

Abstract
The Na+/dicarboxylate co-transporter, NaDC-1, from the kidney and small intestine, transports three sodium ions together with one divalent anion substrate, such as succinate2−. A previous study (Pajor, A. M. (2001) J. Biol. Chem. 276, 29961–29968), identified four amino acids, Ser-478, Ala-480, Ala-481, and Thr-482, near the extracellular end of transmembrane helix (TM) 9 that are likely to form part of the permeation pathway of the transporter. All four cysteine-substituted mutants were sensitive to inhibition by the membrane-impermeant reagent [2-(trimethylammonium)ethyl]-methanethiosulfonate (MTSET) and protected by substrate. In the present study, we continued the cysteine scan through extracellular loop 5 and TM10, from Thr-483 to Val-528. Most cysteine substitutions were well tolerated, although cysteine mutations of some residues, particularly within the TM, produced proteins that were not expressed on the plasma membrane. Six residues in the extracellular loop (Thr-483, Thr-484, Leu-485, Leu-487, Ile-489, and Met-493) were sensitive to chemical labeling by MTSET, depending on the conformational state of the protein. Transport inhibition by MTSET could be prevented by substrate regardless of temperature, suggesting that the likely mechanism of substrate protection is steric hindrance rather than large-scale conformational changes associated with translocation. We conclude that extracellular loop 5 in NaDC-1 appears to have a functional role, and it is likely to be located in or near the substrate translocation pore in the protein. Conformational changes in the protein affect the accessibility of the residues in extracellular loop 5 and provide further evidence of large-scale changes in the structure of NaDC-1 during the transport cycle.
The Na+/dicarboxylate co-transporter, NaDC-1, is found on the apical membrane of the epithelial cells of the renal proximal tubule and the small intestine (1). NaDC-1 carries a broad range of divalent anion substrates including dicarboxylates, such as succinate and α-ketoglutarate, and protonated tricarboxylates, such as citrate2−. NaDC-1 is involved in regulating concentrations of citric acid cycle intermediates in kidney cells and in the urine. Consequently, NaDC-1 activity affects the development of kidney stones (2) and regulation of blood pressure (3). NaDC-1 has also been implicated in life span determination; the Drosophila homolog, Indy, is a longevity gene also expressed in the gastrointestinal tract (4). NaDC-1 belongs to the SCL13 gene family that includes sodium-coupled transporters for sulfate and di- and tricarboxylates in vertebrates (5) and the sodium-independent malate transporter from plant vacuoles (6).
The identification of the substrate and cation binding sites in NaDC-1 and the transmembrane helices (TMs)1 involved in forming the permeation pathway remains an area of active investigation. Our previous studies have shown that the carboxyl-terminal half of NaDC-1, TM7–TM11, contains residues that determine substrate specificity (7). Chimera studies suggest that TM10 and the adjacent loops contain amino acids that determine differences in Km for citrate and sodium (8). Recently, we have found that TM9 is involved in the conformational changes seen during the transport cycle and likely forms part of the permeation pathway through the transporter (9). The extracellular end of TM9 in NaDC-1 contains four conformationally sensitive residues, Ser-479, Ala-480, Ala-481, and Thr-482 (9). Cysteine substitutions of these amino acids resulted in proteins that were sensitive to inhibition by the membrane-impermeant methanethiosulfonate reagent [2-(trimethylammonium)ethyl]-methanethiosulfonate (MTSET). In the present study, the cysteine scan was continued through the extracellular loop between TM9 and TM10 (extracellular loop 5) and in TM10, from Thr-483 to Val-528, in order to identify conformationally sensitive residues. As described previously, the membrane-impermeant reagent MTSET was used to react with accessible thiolate anions (10), and the functional consequences of the labeling were measured.
The major finding of this study is that MTSET-accessible residues are found in the loop between TM9 and TM10 and not in the transmembrane region. Eleven of the cysteine-substituted mutants were sensitive to MTSET, with either increased or decreased activity after chemical labeling. The most sensitive cysteine mutants were T483C, T484C, L485C, L487C, I489C, and M493C, all of which exhibited conformational differences in their sensitivity to MTSET labeling as well as substrate protection. All six cysteine-substituted mutants were most accessible to MTSET in the presence of sodium, the conformational state with the highest affinity for substrate. Because the conformational changes in accessibility of TM9 are also continued in the extracellular loop between TM9 and TM10, both TM9 and the loop are likely to contribute residues that line the permeation pathway in NaDC-1.
EXPERIMENTAL PROCEDURES
Subcloning and Mutagenesis
The rabbit NaDC-1 (GenBank™ U12186) contains an endogenous cysteine at position 476 that is sensitive to labeling by p-chloromercuribenezesulfonate (11). Therefore, the parental transporter for this project was the C476S mutant of rabbit NaDC-1 in pcDNA3.1 vector (Invitrogen) for expression in mammalian cells. The mutants were made using the oligonucleotide-directed method of Kunkel (12) with single-stranded C476S as a template. Positive mutants were identified by sequencing.
Expression of NaDC-1 Mutants in HRPE Cells
HRPE cells derived from human retinal pigment epithelial cells transformed with SV40 (AG 06096; Coriell Institute) were cultured in modified Eagle’s medium containing Glutamax and 25 mm HEPES (Invitrogen) supplemented with 15% non-inactivated fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in 5% CO2. For 6-well plates, 3 × 105 cells were plated per well and transiently transfected the following day with 3 μl of FuGENE 6 (Roche Applied Science) and 1 μg of plasmid DNA (3:1 ratio), according to the manufacturer’s directions. For 24-well plates, 1.2 × 105 cells were plated per well, and each well of cells was transfected with 1.8 μl of FuGENE 6 and 0.6 μg of plasmid DNA (9:3 ratio). The optimal ratio of FuGENE 6 to DNA was determined in preliminary experiments (results not shown).
Transport Assays
Transport assays were carried out 48 h after transfections. The sodium buffer contained 120 mm NaCl, 5 mm KCl, 1.2 mm MgSO4, 1.2 mm CaCl2, 5 mm D-glucose, 25 mm HEPES, pH adjusted to 7.4 with 1 m Tris. For the assays, each well was washed twice with sodium buffer and then incubated with sodium buffer containing radio-tracer succinate for 30 min. The uptakes were stopped, and radioactivity was washed away with four washes of sodium buffer. The wash volume was 3 ml for 6-well plates and 1 ml for 24-well plates. The uptake volume was 1 ml for 6-well plates and 0.25 ml for 24-well plates. Cells were dissolved in 1% SDS, transferred to scintillation vials, and counted. The initial expression experiments were done using [14C]succinate (44 mCi/mmol; PerkinElmer Life Sciences), but it became unavailable part way through the project. Later experiments were done using [3H]succinate (40 Ci/mmol; ViTrax). There were no apparent differences between experiments done with [14C]succinate and [3H]succinate. All experiments are expressed as a percentage of the activity in the parental C476S transporter from the same transfection experiment or as a percentage of the activity with and without MTSET. For all experiments, uptakes in vector-transfected cells were subtracted from uptakes in NaDC-1-transfected cells. Statistical analysis with one-way analysis of variance or Student’s t test was done using the Sigma Stat program (Jandel).
Chemical Labeling with MTSET
For experiments involving MTSET (Toronto Research Chemicals), the MTSET was pre-weighed and kept on ice. The appropriate buffer was added just before use. The MTSET was prepared in sodium or choline buffer (identical to sodium buffer except with choline chloride replacing NaCl), with or without 10 mm succinate. The cells were first washed three times with choline buffer and then incubated with the MTSET solution for 10 min. The MTSET solution was removed with three washes of sodium buffer. Control groups were preincubated in sodium buffer without MTSET. The transport activity was measured as described above. Each MTSET experiment contained four groups: vector controls with and without MTSET and plasmids containing NaDC-1 mutants with and without MTSET.
Because chemical modification by MTSET produces irreversible transport inhibition, the concentration dependence of MTSET inhibition was modeled according to reversible non-competitive inhibition, which resembles irreversible inhibition kinetically (13). The data were fitted to the following equation: %i = (%imax × [I])/(IC50 + [I]), where %i is the percentage of inhibition at a given inhibitor concentration, %imax is the maximal percentage of inhibition, IC50 is the concentration of inhibitor that produces half-maximal inhibition, and [I] is the concentration of inhibitor. Pseudo first order rates of inactivation, k (in min−1 mm−1), were estimated from the IC50 values using 0.5 = e−t × [IC50] × k, where 0.5 represents 50% activity remaining after MTSET treatment, and t is the time of MTSET inactivation in minutes.
Cell Surface Biotinylations
Cell surface biotinylations of NaDC-1 mutants expressed in HRPE cells were done using the membrane-impermeant reagent, Sulfo-NHS-LC-biotin (Pierce), which was shown in our previous study to distinguish between NaDC-1 mutants that were intracellular and those that were on the cell surface (14). The biotinylation procedure was a modification of the procedure described by Chen et al. (15). Briefly, each well of cells in 6-well plates was washed three times with 3 ml of phosphate-buffered saline, pH 9, containing 1 mm Ca2+ and Mg2+ (PBS/CM) and then incubated for 30 min at room temperature with 0.5 ml of 1.5 mg/ml Sulfo-NHS-LC-biotin (Pierce) prepared in PBS/CM. The wells were washed once and then incubated on ice for 20 min with 3 ml of cold Quench buffer (PBS/CM with 100 mm glycine). The cells were then treated with 0.5 ml of lysis buffer (1% Triton X-100, 150 mm NaCl, 5 mm EDTA, and 20 mm Tris, pH 7.5, supplemented with 10 μg/ml pepstatin, 10 μg/ml leupeptin, and 0.5 mm phenylmethylsulfonyl fluoride) on ice with gentle rocking for 30 min. The contents of 2 wells were combined in microcentrifuge tubes and pelleted, and the supernatants were transferred to new tubes. The samples were incubated with 50 μl of Immunopure Immobilized Streptavidin (Pierce) overnight at 4 °C with end-over-end rotation. The next day, the beads were washed according to the manufacturer’s directions, and the eluted samples were used in Western blotting as described previously (16). Blots were incubated with a 1:1000 dilution of anti-NaDC-1 antibodies (14). Antibody binding was detected using the Supersignal West Pico chemiluminescent substrate kit (Pierce). Images were acquired with a Kodak Image Station 440CF imager using six captures of 5 min each to avoid saturation of the image. Mass and quantity of the protein bands were determined using Image 1D analysis software (Eastman Kodak Co.).
Labeling of Cysteine Mutants with MTSEA-biotin
HRPE cells were preincubated with sodium uptake buffer with or without 1 mm MTSET for 10 min at room temperature. The MTSET solution was washed away by three washes with 3 ml of PBS/CM, pH 7.5. For each experiment, a 200 mm stock solution of MTSEA-biotin (Toronto Research Chemicals) was prepared in Me2SO and kept cold and dark. The MTSEA-biotin was diluted to 2 mm with PBS/CM, pH 7.5, just before use and added to the cells for 30 min at room temperature with rocking. The cells were rinsed three times with 3 ml of cold PBS/CM, pH 7.5. The remaining steps were identical to those in the Sulfo-NHS-LC biotinylation protocol. The cells were lysed and centrifuged, and biotinylated proteins were precipitated with Immunopure Immobilized Streptavidin (Pierce). The eluted samples were used in Western blotting as described above.
RESULTS
Cysteine-scanning Mutagenesis of NaDC-1
The secondary structure model of the rabbit NaDC-1 is shown in Fig. 1A. There are 11 predicted transmembrane helices, and the carboxyl terminus, containing a conserved N-glycosylation site, is on the outside of the cell (17). Forty-three amino acids between Thr-483 and Val-528 (Fig. 1B), located in extracellular loop 5 between transmembrane helices (TM) 9 and 10 and within TM10, were mutated to cysteine one at a time. The sequence alignment of this region with sequences from selected members of the SLC13 family is shown in Fig. 1B. The start of TM10 is predicted to be the conserved leucine residue at position 502.
Fig. 1.
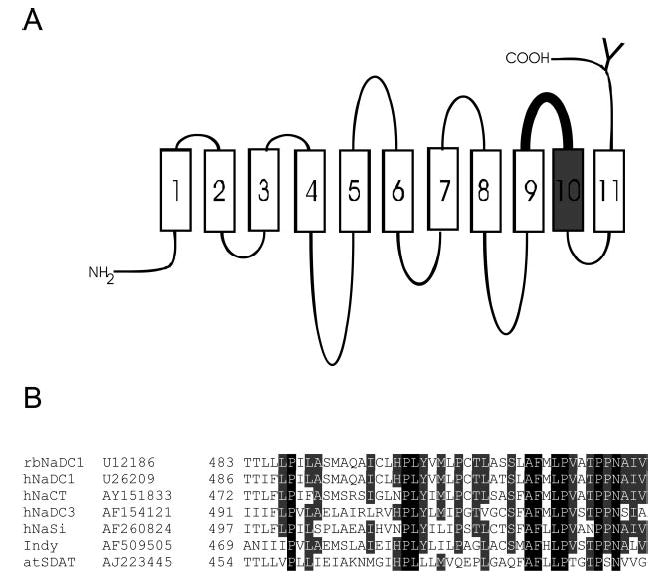
A, secondary structure model of NaDC-1 highlighting the region of the protein mutated in the cysteine scan. NaDC-1 contains 11 TMs, an intracellular amino terminus, and an extracellular carboxyl terminus. The conserved N-glycosylation site is shown by a Y. The 43 cysteine-substituted mutants in this study were made from Thr-483 to Val-528 including extracellular loop 5 (shown in bold) and TM10 (shown in gray). The outside of the cell is at the top of the figure. B, sequence alignments of the 43 amino acids mutated in this study among members of the SLC13 gene family. Names of the transporters and GenBank™ accession numbers are shown at the left. The NaDC and NaCT proteins are Na+/di- or tricarboxylate transporters, NaSi-1 is a Na+/sulfate transporter, Indy is the sodium-independent dicarboxylate exchanger from Drosophila, and atSDAT is a malate exchanger from Arabidopsis vacuoles (see reviews, Refs. 1 and 5).
As in our previous studies (9, 18, 19), the parental transporter for this study was the C476S mutant of NaDC-1. Although the wild-type NaDC-1 is insensitive to methanethiosulfonate reagents, the endogenous cysteine at position 476 mediates inhibition by another cysteine selective reagent, p-chloromercuribenezesulfonate. The C476S mutant still contains 10 endogenous cysteines. As in any mutagenesis project, there is a possibility that the introduction of new cysteines could produce changes in protein conformation that expose the remaining endogenous cysteines, although this does not appear to be the case in NaDC-1. In the TM9 cysteine scan study, we found that there was no difference in MTSET sensitivity of cysteine-substituted mutants made in a background containing only three endogenous cysteines (3C mutant) or in the C476S mutant containing 10 endogenous cysteines (9). Because the replacement of endogenous cysteines can produce a reduction in protein expression, the risk of exposing endogenous cysteines has to be balanced against having measurable transport activity. We found previously that there is a correlation between the number of endogenous cysteines in NaDC-1 and the amount of protein expressed at the plasma membrane, and the transport activity of the 3C mutant is only about 25% of that of the C476S mutant (11). It is possible that removing endogenous cysteines changes the structure of the transporter, which then results in decreased protein expression.
Transport Activity and Expression of Cysteine-substituted Mutants
The cysteine mutants of NaDC-1 were transiently transfected into the HRPE cell line derived from human retinal pigment epithelium. This cell line has low background dicarboxylate transport activity (20), and it is useful for plate transport assays because the cells are strongly attached to the plastic culture dishes.
The cell surface expression of the mutants was measured by biotinylation with a membrane-impermeant reagent, Sulfo-NHS-LC-biotin (Fig. 2). Single Western blots are shown in Fig. 2, and the results of scans of multiple blots are shown in Fig. 3. The protein expression of many of the mutant transporters was similar to that of the C476S parental transporter. The transport activity of the mutants is also shown in Fig. 3 for comparison with protein expression. The mutants were initially screened for functional activity in 24-well plates, and those that had activity less than ~30% of the activity of the C476S control were assayed in 6-well plates. Most of the mutants exhibited some measurable transport activity (Fig. 3). Eight of the mutants were either completely inactive or had activity that was less than 6% of the parental transporter activity: L485C and P488C (in the loop) and S512C, A515C, A521C, T522C, P523C, and P524C (in TM10). Most of the inactive mutants found in TM10 (S512C, T522C, and P523C) had little or no cell surface expression, suggesting that the mutations altered protein trafficking or stability. Interestingly, a few mutants exhibited measurable transport activity but greatly reduced cell surface expression, including L487C, H500C, and V520C, suggesting that these mutations might increase activity or decrease Km.
Fig. 2. Western blots of cell surface biotinylated cysteine-substituted mutants.
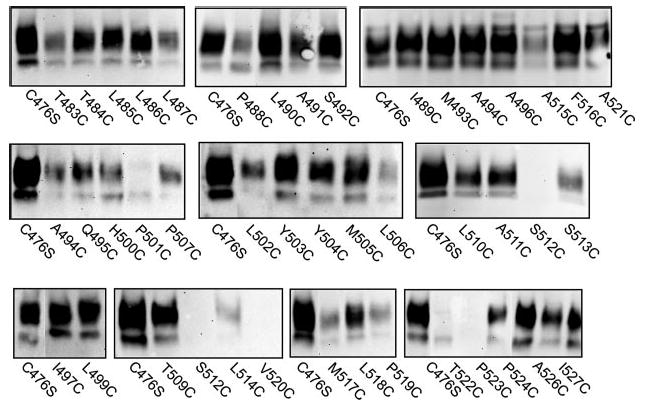
Intact HRPE cells were treated with Sulfo-NHS-LC-biotin as described under “Experimental Procedures.” Western blots were probed with anti-NaDC-1 antibodies (1:1000 dilution) followed by horseradish peroxidase-linked anti-rabbit immunoglobulin at a dilution of 1:5000. Each blot contains an internal control of the parental transporter, C476S. The two protein bands represent differently glycosylated forms of the transporter (14).
Fig. 3. Activity and expression of cysteine-substituted mutants of NaDC-1 expressed in HRPE cells.
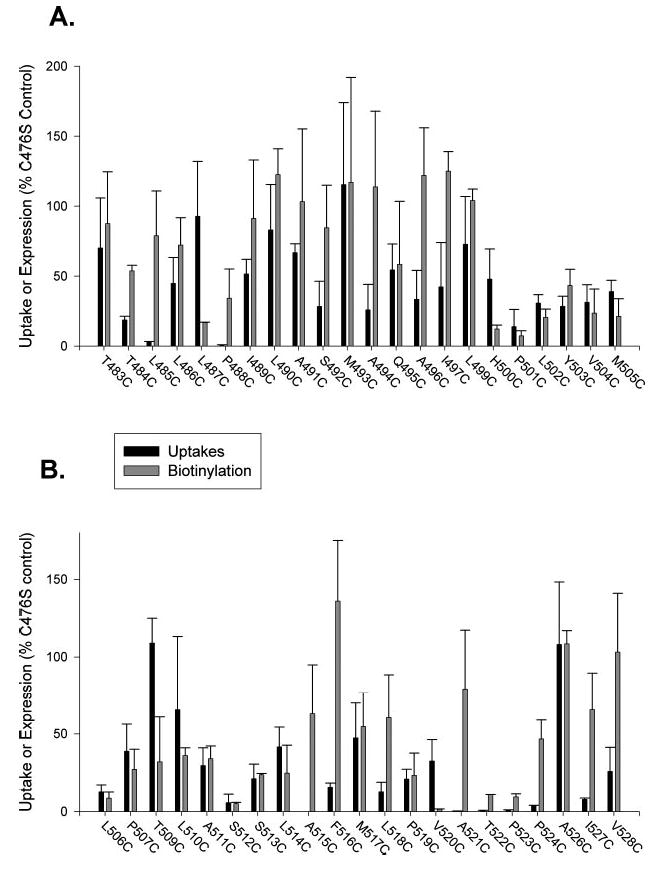
A, mutants T483C–M505C. B, Mutants L506C–V528C. Uptakes of [14C]succinate were measured and expressed as a percentage of the parental transporter, C476S, from the same transfection experiment. The gaps in numbering of mutants refer to positions 498 and 507, endogenous cysteines in NaDC-1 that were present in all mutants, and the conserved Asn-525, which we were unable to mutate to cysteine. Bars represent means ± range or S.E. (n = 2–8 separate transfections). For measurement of protein expression, Western blots, such as the ones shown in Fig. 2, were quantitated using Image software, the intensities of both protein bands were added together (because glycosylation does not affect transport activity), and the abundance of each mutant transporter was expressed as a percentage of the C476S control in the same blot. The bars represent means ± range or S.E. (n = 2–4 blots).
Sensitivity to MTSET
Thirty-nine mutants with measurable transport activity were screened for their sensitivity to a 1 mm concentration of MTSET. The parental C476S mutant (Fig. 4) is insensitive to MTSET (Fig. 4), MTSEA, and (2-sulfonato-ethyl)methanethiosulfonate (data not shown) when expressed in HRPE cells and in Xenopus oocytes, as described in our previous study (9). The wild-type NaDC-1 is also insensitive to MTSET when expressed in HRPE cells (data not shown) and Xenopus oocytes (9). Only 11 of 39 of the cysteine-substituted mutants exhibited functional consequences of pretreatment with MTSET. All of the sensitive mutants were located in the putative loop between TM9 and TM10 or at the extracellular surface of TM10 (Y503C), whereas none of the substituted cysteine mutants in the transmembrane helix were sensitive to MTSET (data not shown). Two mutants, A494C and L499C, consistently showed a stimulation in activity after pretreatment with MTSET (Fig. 4). Two mutants, A491C and Q495C, were only moderately inhibited (about 30%) by pretreatment with 1 mm MTSET, suggesting that either they are not accessible to the MTSET or they are accessible to the MTSET, but the functional consequences of chemical modification are minimal (Fig. 4). The Y503C mutant was also only moderately affected by MTSET, with ~40% inhibition (Fig. 5). This inhibition appears to be maximal because treatment of A491C, Q495C, and Y503C with concentrations of MTSET of up to 2.5 mm and incubations of up to 1 h did not produce further inhibition (data not shown). Six of the mutants (T483C, T484C, L485C, L487C, I489C, and M493C) were almost completely inactive after preincubation with 1 mm MTSET. These six mutants were then studied in more detail.
Fig. 4. Effect of 1 mm MTSET on succinate transport by cysteine substituted mutants.
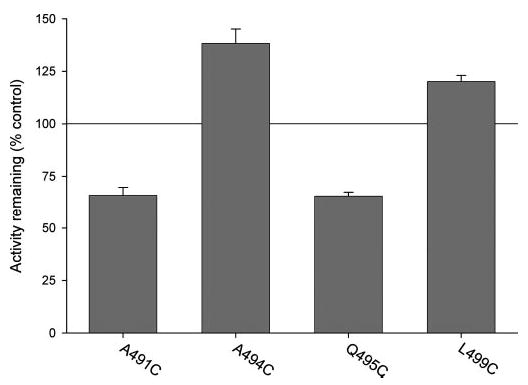
HRPE cells expressing cysteine-substituted mutants were preincubated for 10 min with 1 mm MTSET in sodium buffer or with sodium buffer alone (control). The preincubation solutions were washed away, and the remaining uptake activity of [3H]succinate was measured. Uptake activities in cells pretreated with MTSET are expressed as a percentage of the uptakes in controls pretreated with sodium buffer alone.
Fig. 5. Effects of cations and substrate on sensitivity of cysteine-substituted mutants to MTSET.
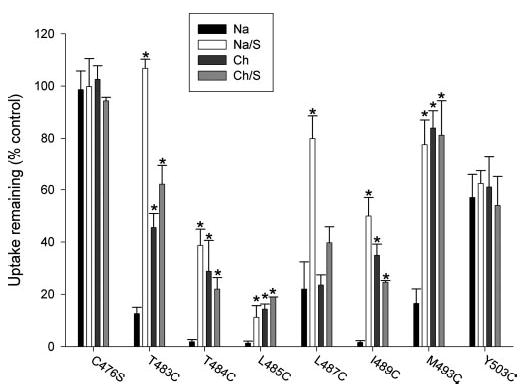
HRPE cells expressing cysteinesubstituted mutants were preincubated for 10 min with sodium (Na) or choline buffer (Ch) with or without 10 mm succinate (S) and with or without MTSET. The preincubation solutions were washed away, and the remaining uptake activity of [3H]succinate was measured. Uptake activities of cells pretreated with MTSET are shown as a percentage of the uptakes in control cells pretreated with the same buffers without MTSET. MTSET concentrations were 10 μM (T484C, L485C, I489C, and M493C) or 1 mm (C476S, T483C, L487C, and Y503C), depending on the sensitivity of each mutant. Bars represent means ± S.E. (n = 2–6 separate experiments). *, significant difference compared with sodium group (p < 0.05).
The substrate affinity of the cysteine-substituted mutants did not appear to be affected by the cysteine mutation. The C476S parental transporter had a Km for succinate of 399 μm, compared with 238 μm in our previous study (9). The Km for succinate in T483C was 410 ± 28 (n = 2); in L487C, the Km was 227 μm; and in M493C, the Km was 650 μm (data not shown). The remaining mutants, T484C, L485C, and I489C, were assayed in 6-well plates because of low activity (see Fig. 3). For those mutants, the signal above background at high substrate concentrations was too low for accurate kinetic measurements.
Effects of Cations and Substrate on MTSET Sensitivity
To determine whether the substituted cysteines are accessible to MTSET in different conformational states, the preincubation with MTSET was done in either sodium or choline buffer, with or without succinate. As shown previously, NaDC-1 has an ordered binding mechanism in which sodium binds first and triggers a conformational change that increases substrate affinity, then substrate binds, and translocation occurs (9). Therefore, the transporter is likely to be in different conformational states when incubated in the presence or absence of sodium or substrate. As shown in Fig. 5, the Y503C mutant, located near the extracellular end of TM10, reacted with MT-SET at the same rate, regardless of the preincubation conditions. Therefore, Y503C probably has similar accessibility to the extracellular medium in all conformational states of the transporter. In contrast, the other mutants that were inhibited by labeling with MTSET (T483C, T484C, L485C, L487C, I489C, and M493C) exhibited differences in apparent accessibility depending on the preincubation conditions (Fig. 5). Five of the mutants (T483C, T484C, L485C, I489C, and M493C) were most sensitive to inhibition by MTSET when the prein-cubation was done in sodium buffer, suggesting that the accessibility of these substituted cysteines is greatest in the conformational state or states seen in the presence of sodium. The L487C mutant was equally sensitive to inhibition by MTSET in sodium or choline, suggesting that this residue is exposed in the conformational states seen in the absence as well as the presence of sodium. The six conformationally sensitive mutants also exhibited substrate protection because there was decreased inhibition by MTSET in the presence of sodium and 10 mm succinate (Fig. 5).
Concentration Dependence of MTSET Inhibition
The concentration dependence of MTSET inhibition for the six sensitive mutants, T483C, T484C, L485C, L487C, I489C, and M493C, was determined by measuring the succinate transport activity before and after a 10-min preincubation with increasing concentrations of MTSET (Fig. 6). Three of the mutants, T484C, L485C, and I489C, were very sensitive to inhibition by MTSET, with estimated IC50 values of <0.5 μM. M493C had an IC50 of ~6 μm. T483C and L487C were relatively insensitive to MTSET with IC50 values of 0.3 and 0.7 mm, respectively. For comparison, the pseudo first order rate constants (k values) were estimated from the IC50 values (note that Fig. 6 shows a single experiment; the k values represent mean ± range of two experiments). The most reactive residues, I489C, T484C, and L485C, had the largest estimated k values of about 180 min−1 mm−1. M493C had an intermediate value of 11 ± 0.3 min−1 mm−1. The least reactive residues were T483C (0.2 ± 0.04 min−1 mm−1) and L487C (0.1 ± 0.02 min−1 mm−1).
Fig. 6. Concentration dependence of MTSET inhibition of succinate transport by cysteine-substituted mutants.
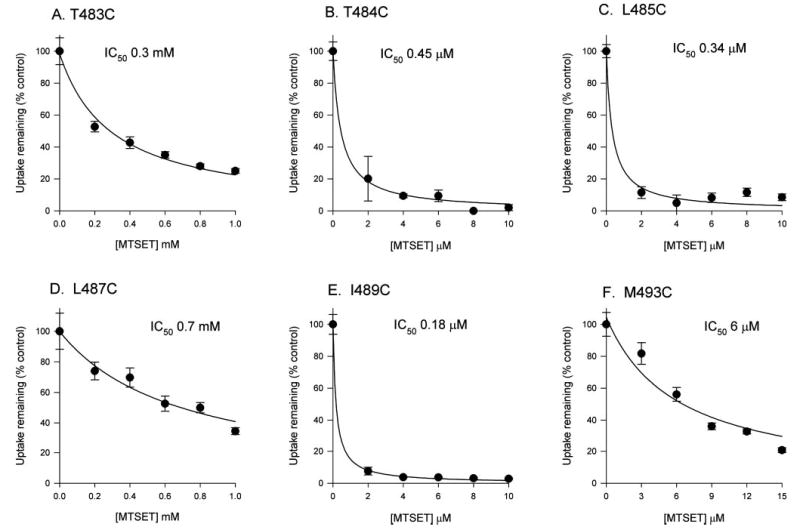
HRPE cells were preincubated in Na+ buffer only (control) or with increasing concentrations of MTSET in sodium buffer. The succinate transport activity was then measured and expressed as a percentage of the transport activity in the control group. Each point represents the mean ± S.E. (n = 4 wells).
Effect of Temperature on MTSET Inhibition
There are several possible explanations to account for substrate protection of MTSET labeling. For example, the conformational change that occurs after substrate binding could occlude the substituted cysteine and make it inaccessible to MTSET. Alternately, substrate binding could physically prevent access of the MTSET to the substituted cysteine. Because large-scale conformational changes in transporters are reduced at low temperature, we compared the extent of substrate protection at room temperature and on ice. The remaining transport activity after MTSET labeling was measured at room temperature. As shown in Fig. 7, there was very little effect of temperature on MTSET inhibition in the absence of substrate. There was also no effect of temperature on substrate protection from MTSET in the T483C and M493C mutants. However, the T484C, L485C, L487C, and I489C mutants had a significant decrease in the effect of MTSET in the presence of substrate when the incubation was carried out on ice as compared with at room temperature (Fig. 7). The result of our experiment suggests that the rate of inactivation by MTSET in the presence of substrate is likely to be lower in the cold than at room temperature. If the major effect of substrate protection is a conformational change due to translocation, then we would expect to see a decrease in substrate protection in the cold, i.e. increased sensitivity to MTSET. For example, in a similar experiment with the Na+/serotonin transporter, cold temperature abolished substrate protection of MTSEA binding at Cys-357 (21). Instead, T484C, L485C, L487C, and I489C exhibited increased substrate protection in the cold. Therefore, the major effect of substrate protection in these mutants is likely to be related to substrate binding, thereby producing steric hindrance of MTSET labeling.
Fig. 7. Temperature dependence of MTSET labeling.
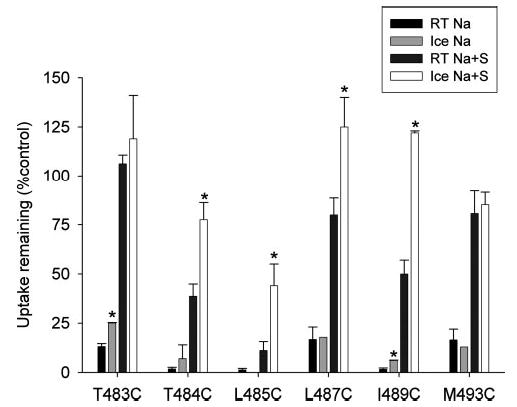
HRPE cells transiently expressing MTSET-sensitive cysteine mutants were preincubated in sodium buffer (Na) with or without 10 mm succinate (S) and with or without MTSET. The MTSET concentrations were 10 μm (T484C, L485C, I489C, and M493C) and 1 mm (T483C and L487C). The preincubations were done at room temperature (RT) or on ice for 10 min. After the pretreatment, cells were washed and allowed to come to room temperature, and then uptakes of [3H]succinate were measured as described previously. Data shown are means ± range or S.E. (n = 2–6 experiments). *, significant differences (p < 0.05) between groups incubated at room temperature and on ice.
Labeling of Substituted Cysteines with MTSEA-biotin
Most of the experiments in this study have examined the functional effects of chemical modification with MTSET. Residues that show no functional consequence of MTSET treatment could still be accessible to chemical labeling by this reagent. Therefore, we also examined whether some of the substituted cysteines could be labeled with a biotin derivative, MTSEA-biotin. Labeling was done after preincubation with or without MTSET. The parental transporter, C476S, and a cysteine mutant that was very sensitive to MTSET in our previous study, T482C (9), were used as controls in each experiment. As shown in Fig. 8, there is some residual labeling of C476S with MTSEA-biotin, but this is not affected by preincubation with MTSET. The C476S mutant contains 10 endogenous cysteines, but the results suggest that these cysteines are not accessible to MTSET. This result rules out the possibility that MTSET labels the endogenous cysteines in C476S without any functional consequences. The positive control mutant, T482C, was strongly labeled with MTSEA-biotin, and this was inhibited by preincubation in MTSET. M493C was sensitive to MTSET and also showed MTSET-blockable labeling by MTSEA-biotin. Interestingly, L499C exhibits MTSET-blockable labeling by MTSEA-biotin, and it is one of the residues that was activated after treatment with MTSET. Two residues that exhibit only partial inhibition by MTSET, Q495C and Y503C, were not strongly labeled by MTSEA-biotin despite expression on the plasma membrane that was ~50% of control (Sulfo-NHS-LC-biotin; Fig. 3). It appears that the sensitivity of MTSEA-biotin is lower than that of Sulfo-NHS-LC-biotin. Three residues that are insensitive to MTSET and predicted to be in transmembrane helix 10 (M505C, T509C, and A511C) also had little labeling by MTSEA-biotin compared with the C476S control and little or no difference with MTSET preincubation.
Fig. 8. Labeling of cysteine-substituted mutants with MTSEA-biotin.
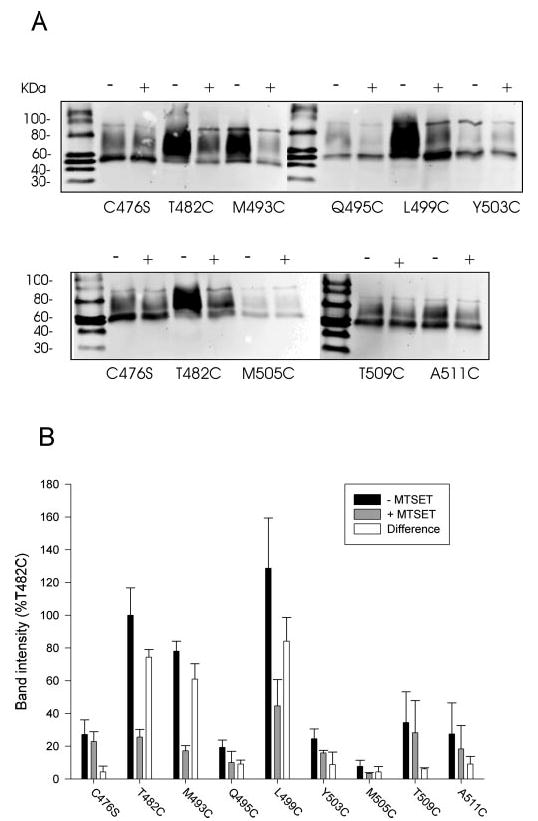
A, HRPE cells expressing some of the cysteine mutants were first preincubated with sodium buffer with or without 1 mm MTSET to label extracellularly accessible cysteines. The preincubation solution was washed away, and the cell monolayers were biotinylated with MTSEA-biotin and transferred to Western blots as described under “Experimental Procedures.” The blots were treated with anti-NaDC-1 antibodies (1:1000 dilution). The negative control mutant, C476S, and the positive control, T482C, were included in each biotinylation experiment. +, pretreatment with MTSET; −, pretreatment with sodium buffer. The chemiluminescent size standards, Magic Mark Western Standards (Invitrogen), are shown, and mass (in kDa) is indicated to the left. B, summary of MTSEA biotinylation results. Western blots, such as those shown in A, were quantitated and expressed as a percentage of the T482C intensity from the same blot. The difference between the signal in the presence and absence of MTSET, which represents the MTSET-inhibitable binding of MTSEA-biotin, is also shown. Bars represent mean ± range (n = 2 blots).
DISCUSSION
The transport cycle of secondary active transporters such as the Na+/dicarboxylate co-transporter, NaDC-1, involves several conformational changes induced by sodium and substrate binding and a large-scale conformational change to allow translocation from one side of the membrane to the other. Our previous study identified TM9 in NaDC-1 as a potential component of the substrate permeability pathway and a participant in the conformational changes that occur during transport (9). Four amino acids at the extracellular face of TM9 (Ser-478, Ala-480, Ala-481, and Thr-482) were found to show differences in accessibility to the membrane-impermeant cysteine reagent, MTSET, during the transport cycle. In the present study, we have continued the cysteine scan from Thr-483 to Val-528, including extracellular loop 5 and TM10, and examined the sensitivity of these mutants to MTSET. The main findings are that extracellular loop 5 is also likely to participate in the conformational changes during the transport cycle of NaDC-1 because residues in this loop exhibit differences in MTSET accessibility with different conformational states produced by the presence or absence of sodium and substrate. In contrast, cysteine mutations within TM10 were insensitive to inhibition by MTSET.
Most of the cysteine substitutions made in this study were well tolerated, with many mutants retaining at least 50% of the wild-type activity and protein expression. Some conserved residues, particularly within the putative TM10, were sensitive to substitution with cysteine, and the resulting proteins were not well expressed on the plasma membrane. The alignment of sequences from representative members of the SLC13 family (Fig. 1B) shows several residues that are conserved in all members of the family, including homologs from Drosophila and plants. The highly conserved residues seemed to correlate well with the greatest decrease in transport activity. For ex ample, cysteine mutants of two highly conserved residues, Ala-515 and Phe-516, were expressed at the plasma membrane but had very low activity, suggesting that these residues may have functional roles.
Eleven of the 43 cysteine-substituted amino acids in NaDC-1 were functionally affected by chemical modification with MTSET, verifying that these residues are either located on the outside of the cell or in an aqueous pore accessible from the outside. MTSET is a water-soluble reagent that does not cross the plasma membrane, and it labels cysteines that are accessible from the outside of the cell. In our secondary structure model (Fig. 9), ten of the MTSET-sensitive residues are found in extracellular loop 5 between TM9 and TM10, and one residue, Tyr-503, is located at the extracellular end of TM10. In general, there was good correlation between the functional effects of MTSET and the accessibility to labeling by MTSEA biotinylation, with a few exceptions. For example, the Q495C and Y503C mutants are both accessible to MTSET, although the inhibition was modest (at most 40%). These residues were not labeled by MTSEA-biotin. This might indicate that there are constraints on the size of the molecules that can reach these residues. Alternately, there could be differences in assay sensitivity between the Sulfo-NHS-LC biotinylations, which label all extracellular lysines, and MTSEA biotinylations, which would label a single cysteine. The addition of more biotin molecules to the protein may result in more effective precipitation with streptavidin beads.
Fig. 9. Model showing MTSET-sensitive residues in TM9 and TM10 and the connecting extracellular loop 5.
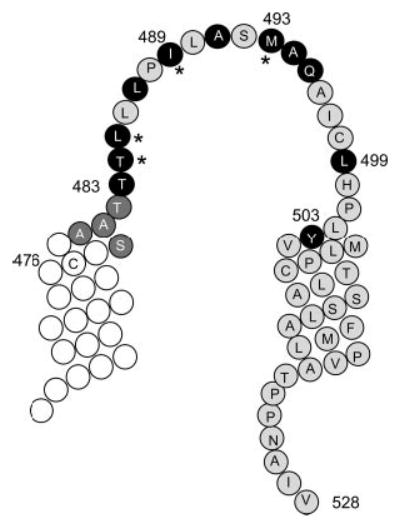
Four residues at the top of TM9 (from Ser-478 to Thr-482, shown as dark gray circles with white letters) were identified in our previous study as conformationally sensitive (9). Residues sensitive to MTSET modification in the present study are shown in black circles with white letters. The asterisks denote positions of cysteine-substituted amino acids that are very sensitive to inhibition by MTSET (IC50 < 6 μM).
The MTSET accessibility of five of the mutants in this study (T483C, T484C, L485C, I489C, and M493C) seems to parallel the accessibility of the substrate binding site to the outside of the cell. As illustrated in Fig. 10, the transport cycle of NaDC-1 involves ordered binding, with the three sodium ions binding first, triggering a conformational change that increases affinity for succinate, followed by substrate binding (22). The binding of substrate is then followed by a conformational change to reorient the substrate and cation binding sites to the inside of the cell, after which the substrate and cations are released. At present, we have little information on the order of substrate release on the inside of the cell, but it appears that at least one of the sodium ions is released last (22). Similar to the exposure of the substrate binding site, the five cysteine-substituted mutants have the highest accessibility to MTSET in the conformational state produced in the presence of sodium and low accessibility in the conformational state or states seen in the absence of sodium (Fig. 10). The results suggest that these five residues, found in the extracellular loop, are likely to be found near the substrate binding site or in the water-filled cavity that contains the substrate binding site. At present, we have no information on whether these residues exhibit conformationally sensitive differences in accessibility from the inside of the cell, which would be predicted from the alternating access model of transport. The L487C mutant, also located in extra-cellular loop 5, was equally sensitive to MTSET labeling in sodium and choline but exhibited substrate protection, suggesting that this residue is accessible in early conformational states of the transport cycle.
Fig. 10. Simplified model of coupled sodium and dicarboxylate transport by NaDC-1.
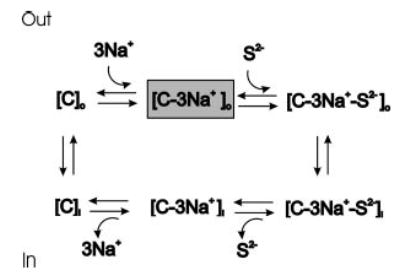
The C refers to the transporter; S2− is the divalent anion substrate; subscripts o and i refer to accessibility of binding sites from the outside and inside of the cell, respectively. The transport cycle includes the ordered binding of three sodium ions to the transporter, which increases the affinity for substrate, followed by substrate binding. The fully loaded carrier undergoes a conformational change that reorients the binding sites to the inside of the cell. The substrate and sodium are then released on the inside, and the empty carrier undergoes another conformational change to reorient the binding sites to the outside of the cell. The gray box shows the conformational state with highest affinity for substrate, seen in the presence of sodium.
Based on the differences in sensitivity to MTSET among the residues in extracellular loop 5 (see Fig. 9), it is likely that this loop does not simply extend to the outside of the cell. It is possible that the differences in accessibility to MTSET are determined by the local environment adjacent to individual residues or by structure in the loop, such as an α-helix or a re-entrant loop. There are examples of transport proteins, such as the anion exchanger AE1, that contain functionally important loops in contact with transmembrane helices rather than being embedded in the lipid bilayer (23). Other examples of sodium-coupled transport proteins with functionally important loop regions include the γ-aminobutyric acid and serotonin transporters (24–26). There is some evidence that the reentrant loops in the glutamate transporters could form part of the aqueous pore through the transporter (18, 27).
Most of the MTSET-sensitive residues in NaDC-1 were protected from chemical modification by the presence of substrate. This can be explained either by a steric hindrance of binding of MTSET when substrate is bound or by a conformational change upon substrate binding that moves the cysteine away from MTSET. Because there was no reduction of substrate protection when the experiments were done in the cold, which should inhibit large-scale conformational changes in NaDC-1, the likeliest explanation is that the binding of substrate interferes with binding of MTSET and verifies that extracellular loop 5 is a key region of the protein in substrate permeation. This finding is similar to that of previous studies of the serotonin and glutamate, which identified several key residues that can be protected from methanethiosulfonate reagents by substrate regardless of temperature (24, 28, 29). In contrast, one of the endogenous cysteines found in the serotonin transporter does not show substrate protection in the cold, suggesting that its accessibility is likely to be affected by conformational changes associated with substrate translocation (21).
In conclusion, we have found that the fifth extracellular loop in NaDC-1, located between TM9 and TM10, participates in the conformational changes during the transport cycle. Six amino acids in the loop are sensitive to inhibition by MTSET, and they exhibit differences in accessibility in different conformational states of the transporter. The accessibility of five of these residues (T483C, T484C, L485C, I 489C, and M493C) appears to follow the accessibility of the substrate binding site. Substrate protection was seen in all MTSET-sensitive mutants, which appears to be due to a steric hindrance of MTSET binding rather than inaccessibility of the substituted cysteine as a consequence of large-scale conformational change. Therefore, in addition to TM9 identified in our previous study, the extracellular loop adjacent to it also participates in conformational changes seen during the transport cycle. Our findings suggest that extracellular loop 5 either transmits structural information to other parts of the protein during conformational changes or that residues in the loop are located within the large central water-filled cavity that contains the substrate binding site.
Footnotes
This work was supported National Institutes of Health Grant DK46269.
The abbreviations used are: TM, transmembrane helix; MTSET, [2-(trimethylammonium)ethyl]-methanethiosulfonate; HRPE, human retinal pigment epithelium; PBS/CM, phosphate-buffered saline, pH 9, containing 1 mm Ca23 and Mg23; MTSEA, (2-aminoethyl)methanethio-sulfonate.
References
- 1.Pajor AM. J Membr Biol. 2000;175:1–8. doi: 10.1007/s002320001049. [DOI] [PubMed] [Google Scholar]
- 2.Pajor AM. Semin Nephrol. 1999;19:195–200. [PubMed] [Google Scholar]
- 3.He W, Miao FJP, Lin DCH, Schwander RT, Wang Z, Gao J, Chen JL, Tian H, Ling L. Nature. 2004;429:188–193. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 4.Rogina B, Reenan RA, Nilsen SP, Helfand SL. Science. 2000;290:2137–2140. doi: 10.1126/science.290.5499.2137. [DOI] [PubMed] [Google Scholar]
- 5.Markovich D, Murer H. Pflugers Arch. 2004;447:594–602. doi: 10.1007/s00424-003-1128-6. [DOI] [PubMed] [Google Scholar]
- 6.Emmerlich V, Linka N, Reinhold T, Hurth MA, Traub M, Martinoia E, Neuhaus HE. Proc Natl Acad Sci U S A. 2003;100:11122–11126. doi: 10.1073/pnas.1832002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pajor AM, Sun N, Bai L, Markovich D, Sule P. Biochim Biophys Acta. 1998;1370:98–106. doi: 10.1016/s0005-2736(97)00249-6. [DOI] [PubMed] [Google Scholar]
- 8.Kahn ES, Pajor AM. Biochemistry. 1999;38:6151–6156. doi: 10.1021/bi9827722. [DOI] [PubMed] [Google Scholar]
- 9.Pajor AM. J Biol Chem. 2001;276:29961–29968. doi: 10.1074/jbc.M011387200. [DOI] [PubMed] [Google Scholar]
- 10.Danielson MA, Bass RB, Falke JJ. J Biol Chem. 1997;272:32878–32888. doi: 10.1074/jbc.272.52.32878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pajor AM, Krajewski SJ, Sun N, Gangula R. Biochem J. 1999;344:205–209. [PMC free article] [PubMed] [Google Scholar]
- 12.Kunkel TA. Proc Natl Acad Sci U S A. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Segel, I. H. (1975) Enzyme Kinetics, John Wiley and Sons, New York
- 14.Pajor AM, Sun N, Valmonte HG. Biochem J. 1998;331:257–264. doi: 10.1042/bj3310257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J-G, Liu-Chen S, Rudnick G. Biochemistry. 1997;36:1479–1486. doi: 10.1021/bi962256g. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Pajor AM. Am J Physiol Cell Physiol. 2003;285:C1188–C1196. doi: 10.1152/ajpcell.00162.2003. [DOI] [PubMed] [Google Scholar]
- 17.Zhang FF, Pajor AM. Biochim Biophys Acta. 2001;1511:80–89. doi: 10.1016/s0005-2736(00)00385-0. [DOI] [PubMed] [Google Scholar]
- 18.Seal RP, Amara SG. Neuron. 1998;21:1487–1498. doi: 10.1016/s0896-6273(00)80666-2. [DOI] [PubMed] [Google Scholar]
- 19.Yao X, Pajor AM. Biochemistry. 2002;41:1083–1090. doi: 10.1021/bi0156761. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, Fei YJ, Kekuda R, Yang-Feng TL, Devoe LD, Leibach FH, Prasad PD, Ganapathy ME. Am J Physiol Cell Physiol. 2000;278:C1019–C1030. doi: 10.1152/ajpcell.2000.278.5.C1019. [DOI] [PubMed] [Google Scholar]
- 21.Androutsellis-Theotokis A, Ghassemi F, Rudnick G. J Biol Chem. 2001;276:45933–45938. doi: 10.1074/jbc.M107462200. [DOI] [PubMed] [Google Scholar]
- 22.Yao X, Pajor AM. Am J Physiol Renal Fluid Electrolyte Physiol. 2000;279:F54–F64. doi: 10.1152/ajprenal.2000.279.1.F54. [DOI] [PubMed] [Google Scholar]
- 23.Hamasaki N, Abe Y, Tanner MJA. Biochemistry. 2002;41:3852–3854. doi: 10.1021/bi011918l. [DOI] [PubMed] [Google Scholar]
- 24.Zomot E, Kanner B. J Biol Chem. 2003;278:42950–2958. doi: 10.1074/jbc.M209307200. [DOI] [PubMed] [Google Scholar]
- 25.Tamura S, Nelson H, Tamura A, Nelson N. J Biol Chem. 1995;270:28712–8715. doi: 10.1074/jbc.270.48.28712. [DOI] [PubMed] [Google Scholar]
- 26.Stephan MM, Chen MA, Penado KMY, Rudnick G. Biochemistry. 1997;36:1322–328. doi: 10.1021/bi962150l. [DOI] [PubMed] [Google Scholar]
- 27.Grunewald M, Menaker D, Kanner BI. J Biol Chem. 2002;277:26074–6080. doi: 10.1074/jbc.M202248200. [DOI] [PubMed] [Google Scholar]
- 28.Leighton BH, Seal RP, Shimamoto K, Amara SG. J Biol Chem. 2002;277:29847–9855. doi: 10.1074/jbc.M202508200. [DOI] [PubMed] [Google Scholar]
- 29.Henry LK, Adkins EM, Han Q, Blakely RD. J Biol Chem. 2003;278:37052–7063. doi: 10.1074/jbc.M305514200. [DOI] [PubMed] [Google Scholar]