchanged in the four experimental groups. Therefore, we suggest that in our experimental paradigm, cell death did not contribute significantly to the changes that were observed in neuronal migration or the numerical density of GABA-positive cells.
In every measurement that showed significant main effects of the cocaine exposure, both the COC-40 and COC-20 groups demonstrated significantly lower values compared to both the SPF-40 and the SAL groups. Moreover, there were no significant differences between COC-20 and COC-40 groups in any of the measurements. The dose-independent nature of the cocaine effect (i.e. COC-20 indistinguishable from COC-40) combined with the lack of a main effect of malnutrition (two-way ANOVA) support our conclusion that the deficits in tangential neuronal migration and GABAergic neuronal numerical density were direct effects of prenatal cocaine exposure and not the indirect effects of malnutrition.
Prenatal cocaine exposure, from implantation to birth, can cause permanent alterations in cortical GABAergic circuitry in rabbits (X.-H. Wang et al., 1995; Jones et al., 1996; Stanwood et al., 2001a,b). These permanent changes are specific to dopamine-rich areas of the cerebral cortex such as the anterior cingulate cortex, medial prefrontal cortex, entorhinal cortex and the piriform cortex (Stanwood et al., 2001b), and are additionally reflected by changes in the morphology of apical dendrites of projection neurons intrinsic to those areas (Jones et al., 1996). The correlation between dopaminergic inputs and prenatal cocaine-induced changes in GABA circuitry reported by those investigators present a compelling case for a role for the dopaminergic system in mediating cocaine’s effects. The ganglionic eminence and striatal differentiating fields receive rich dopaminergic inputs and express dopamine receptors as early as E13 (Ohtani et al., 2003). Therefore, the GABAergic neurons embark upon their migratory route in a dopamine-rich milieu. In fact, cocaine exposure reduces coupling of fetal dopaminergic receptors to their G-protein partners and attenuates dopaminergic signaling (H.Y. Wang et al., 1995; Friedman et al., 1996; Zhen et al., 2001). In contrast, our data show deficits in GABA neuron distribution in the dopamine-rich prefrontal region as well as the relatively dopamine-sparse somatosensory region of the E15 cerebral wall. In other words, during the embryonic period the effects of prenatal cocaine exposure on GABA neuron distribution may not be specific to dopamine-rich regions of the cerebral wall, but may be more global. However, a direct link between prenatal cocaine exposure and dopaminergic mechanisms in neuronal migration remains to be established.
The number of GABAergic neurons increased in all laminae of the anterior cingulate cortex (X.-H. Wang et al., 1995) and medial prefrontal cortex (Stanwood et al., 2001b) but not the visual cortex of adult rabbits exposed to cocaine prenatally (Stanwood et al., 2001b). However, the number or laminar distribution of parvalbumin containing neurons, which are a subset of the GABAergic neurons were unaffected in both areas (Wang et al., 1996). Our data show deficits in GABAergic neurons in the embryonic mouse cerebral wall. We do not know if the deficits persist into adulthood, nor do we know if changes occur in parvalbumin positive interneurons. Therefore, a direct comparison between our data and the data from the mature rabbit is not possible. However, when data on the long-term effects of prenatal cocaine exposure on GABA-positive cell numerical density and distribution in our mouse model become available, a comparison would be informative. A recent report (Morrow et al., 2003) showed decreased length and density of GABA-positive axo-axonic contacts in the medial prefrontal cortex of adult rats exposed to intravenous cocaine in the embryonic period. However, the number of parvalbumin containing neurons, which give rise to the axo-axonic contacts, was unaffected.
Therefore, whether the more global deficits in cortical GABA neuron numbers in our study are species-specific or specific to route of prenatal cocaine administration (subcutaneous versus intravenous), or whether such changes are permanent is unknown. However, the decreased GABAergic neuronal migration and deficits in GABAergic neuron numbers suggest a developmental mechanism for cocaine-induced alterations in cortical inhibitory circuitry.
In summary, our data show that prenatal cocaine exposure decreases neuronal migration from the ganglionic eminence to the cerebral wall and decreases the density of GABAergic neurons in multiple regions of the cerebral wall. GABAergic neurons produced in the ganglionic eminence are critical components of inhibitory circuitry in multiple regions of the telencephalon including the cerebral cortex, hippocampus, amygdala and nucleus accumbens (Nery et al., 2002). Many of these regions are critical components of the reward-mediating pathways and mechanisms. Therefore, disruption of neuronal migration from the ganglionic eminence could produce widespread deficits in inhibitory circuitry in multiple areas of the brain. Whether such deficits may contribute to the structural and functional neurological changes associated with prenatal cocaine exposure evident in experimental animals and humans will be an important topic for further study.
Footnotes
Supported by USPHS grants HD 05515 (J.E.C.), DA 08648 (B.E.K.), DA 00354 (B.E.K.), MH57748 (S.A.T.) and NS 43426 (P.G.B).
References
- Akbari HM, Kramer HK, Whitaker-Azmitia PM, Spear LP, Azmitia EC. Prenatal cocaine exposure disrupts the development of the serotonergic system. Brain Res. 1992;572:57–63. doi: 10.1016/0006-8993(92)90450-n. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JLR. Interneuron migration from basal forebrain to neocortex: sependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Mione MC, Yun K, Rubenstein JLR. Differential origins of neocortical projection and local circuit neurons: role of Dlx genes in neocortical interneuronogenesis. Cereb Cortex. 1999;9:646–654. doi: 10.1093/cercor/9.6.646. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Marin O, Horn C, Jennings K, Rubenstein JL. Distinct cortical migrations from the medial and lateral ganglionic eminences. Development. 2001;128:353–363. doi: 10.1242/dev.128.3.353. [DOI] [PubMed] [Google Scholar]
- Anderson-Brown T, Slotkin TA, Seidler FJ. Cocaine acutely inhibits DNA synthesis in developing rat brain regions: evidence for direct actions. Brain Res. 1990;537:197–202. doi: 10.1016/0006-8993(90)90358-i. [DOI] [PubMed] [Google Scholar]
- Ang ES, Jr, Haydar TF, Gluncic V, Rakic P. Four-dimensional migratory coordinates of GABAergic interneurons in the developing mouse cortex. J Neurosci. 2003;23:5805–5815. doi: 10.1523/JNEUROSCI.23-13-05805.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar TN, Schaffner AE, Scott CA, O’Connell C, Barker JL. Differential response of cortical plate and ventricular zone cells to GABA as a migration stimulus. J Neurosci. 1998;18:6378–6387. doi: 10.1523/JNEUROSCI.18-16-06378.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar TN, Scott CA, Greene CL, Wen X, Smith SV, Maric D, Liu QY, Colton CA, Barker JL. Glutamate acting at NMDA receptors stimulates embryonic cortical neuronal migration. J Neurosci. 1999;19:4449–4461. doi: 10.1523/JNEUROSCI.19-11-04449.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhide PG. Cell cycle kinetics in the embryonic mouse corpus striatum. J Comp Neurol. 1996;374:506–522. doi: 10.1002/(SICI)1096-9861(19961028)374:4<506::AID-CNE3>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Bhide PG, Frost DO. Stages of growth of hamster retinofugal axons: implications for developing axonal pathways with multiple targets. J Neurosci. 1991;11:485–504. doi: 10.1523/JNEUROSCI.11-02-00485.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibb JA, Chen J, Taylor JR, Svenningsson P, Nishi A, Snyder GL, Yan Z, Sagawa ZK, Ouimet CC, Nairn AC, Nestler EJ, Greengard P. Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature. 2001;410:376–380. doi: 10.1038/35066591. [DOI] [PubMed] [Google Scholar]
- Bongarzone ER, Howard SG, Schonmann V, Campagnoni AT. Identification of the dopamine D3 receptor in oligodendrocyte precursors: potential role in regulating differentiation and myelin formation. J Neurosci. 1998;18:5344–5353. doi: 10.1523/JNEUROSCI.18-14-05344.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases O, Seif I, Grimsby J, Gaspar P, Chen K, Pournin S, Muller U, Aguet M, Babinet C, Shih JC, De Maeyer E. Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science. 1995;268:1763–1766. doi: 10.1126/science.7792602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases O, Vitalis T, Seif I, De Maeyer E, Sotelo C, Gaspar P. Lack of barrels in the somatosensory cortex of monoamine oxidase A-deficient mice: role of a serotonin excess during the critical period. Neuron. 1996;16:297–307. doi: 10.1016/s0896-6273(00)80048-3. [DOI] [PubMed] [Google Scholar]
- Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- Corbin JG, Gaiano N, Machold RP, Langston A, Fishell G. The Gsh2 homeodomain gene controls multiple aspects of telen-cephalic development. Development. 2000;127:5007–5020. doi: 10.1242/dev.127.23.5007. [DOI] [PubMed] [Google Scholar]
- Denaxa M, Chan CH, Schachner M, Parnavelas JG, Karagogeos D. The adhesion molecule TAG-1 mediates the migration of cortical interneurons from the ganglionic eminence along the corticofugal fiber system. Development. 2001;128:4635–4644. doi: 10.1242/dev.128.22.4635. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Greer PL, Morabito MA, Orlando LR, Tsai LH. The cyclin-dependent kinase 5 activators p35 and p39 interact with the alpha-subunit of Ca2+/calmodulin-dependent protein kinase II and alpha-actinin-1 in a calcium-dependent manner. J Neurosci. 2002;22:7879–7891. doi: 10.1523/JNEUROSCI.22-18-07879.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman E, Wang HY. Prenatal cocaine exposure alters signal transduction in the brain D1 dopamine receptor system. Ann N Y Acad Sci. 1998;846:238–247. doi: 10.1111/j.1749-6632.1998.tb09741.x. [DOI] [PubMed] [Google Scholar]
- Friedman E, Yadin E, Wang HY. Effect of prenatal cocaine on dopamine receptor-G protein coupling in mesocortical regions of the rabbit brain. Neuroscience. 1996;70:739–747. doi: 10.1016/s0306-4522(96)83011-9. [DOI] [PubMed] [Google Scholar]
- Gilmore EC, Ohshima T, Goffinet AM, Kulkarni AB, Herrup K. Cyclin-dependent kinase 5-deficient mice demonstrate novel developmental arrest in cerebral cortex. J Neurosci. 1998;18:6370–6377. doi: 10.1523/JNEUROSCI.18-16-06370.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gressens P, Gofflot F, Van Maele-Fabry G, Misson J-P, Gadisseux J-F, Evrard P, Picard JJ. Early neurogenesis and teratogenesis in whole mouse embryo cultures. Histochemical, immunocytological and ultrastructural study of the premigratory neuronal-glial units in normal mouse embryo and in mouse embryos influenced by cocaine and retinoic acid. J Neuropathol Exp Neurol. 1992a;51:206–219. doi: 10.1097/00005072-199203000-00010. [DOI] [PubMed] [Google Scholar]
- Gressens P, Kosofsky BE, Evrard P. Cocaine-induced disturbances of corticogenesis in the developing murine brain. Neurosci Lett. 1992b;140:113–116. doi: 10.1016/0304-3940(92)90694-3. [DOI] [PubMed] [Google Scholar]
- He N, Song Z, Lidow MS. Cocaine induces cell death within the primate fetal cerebral wall. Neuropathol Appl Neurobiol. 1999;25:504–512. doi: 10.1046/j.1365-2990.1999.00211.x. [DOI] [PubMed] [Google Scholar]
- Jones L, Fischer I, Levitt P. Nonuniform alteration of dendritic development in the cerebral cortex following prenatal cocaine exposure. Cereb Cortex. 1996;6:431–445. doi: 10.1093/cercor/6.3.431. [DOI] [PubMed] [Google Scholar]
- Jones LB, Stanwood GD, Reinoso BS, Washington RA, Wang HY, Friedman E, Levitt P. In utero cocaine-induced dysfunction of dopamine D1 receptor signaling and abnormal differentiation of cerebral cortical neurons. J Neurosci. 2000;20:4606–4614. doi: 10.1523/JNEUROSCI.20-12-04606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosofsky BE, Wilkins AS, Gressens P, Evrard P. Transplacental cocaine exposure: A mouse model demonstrating neuroanatomic and behavioral abnormalities. J Child Neurol. 1994;9:234–241. doi: 10.1177/088307389400900303. [DOI] [PubMed] [Google Scholar]
- Kwon YT, Tsai LH. A novel disruption of cortical development in p35−/− mice distinct from reeler. J Comp Neurol. 1998;395:510–522. doi: 10.1002/(sici)1096-9861(19980615)395:4<510::aid-cne7>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Lauder JM. Neurotransmitters as morphogens. Prog Brain Res. 1988;73:365–387. doi: 10.1016/S0079-6123(08)60516-6. [DOI] [PubMed] [Google Scholar]
- Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci. 1999;99:7881–7888. doi: 10.1523/JNEUROSCI.19-18-07881.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt P, Harvey JA, Friedman E, Simansky K, Murphy EH. New evidence for neurotransmitter influences on brain development. Trends Neurosci. 1997;20:269–274. doi: 10.1016/s0166-2236(96)01028-4. [DOI] [PubMed] [Google Scholar]
- Lidow MS. Prenatal cocaine exposure adversely affects development of the primate cerebral cortex. Synapse. 1995;21:332–341. doi: 10.1002/syn.890210408. [DOI] [PubMed] [Google Scholar]
- Lidow MS, Song ZM. Effect of cocaine on cell proliferation in the cerebral wall of monkey fetuses. Cereb Cortex. 2001;11:545–551. doi: 10.1093/cercor/11.6.545. [DOI] [PubMed] [Google Scholar]
- Lidow MS, Bozian D, Song Z. Cocaine affects cerebral neocortical cytoarchitecture in primates only if administered during neocortical neuronogenesis. Brain Res Dev Brain Res. 2001;128:45–52. doi: 10.1016/s0165-3806(01)00139-0. [DOI] [PubMed] [Google Scholar]
- Marin O, Anderson SA, Rubenstein JL. Origin and molecular specification of striatal interneurons. J Neurosci. 2000;20:6063–6076. doi: 10.1523/JNEUROSCI.20-16-06063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Yaron A, Bagri A, Tessier-Lavigne M, Rubenstein JL. Sorting of striatal and cortical interneurons regulated by semaphorin- neuropilin interactions. Science. 2001;293:872–875. doi: 10.1126/science.1061891. [DOI] [PubMed] [Google Scholar]
- Mason HA, Ito S, Corfas G. Extracellular signals that regulate the tangential migration of olfactory bulb neuronal precursors: inducers, inhibitors, and repellents. J Neurosci. 2001;21:7654–7663. doi: 10.1523/JNEUROSCI.21-19-07654.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayes LC. Developing brain and in utero cocaine exposure: effects on neural ontogeny. Dev Psychopathol. 1999;11:685–714. doi: 10.1017/s0954579499002278. [DOI] [PubMed] [Google Scholar]
- Meyer JS, Shearman LP, Collins LM, Maguire RL. Cocaine binding sites in fetal rat brain: implications for prenatal cocaine action. Psychopharmacology. 1993;112:445–451. doi: 10.1007/BF02244892. [DOI] [PubMed] [Google Scholar]
- Molliver ME. Role of monoamines in the development of the neocortex. Neurosci Res Program Bull. 1982;20:492–507. [PubMed] [Google Scholar]
- Morrow BA, Elsworth JD, Roth RH. Axo-axonic structures in the medial prefrontal cortex of the rat: reduction by prenatal exposure to cocaine. J Neurosci. 2003;23:5227–5234. doi: 10.1523/JNEUROSCI.23-12-05227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–150. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Alifragis P, Wong RO, Parnavelas JG. Ventricle-directed migration in the developing cerebral cortex. Nat Neurosci. 2002;5:218–224. doi: 10.1038/nn813. [DOI] [PubMed] [Google Scholar]
- Nery S, Fishell G, Corbin JG. The caudal ganglionic eminence is a source of distinct cortical and subcortical cell populations. Nat Neurosci. 2002;5:1279–1287. doi: 10.1038/nn971. [DOI] [PubMed] [Google Scholar]
- Nery S, Corbin JG, Fishell G. Dlx2 progenitor migration in wild type and nkx2.1 mutant telencephalon. Cereb Cortex. 2003;13:895–903. doi: 10.1093/cercor/13.9.895. [DOI] [PubMed] [Google Scholar]
- Ohtani N, Goto T, Waeber C, Bhide PG. Dopamine modulates cell cycle in the lateral ganglionic eminence. J Neurosci. 2003;23:2840–2850. doi: 10.1523/JNEUROSCI.23-07-02840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnavelas JG. The origin and migration of cortical neurones: new vistas. Trends Neurosci. 2000;23:126–131. doi: 10.1016/s0166-2236(00)01553-8. [DOI] [PubMed] [Google Scholar]
- Powell EM, Mars WM, Levitt P. Hepatocyte growth factor/scatter factor is a motogen for interneurons migrating from the ventral to dorsal telencephalon. Neuron. 2001;30:79–89. doi: 10.1016/s0896-6273(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Powell EM, Campbell DB, Stanwood GD, Davis C, Noebels JL, Levitt P. Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. J Neurosci. 2003;23:622–631. doi: 10.1523/JNEUROSCI.23-02-00622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinoso BS, Undie AS, Levitt P. Dopamine receptors mediate differential morphological effects on cerebral cortical neurons in vitro. J Neurosci Res. 1996;43:439–453. doi: 10.1002/(SICI)1097-4547(19960215)43:4<439::AID-JNR5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Shearman LP, Collins LM, Meyer JS. Characterization and localization of [125I]RTI-55-labeled cocaine binding sites in fetal and adult rat brain. J Pharmacol Exp Ther. 1996;277:1770–1783. [PubMed] [Google Scholar]
- Stanwood GD, Washington RA, Levitt P. Identification of a sensitive period of prenatal cocaine exposure that alters the development of the anterior cingulate Cortex. Cereb Cortex. 2001a;11:430–440. doi: 10.1093/cercor/11.5.430. [DOI] [PubMed] [Google Scholar]
- Stanwood GD, Washington RA, Shumsky JS, Levitt P. Prenatal cocaine exposure produces consistent developmental alterations in dopamine-rich regions of the cerebral cortex. Neuroscience. 2001b;106:5–14. doi: 10.1016/s0306-4522(01)00256-1. [DOI] [PubMed] [Google Scholar]
- Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- Theiler K (1972) The house mouse. Development and normal stages from fertilization to 4 weeks of age. Berlin: Springer.
- Tobet SA, Chickering TW, Hanna I, Crandall JE, Schwarting GA. Can gonadal steroids influence cell position in the developing brain? Horm Behav. 1994;28:320–327. doi: 10.1006/hbeh.1994.1028. [DOI] [PubMed] [Google Scholar]
- Vitalis T, Cases O, Callebert J, Launay JM, Price DJ, Seif I, Gaspar P. Effects of monoamine oxidase A inhibition on barrel formation in the mouse somatosensory cortex: determination of a sensitive developmental period. J Comp Neurol. 1998;393:169–184. doi: 10.1002/(sici)1096-9861(19980406)393:2<169::aid-cne3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Wang HY, Runyan S, Yadin E, Friedman E. Prenatal exposure to cocaine selectively reduces D1 dopamine receptor-mediated activation of striatal Gs proteins. J Pharmacol Exp Ther. 1995;273:492–498. [PubMed] [Google Scholar]
- Wang X-H, Levitt P, Grayson DR, Murphy EH. Intrauterine cocaine exposure of rabbits: Persistent elevation of GABA-immunoreactive neurons in anterior cingulate cortex but not visual cortex. Brain Res. 1995;689:32–46. doi: 10.1016/0006-8993(95)00528-x. [DOI] [PubMed] [Google Scholar]
- Wang X-H, O’Brien-Jenkins A, Choi L, Murphy EH. Altered neuronal distribution of parvalbumin in anterior cingulate cortex of rabbits exposed in utero to cocaine. Exp Brain Res. 1996;112:359–371. doi: 10.1007/BF00227942. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Garcia-Verdugo JM, Herrera DG, Alvarez-Buylla A. Young neurons from medial ganglionic eminence disperse in adult and embryonic brain. Nat Neurosci. 1999;2:461–466. doi: 10.1038/8131. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez-Buylla A. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development. 2001;128:3759–3771. doi: 10.1242/dev.128.19.3759. [DOI] [PubMed] [Google Scholar]
- Wilkins AS, Genova LM, Posten W, Kosofsky BE. Transplacental cocaine exposure 1: A rodent model. Neurotoxicol Teratol. 1998;20:215–226. doi: 10.1016/s0892-0362(97)00125-6. [DOI] [PubMed] [Google Scholar]
- Wu W, Wong K, Chen JH, Jiang ZH, Dupuis S, Wu JY, Rao Y. Directional guidance of neuronal migration in the olfactory system by the protein Slit. Nature. 1999;400:331–336. doi: 10.1038/22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen X, Torres C, Wang HY, Friedman E. Prenatal exposure to cocaine disrupts D1A dopamine receptor function via selective inhibition of protein phosphatase 1 pathway in rabbit frontal cortex. J Neurosci. 2001;21:9160–9167. doi: 10.1523/JNEUROSCI.21-23-09160.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]