

Abstract
The serine protease prohormone convertase 2 (PC2), principally involved in the processing of polypeptide hormone precursors in neuroendocrine tissues, requires interaction with the neuroendocrine protein 7B2 to generate an enzymatically active form. 7B2 null mice express no PC2 activity and release large quantities of uncleaved ACTH, resulting in a lethal endocrine condition that resembles pituitary Cushing's (Westphal, C. H., Muller, L., Zhou, A., Bonner-Weir, S., Schambelan, M., Steiner, D. F., Lindberg, I. & Leder, P. (1999) Cell 96, 689). Here, we have compared the 7B2 and PC2 null mouse models to determine why the 7B2 null, but not the PC2 null, exhibits a lethal disease state. Both 7B2 and PC2 nulls contained highly elevated pituitary adrenocorticotropic hormone (ACTH); the neurointermediate lobe content of ACTH in 7B2 nulls was 13-fold higher than in WT mice; that of the PC2 null was 65-fold higher. However, circulating ACTH levels were much higher in the 7B2 null than in the PC2 null. Because hypothalamic inhibitory dopaminergic control represents the major influence on intermediate lobe proopiomelanocortin-derived peptide secretion, dopamine levels were measured, and they revealed that 7B2 null pituitaries contained only one-fourth of WT pituitary dopamine. Adrenalectomized 7B2 null animals survived past the usual time of death at 5 weeks; a month after adrenalectomy, they exhibited normal levels of pituitary dopamine, circulating ACTH, and corticosterone. Elevated corticosterone, therefore, seems to play a central role in the lethal phenotype of the 7B2 null, whereas a 7B2-mediated dopaminergic deficiency state may be involved in the actual ACTH hypersecretion phenomenon. Interestingly, adrenalectomized 7B2 nulls also developed unexpectedly severe obesity.
A wide spectrum of peptide hormones, active proteins, and neuropeptides are generated in animal cells by means of proteolytic processing of inactive precursor proteins. Serine proteinases related to the bacterial enzyme subtilisin now are thought to be the major enzymes responsible for these proteolytic cleavage events (reviewed in refs. 1–3).
Seven members of this enzyme family have now been identified in mammals. These proteinases cleave proprotein substrates at paired basic residues to generate mature secreted or cell surface proteins and/or biologically active peptides. The prohormone convertase 1 (PC1/PC3) and prohormone convertase 2 (PC2) principally are involved in the processing of precursors to polypeptide hormones and neuropeptides in neuroendocrine tissues (2, 4). Among the convertases, PC2 differs significantly from other members of this serine protease family. Despite an autocatalytic activation mechanism which resembles that of other enzymes (4, 5), proPC2 must interact with the small neuroendocrine protein 7B2 in the secretory pathway (6) for the generation of a proPC2 form capable of maturation to active PC2 (7, 8).
First identified as a neuroendocrine-specific protein (9), 7B2 is highly conserved throughout vertebrate evolution (10–14). 7B2 is synthesized as a 185-aa precursor protein and cleaved during transit in the secretory pathway into two fragments: an amino-terminal domain (21-kDa 7B2) and a carboxyl-terminal domain. As discussed above, 7B2 must interact with proPC2 (6) to mature to the active form (7). However, 7B2 also has an opposing biological function: intact 7B2 as well as its 31-residue carboxyl-terminal domain represent potent inhibitors of active PC2 (15, 16). Thus, 7B2 represents an interesting polyfunctional convertase-binding protein.
As PC1, PC2, and 7B2 are involved in the generation of the mature forms of many neuropeptides and peptide hormones, the disappearance of these gene products would be expected to yield major alterations in endocrine function, metabolism, and/or behavior. Indeed, although PC2 null mice did not exhibit major alterations in glucocorticoid levels, growth, or reproduction, they were chronically hypoglycemic with severe defects in proglucagon processing to glucagon (17, 18), processing of prosomatostatin to SS-14 (17), and processing of proinsulin to insulin in the islets of Langerhans (19). They also have significant deficits in opioid peptide levels in the central nervous system (20, 21).
The 7B2 null mouse completely lacks PC2 activity and thus shares the above processing phenotypes with the PC2 nulls along with the associated hypoglycemia, hyperproglucagonemia, hyperproinsulinemia, and altered-islet cell morphology (22). However, unlike the PC2 null, 7B2 nulls die between 5 and 6 weeks of age because of a lethal form of Cushing's disease brought about by excess adrenocorticotropic hormone (ACTH) secretion from the pituitary neurointermediate lobe (22).
Pituitary ACTH is derived from the opioid peptide precursor proopiomelanocortin (POMC), known to be processed by PC1 and PC2 (reviewed in refs. 5, 23, and 24). The regulation of POMC processing differs between the anterior and neurointermediate lobes of the pituitary (reviewed in ref. 25). Both the anterior as well as the intermediate lobe of the pituitary synthesize POMC; in the anterior lobe, POMC is cleaved by PC1 to yield ACTH, the final corticotropic product in this lobe, and other peptides such as β-lipotrophin. In the intermediate lobe, PC2 cleaves ACTH internally into the noncorticotropic peptides α-melanocyte stimulating hormone (α-MSH) and corticotropin-like intermediate lobe peptide (CLIP; refs. 26–28). In the 7B2 null pituitary, inactive PC2 fails to generate α-MSH in the intermediate pituitary, resulting in very high levels of intact ACTH in this lobe (22). Interestingly, 7B2 null animals develop a lethal form of Cushing's, whereas PC2 nulls remain generally healthy with a normal lifespan.
In this report, we have compared the 7B2 and PC2 null models to determine why only 7B2 null animals develop a Cushing's-like disease. Because dopaminergic input represents the primary control of peptide release from the intermediate lobe, we have studied the effect of 7B2 deletion on pituitary dopamine levels. We show the rescue of the lethal phenotype in the 7B2 null mouse by adrenalectomy, thereby confirming the crucial role of excess corticosterone in the development of the Cushing's-like disease. We find that the 7B2 null, but not the PC2 null nor the adrenalectomized 7B2 null, exhibits low levels of pituitary dopamine. Finally, we report that 7B2 null animals lacking adrenals exhibit late-onset obesity.
Materials and Methods
Animals.
Five-week-old 129/Sv 7B2 and 129/Sv-C57BL/6J PC2 WT and null animals (obtained by breeding 7B2 and PC2 heterozygote mice) were used for the pituitary ACTH, serum ACTH, and pituitary dopamine analyses. Three-week-old 129/Sv 7B2 WT and null animals were submitted to bilateral adrenalectomy. The original 7B2 null mutant mice were generated by embryonic stem-targeting technology, using a transposon-based system (29, 30). The PC2 null mutant mouse line was generated as described (17). Mice were housed in an Association for Assessment and Accreditation of Laboratory Animal Care-approved animal care facility; all procedures were approved by the Louisiana State University Health Sciences Center (LSUHSC) animal care committee.
Serum ACTH Assay.
Serum was prepared from trunk blood obtained from 5- to 6-week old animals killed by decapitation at the same time of the day (11 a.m.–2 p.m.). Clotted blood was centrifuged briefly to separate the serum from cells. Sera were collected individually in fresh tubes and stored at −70°C until use. Fifty μl of 7B2 null, PC2 null, 7B2 WT, and PC2 WT sera was assayed in duplicate by using the two-site Nichols human ACTH1–39 assay kit (Nichols Institute, San Juan Capistrano, CA), which is specific for intact ACTH1–39 does not recognize ACTH-cleavage products.
Analysis of Anterior Lobe (AP) and Neurointermediate Lobe (NIL) ACTH by RIA.
Pituitaries from WT and null animals of the same age and sex were removed and dissected into APs and NILs under a stereomicroscope. Separate lobes were individually homogenized by means of sonication in 250 μl of ice-cold 5 N acetic acid with 2 mg/ml BSA. The samples were stored frozen at −70°C before ACTH analysis. Ten μl of a 1/400 dilution of 7B2 null or PC2 null AP or NIL, or 10 μl of a 1/200 dilution of 7B2 or PC2 WT APs or NILs were assayed in duplicate with the Nichols human ACTH1–39 assay kit.
Adrenalectomy.
Eight 7B2 null males and twelve 7B2 WT males, all three-week-old animals, were anesthetized with halothane or ketamine (100 μl/100 g of mouse weight of a 100 mg/ml source; Schering-Plough) and subjected to bilateral adrenalectomy. Mice then were given water containing 0.9% saline to drink to counter the effect of mineralocorticoid removal. A prophylactic antibiotic (1% sulfamethazine sodium) was added to the drinking water. Adrenalectomized animals were killed 4 weeks after surgery. Only male 7B2 nulls are included in the data as female nulls were often too debilitated to survive surgery.
Dopamine.
Six pituitaries from 7B2 null and nine from WT mice were harvested and kept at -80°C until use. Each pituitary then was sonicated in 60 μl of 0.5 N HClO4 containing 10 mg/ml sodium bisulfite. Six PC2 null and WT animals as well as six adrenalectomized (ADX) 7B2 null and WT mice were treated and analyzed similarly. After centrifugation, 40 μl of each supernatant was injected at sensitivity 2 and at 0.5 V onto a liquid chromatography column (MF-6213 Phase II, BAS, West Lafayette, IN; ODS 3 μM, 100 × 3.2 mm) combined with a BAS electrochemical detector for catecholamine analysis. Citric acetate buffer (17.6 mM, pH 4) containing 50 mg/liter of sodium octyl sulfate and 10% methanol was used as the mobile phase. No differences were observed between females and males, and thus data have been pooled.
Bromocriptine Treatment.
Two 4-week-old 7B2 null males and littermate WT controls were treated for 3 days with a daily injection of 10 mg/kg 2-bromo-α-ergocryptine in PBS containing 15% ethanol. Blood was collected from the tail 1 h after the injections and also at the time of killing by rapid decapitation and kept from clotting by the addition of EDTA. Plasma samples were frozen at −80°C before assay for ACTH with the Nichols human ACTH1–39 assay kit.
Results
ACTH Levels and Distribution in 7B2 and PC2 Null Pituitaries.
By 5 to 6 weeks of age, 7B2 null animals show high-plasma ACTH and corticosteroid levels, with adrenocortical hyperplasia, and develop a lethal form of pituitary Cushing's syndrome (22); PC2 null animals do not (17). To understand why only the 7B2 null mutation results in a lethal disease state, we compared ACTH levels in 7B2 and PC2 null pituitaries using an RIA that measures intact ACTH (Fig. 1). Interestingly, PC2 WT females (which represent a mixture of the mouse strains C57BL/6J and 129/Sv) showed higher levels of pituitary ACTH than the 7B2 WT females (pure 129/Sv), indicating possible strain differences in pituitary ACTH content (Fig. 1). Increased pituitary ACTH in both nulls is expected because PC2 is not functional in PC2 or 7B2 null animals; consequently ACTH, generated from POMC by PC1, cannot be internally cleaved to α-MSH and CLIP and accumulates. Of note is the fact that this excess ACTH accumulates to an even greater degree in the PC2 null NIL than in the 7B2 null NIL; ACTH levels were increased by 65- and 13-fold in the PC2 and 7B2 nulls, respectively, compared with same-strain WT controls.
Figure 1.

Pituitary ACTH levels in isolated pituitary lobes of 7B2 and PC2 null mice and WT controls. The APs and NILs were dissected and separately homogenized before analysis for ACTH content with the Nichols human ACTH1–39 kit.
7B2 null anterior lobe ACTH is increased by 1.3-fold over WT animals, but some of this anterior lobe ACTH may represent contamination with NIL tissue (see below). PC2 null anterior lobe ACTH was not elevated over the WT content. PC2 null anterior lobe ACTH most likely does not represent contamination with NIL tissue, because in situ hybridization with POMC demonstrates that corticotrophs persist in the PC2 null anterior pituitary (see below). Like 7B2 nulls, PC2 null animals show profound elevations over their WT controls in the ACTH content of the NIL, the major site of pituitary PC2 (31). However, because both the PC2 and the 7B2 null animals show highly elevated levels of NIL ACTH, we conclude that a high content of NIL ACTH per se does not result in the development of the Cushing's-like disease.
In Situ Hybridization of POMC mRNA Indicates That POMC Production Is Absent in the 7B2 Null but Not PC2 Null Anterior Lobe.
To determine the effects of PC2 and 7B2 mutations on pituitary POMC expression, in situ hybridization was performed on pituitaries from PC2 and 7B2 mutant mice as well as WT mice. In WT mice of both genotypes, the well characterized pattern of POMC expression was observed with ≈5% of AP cells and all endocrine cells of the NIL found to express POMC (Fig. 2 Top). This pattern was qualitatively and quantitatively similar in the PC2 null (Fig. 2 Bottom Right). In marked contrast, in the AP of 7B2 null mice, POMC-expressing cells were not detectable, whereas POMC mRNA expression in the 7B2 NIL was similar to WT (compare with Fig. 2 Left).
Figure 2.
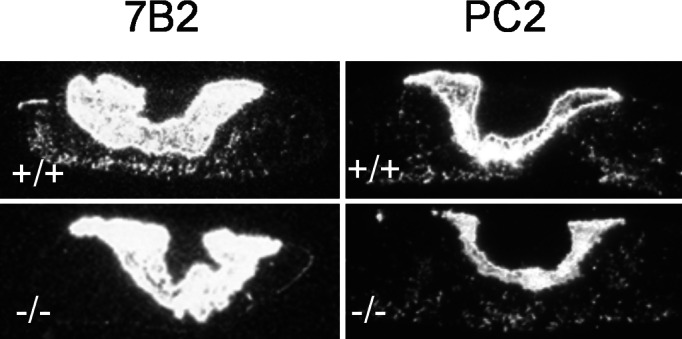
In situ hybridization of POMC mRNA in 7B2 and PC2 null animals. Wild-type animals exhibit POMC mRNA expression in 5% of the AP and in all NIL cells (Top). This pattern is similar in the PC2 null (Bottom Right). In the 7B2 null (Bottom Left), POMC-expressing cells are absent from the AP but are present in the NIL.
Circulating ACTH Is Increased in both the 7B2 and PC2 Null, but Is More Dramatically Increased in the 7B2 Null.
Previous results indicated that the circulating ACTH level in the 7B2 null was dramatically higher than in WT animals (22). Because PC2 nulls exhibit elevated pituitary ACTH but do not develop disease, we assayed sera from these animals for intact ACTH. We found that circulating ACTH in PC2 nulls also was increased compared with WT controls, but not nearly as dramatically as the 7B2 null (Fig. 3A)). Note that circulating ACTH also is increased when the 7B2 null is placed upon the C57BL/6J background (10 ng/ml average for the N5 male nulls); ACTH hypersecretion is thus unlikely to represent a background-specific phenomenon. Corticosterone levels did not vary between PC2 WT animals and PC2 nulls (Fig. 3B). This latter result is consistent with the fact that the PC2 null animals do not develop adrenal hyperplasia or any signs of Cushing's disease. We conclude that the elevation of circulating ACTH in the PC2 null is not sufficient to stimulate adrenal hyperactivity.
Figure 3.

(A and B) Circulating ACTH is somewhat increased in the PC2 null but is dramatically increased in the 7B2 null (A); corticosterone is increased only in the 7B2 null (B). Serum for both assays was prepared from 5-week-old animals killed by rapid decapitation at the same time of day. Note that circulating ACTH levels in the PC2 and 7B2 WT mice were not significantly different, whereas 7B2 null male and female ACTH levels were significantly increased over WT (***, P < 0.0001, Student's t test). A significant difference between PC2 null male animals and controls is shown by *** (P < 0.0001); differences between PC2 null females and controls are indicated by a single asterisk (*) and reached a significance of only P < 0.012.
Dopamine Analysis.
Dopaminergic control represents the major influence on POMC-derived peptide secretion (32) from the NIL (33). To assess possible alterations of the dopaminergic pathways in the 7B2 null that might lead to abnormally high ACTH secretion, extracts prepared from 7B2 null and WT pituitaries were analyzed by using liquid chromatography combined with electrochemical detection for catecholamines. Table 1 shows that the pituitary dopamine content is reduced by approximately 3/4 in the 7B2 null. In contrast, the PC2 nulls exhibit the same pituitary dopamine levels as their WT controls. These results are consistent with an impairment of dopaminergic control of ACTH secretion in the pituitary of the 7B2 null that does not occur in the PC2 null.
Table 1.
Dopamine contents of 7B2 null, PC2 null, ADX 7B2 null, and wild-type pituitaries
| Pituitaries | Dopamine pmol/pituitary |
|---|---|
| 7B2 WT | 1.4 ± 0.2 |
| 7B2 KO | 0.4 ± 0.1 |
| PC2WT | 1.3 ± 0.3 |
| PC2 KO | 1.2 ± 0.1 |
| 7B2 WTADX | 1.1 ± 0.2 |
| 7B2 KOADX | 1.2 ± 0.3 |
Data indicate means ± SD, n = 6 for 7B2 nulls and n = 9 for controls (P < 0.0001, Student's t test). n = 6 for PC2 nulls and controls (P = 0.15, no significant difference), and n = 6 for adrenalectomized (ADX) 7B2 nulls and controls (P = 0.53, no significant difference).
Bromocriptine Treatment Inhibits ACTH Release from the NIL.
To confirm the role of dopaminergic pathways in the control of intermediate lobe ACTH release, 7B2 null and WT mice received daily injections of bromocriptine, a dopaminergic agonist (34). The levels of circulating ACTH in each of the two 7B2 nulls were reduced by 46 and 51%, respectively, after one injection, and by 75 and 60% after three injections; levels of ACTH in the WT controls did not decrease. These results are consistent with direct dopaminergic control of intermediate lobe ACTH secretion.
Reversal of Lethal Phenotype by Adrenalectomy of 7B2 Nulls.
To assess the contribution of high circulating corticosterone to the early death of the 7B2 null, bilateral adrenalectomy was performed on groups of 3-week-old 7B2 null animals and WT controls. Mice were given water containing 0.9% saline to drink to counter the effect of mineralocorticoid removal. Survival times and weights of all groups were compared. Weight curves were generated from weekly measurements as an indication of general health (Fig. 4; these data showed that 7B2 null male mice lacking adrenals survive longer but develop severe obesity at about 10 weeks of age. Adrenalectomy per se is unlikely to be a factor in the development of obesity, because WT controls that underwent adrenalectomy showed similar weights compared with nonadrenalectomized controls (Fig. 4).
Figure 4.
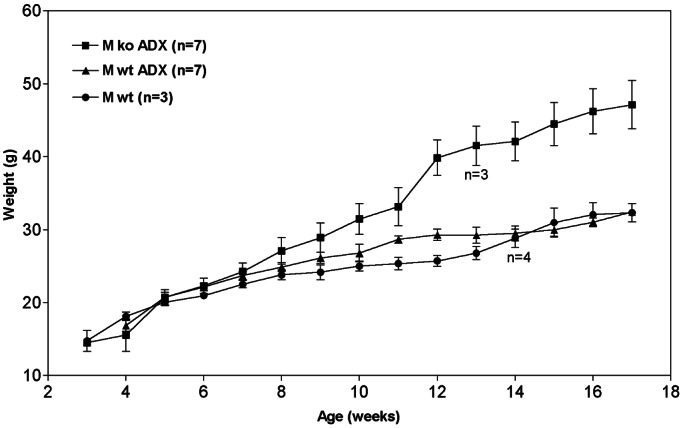
Adrenalectomy reverses the mortality observed at 5–6 weeks and also results in obesity. Mice were weighed once a week until being killed at 12 or 17 weeks (four adrenalectomized 7B2 null mice and three adrenalectomized 7B2 WT mice were killed at 12 weeks of age for pituitary ACTH analysis, hence the drop in numbers at this time).
By using in situ hybridization, POMC mRNA from adrenalectomized 7B2 null mice was found to reappear in the anterior lobe (compare Fig. 2 and Fig. 5A). These data confirm that corticosterone suppresses POMC and ACTH levels in the anterior lobe of the 7B2 null animals. Blood levels of ACTH and corticosterone returned to the normal range when measured at 4 weeks after adrenalectomy of 7B2 nulls (Fig. 5B). The presence of corticosterone in adrenalectomized animals most likely reflects the outgrowth of adrenocortical cells from vestiges left after surgery during the 4-week recovery period. Interestingly, adrenalectomy was able to restore pituitary dopamine levels to normal (Table 1); therefore, increased inhibitory tone most likely accounts for the reduction in circulating ACTH observed in adrenalectomized mice.
Figure 5.

(A and B) Adrenalectomy partially restores POMC expression in the anterior pituitary of the 7B2 null (A); circulating ACTH and corticosterone levels return to the normal range measured at 4 weeks after adrenalectomy (B).
Discussion
The origin of the lethal form of Cushing's-like disease exhibited by the 7B2 null animal (22) has been an enigma, given that the only previously known role for 7B2 is as a binding protein for the convertase PC2 (5), and that the PC2 null animal exhibits no symptoms of the disease (17). To arrive at determining factors for the development of the Cushing's-like illness, we have compared the pituitaries of 7B2 and PC2 null mice.
Our data indicate that the ACTH content in the NILs of both the 7B2 and PC2 null is dramatically higher than in WT animals. Interestingly, the ACTH content of the PC2 null NIL is much higher than that of the 7B2 null NIL; this finding is most likely caused by strain differences, as the WT controls differ in the same fashion, but also may reflect increased rates of secretion vs. retention of ACTH in the gland. Although circulating ACTH levels in the PC2 null (especially PC2 null female) mice do not approach the highly elevated levels of ACTH exhibited by 7B2 nulls, PC2 nulls do exhibit increased circulating ACTH. Nonetheless, the production of corticosterone remains at normal levels in PC2 null mice, suggesting that the adrenal cortex of PC2 nulls is resistant to stimulation by the increase in circulating ACTH. Several lines of evidence show that strain differences in rodents result in differential sensitivity to ACTH (33, 35, 36). Because the 7B2 null mouse was created in a pure 129/Sv background (22), and although the PC2 null has a mixed C57BL/6J and 129/Sv background (17), strain-specific differences in adrenal responsiveness could explain the fact that PC2 null mice have normal corticosterone levels despite the increase in circulating ACTH. The observation that the PC2 null NIL contains more intact ACTH than the 7B2 null, but does not hypersecrete, is interesting and suggests differential control of release in the two null models.
The location of the ACTH hypersecreting cells in the pituitary of 7B2 and PC2 nulls and WT controls was further explored by using POMC in situ hybridization. These experiments support the idea that POMC synthesis is essentially eliminated in the anterior lobe of the 7B2 but not of the PC2 null. It is likely that the shutdown of anterior lobe corticotrophs in 7B2 nulls is mediated by chronically elevated corticosterone, because POMC synthesis in the anterior lobe is well known to be controlled by corticosteroid levels (reviewed in ref. 37). Because PC2 nulls do not exhibit elevated corticosterone levels, they show no such diminution in anterior lobe POMC synthesis. Interestingly, POMC synthesis in the anterior lobe returns upon adrenalectomy of 7B2 nulls, illustrating that corticotrophs are still responsive to changes occurring in the hypothalamic-pituitary-adrenal (HPA) axis.
Many reports have shown that hypothalamic periventricular neurons exert a negative control on the secretion of NIL melanotroph POMC (reviewed in refs. 38–41). Indeed, inhibitory dopaminergic control represents the major influence on POMC-derived peptide secretion in the NIL (32) and is thought to be mediated via D2 receptors that are negatively coupled to adenylate cyclase (42). Several groups have shown that in vitro, either exogenous dopamine or dopamine released from endogenous stores within the intermediate lobe diminishes the spontaneous release of melanotrophic hormones from this lobe (42, 43). We speculated that a diminished pool of dopamine in neurointermediate nerve endings in the 7B2 null might be involved in the enhanced secretion of ACTH. To investigate this idea, we analyzed the pituitary dopamine content of 7B2 and PC2 nulls and controls, as well as adrenalectomized 7B2 nulls and controls. In support of our hypothesis, we found that the pituitary dopamine level of the PC2 null was similar to WT controls, whereas 7B2 nulls contained only a quarter of the normal levels of dopamine. Moreover, adrenalectomized 7B2 nulls—which exhibit normal circulating ACTH levels—also showed normal pituitary dopamine levels. These data lend further credence to a causative role for dopamine deficiency in the ACTH hypersecretion phenomenon observed in the 7B2 null. To confirm direct control by dopamine of ACTH secretion, 7B2 null animals and WT controls received a series of bromocriptine injections followed by determination of plasma ACTH levels. As expected, we found that circulating ACTH in the 7B2 nulls was lowered by bromocriptine treatment. These results imply that the ACTH hypersecretion phenomenon observed in the 7B2 nulls is mediated at least in part through regulation of dopaminergic pathways.
Dopaminergic mechanisms have been implicated previously in Cushing's disease: D2-receptor-deficient mice exhibit unexpectedly elevated ACTH levels with a corresponding increase of corticosterone and consequent hypertrophy of the adrenal gland (44). Moreover, the deletion of the dopamine transporter results in anterior pituitary hypoplasia, dwarfism, and inability to lactate, thus showing a crucial role for this transporter in dopaminergic control of pituitary function (most likely related to prolactin) (45). A subset of patients with Cushing's disease who develop Nelson's disease as a result of bilateral adrenalectomy can be treated successfully with the D2 agonist cabergoline, which effectively lowers circulating ACTH levels (46, 47). Lastly, monoamine oxidase inhibitors such as deprenyl are routinely given to dogs exhibiting pituitary Cushing's signs (48), which supports the idea that monoaminergic systems impact ACTH secretion in Cushing's.
Our data showing rescue of 7B2 null mice by adrenalectomy indicate a role for corticosteroids in the development of disease. The fact that normal pituitary dopamine levels are restored by adrenalectomy indicates that circulating corticosterone plays a major role in the ACTH hypersecretion phenomenon. Exactly how corticosterone could affect dopaminergic tone in the HPA axis is at present obscure. Corticosterone is known to modulate stress-induced dopamine release in the nucleus accumbens (49). Glucocorticoids could influence dopamine synthesis in the 7B2 null by acting on pituitary tyrosine hydroxylase (50). Alternatively, glucocorticoids also might influence dopamine release via neural mechanisms extrinsic to dopaminergic pathways. In addition, glucocorticoids have been shown to modify presynaptic dopaminergic transmission in the nucleus accumbens and in postsynaptic transmission in the dorsal striatum, exhibiting heterogeneous modulation within various dopaminergic projections (51).
Interestingly, adrenalectomized 7B2 nulls exhibit a profound obesity syndrome beginning at 10 weeks, which was an unexpected finding. However, our adrenalectomized animals do possess normal levels of circulating corticosterone, most likely from intense ACTH stimulation of small numbers of adrenal cells remaining after surgery during the 4-week recovery period. The late-onset obesity of the adrenalectomized 7B2 null is reminiscent of the POMC null, the Mc4r null (52, 53), and the fat/fat mouse, and may be related to a common lack of the PC2-generated peptide α-MSH in all of these null animals (54). However, PC2 nulls, which also lack α-MSH, do not exhibit significant obesity, suggesting that other factors also must play a role in the development of this condition. Moreover, the different null models considered (POMC, Mc4r, Mc3r, 7B2, and PC2 nulls) have all been made by using distinct mouse strains, a factor which adds considerable variability to hormonal and metabolic analyses (17, 22, 52, 55–60).
In conclusion, our data indicate that the loss of 7B2, but not PC2, results in the alteration of dopaminergic sensitivity in the intermediate lobe of the pituitary, either by means of a direct effect on dopaminergic mechanisms in the pituitary or hypothalamus, or by means of an indirect effect on adrenal corticosterone secretion. We speculate that once dopaminergic tone is reduced and ACTH hypersecretion is in place, a vicious cycle is set up whereby increased circulating corticosterone maintains ACTH hypersecretion, a process which can be interrupted by adrenalectomy. These data support the investigation of potential 7B2 effects on the regulation of pituitary catecholamine synthesis and/or adrenocortical function.
Acknowledgments
We thank Drs. N. H. Neff and M. Hadjiconstaninou (Department of Pharmacology, Ohio State University) and Dr. J. Porter (Louisiana State University Health Sciences Center) for the initial analyses of dopamine contents of 7B2 null and control mouse pituitaries, G. Hubbard for maintenance of the mouse colonies, and Dr. M. Sarac for mouse injections and critical comments on the manuscript. We also thank Drs. C. Westphal and P. Leder for supplying founder animals for the 7B2 null colony. This work was supported by National Institutes of Health Grants DK49703 (to I.L.), DA08622 (to J.E.P.), and DK13914 and DK20595, and by the Howard Hughes Medical Institute (to D.F.S.) I.L. was supported by a career development award from National Institute on Drug Abuse.
Abbreviations
- ACTH
adrenocorticotropic hormone
- POMC
proopiomelanocortin
- AP
anterior pituitary
- NIL
neurointermediate lobe
- WT
wild type
- α-MSH
α-melanocyte stimulating hormone
- PC2
prohormone convertase 2
References
- 1.Rouille Y, Duguay S J, Lund K, Furuta M, Gong Q, Lipkind G, Oliva A A, Chan S J, Steiner D F. Front Neuroendocrinol. 1995;16:322–361. doi: 10.1006/frne.1995.1012. [DOI] [PubMed] [Google Scholar]
- 2.Seidah N G, Chretien M. Curr Opin Biotechnol. 1997;8:602–607. doi: 10.1016/s0958-1669(97)80036-5. [DOI] [PubMed] [Google Scholar]
- 3.Zhou A, Webb G, Zhu X, Steiner D F. J Biol Chem. 1999;274:20745–20748. doi: 10.1074/jbc.274.30.20745. [DOI] [PubMed] [Google Scholar]
- 4.Steiner D F. Curr Opin Chem Biol. 1998;2:31–39. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- 5.Muller L, Lindberg I. Prog Nucleic Acid Res Mol Biol. 1999;63:69–108. doi: 10.1016/s0079-6603(08)60720-5. [DOI] [PubMed] [Google Scholar]
- 6.Braks J A M, Martens G J M. Cell. 1994;78:263–273. doi: 10.1016/0092-8674(94)90296-8. [DOI] [PubMed] [Google Scholar]
- 7.Zhu X, Lindberg I. J Cell Biol. 1995;129:1641–1650. doi: 10.1083/jcb.129.6.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller L, Zhu X, Lindberg I. J Cell Biol. 1997;139:625–638. doi: 10.1083/jcb.139.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsi K L, Seidah N G, De Serres G, Chretien M. FEBS Lett. 1982;147:261–266. doi: 10.1016/0014-5793(82)81055-7. [DOI] [PubMed] [Google Scholar]
- 10.Martens G J, Bussemakers M J, Jenks B G. Eur J Biochem. 1989;181:75–79. doi: 10.1111/j.1432-1033.1989.tb14695.x. [DOI] [PubMed] [Google Scholar]
- 11.Martens G J M. FEBS Lett. 1988;234:160–164. doi: 10.1016/0014-5793(88)81324-3. [DOI] [PubMed] [Google Scholar]
- 12.Waldbieser G C, Aimi J, Dixon J E. Endocrinology. 1991;128:3228–3236. doi: 10.1210/endo-128-6-3228. [DOI] [PubMed] [Google Scholar]
- 13.Brayton K A, Aimi J, Qiu H, Yazdanparast R, Ghatei M A, Polak J M, Bloom S R, Dixon J E. DNA. 1988;7:713–719. doi: 10.1089/dna.1988.7.713. [DOI] [PubMed] [Google Scholar]
- 14.Mbikay M, Grant S G N, Sirois F, Tadros H, Skowronski J, Lazure C, Seidah N G, Hanahan D, Chretien M. Int J Pept Protein Res. 1989;33:39–45. doi: 10.1111/j.1399-3011.1989.tb00681.x. [DOI] [PubMed] [Google Scholar]
- 15.Martens G J, Braks J A, Eib D W, Zhou Y, Lindberg I. Proc Natl Acad Sci USA. 1994;91:5784–5785. doi: 10.1073/pnas.91.13.5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Horssen A M, Van den Hurk W H, Bailyes E M, Hutton J C, Martens G J M, Lindberg I. J Biol Chem. 1995;270:14292–14296. doi: 10.1074/jbc.270.24.14292. [DOI] [PubMed] [Google Scholar]
- 17.Furuta M, Yano H, Zhou A, Rouille Y, Holst J J, Carroll R, Ravazzola M, Orci L, Furuta H, et al. Proc Natl Acad Sci USA. 1997;94:6646–6651. doi: 10.1073/pnas.94.13.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furuta M, Zhou A, Webb G, Ravazzola M, Orci L, Steiner D F. J Biol Chem. 2001;276:27197–27202. doi: 10.1074/jbc.M103362200. [DOI] [PubMed] [Google Scholar]
- 19.Furuta M, Carroll R, Martin S, Swift H H, Ravazzola M, Orci L, Steiner D F. J Biol Chem. 1998;273:1–7. doi: 10.1074/jbc.273.6.3431. [DOI] [PubMed] [Google Scholar]
- 20.Johanning K, Juliano M A, Juliano L, Lazure C, Lamango N S, Steiner D F, Lindberg I. J Biol Chem. 1998;273:22672–22680. doi: 10.1074/jbc.273.35.22672. [DOI] [PubMed] [Google Scholar]
- 21.Berman Y, Mzhavia N, Polonskaia A, Devi L A. J Biol Chem. 2001;276:1466–1473. doi: 10.1074/jbc.M008499200. [DOI] [PubMed] [Google Scholar]
- 22.Westphal C H, Muller L, Zhou A, Bonner-Weir S, Schambelan M, Steiner D F, Lindberg I, Leder P. Cell. 1999;96:689–700. doi: 10.1016/s0092-8674(00)80579-6. [DOI] [PubMed] [Google Scholar]
- 23.Castro M G, Morrison E. Crit Rev Neurobiol. 1997;11:35–57. doi: 10.1615/critrevneurobiol.v11.i1.30. [DOI] [PubMed] [Google Scholar]
- 24.Seidah N G, Chretien M. Brain Res. 1999;848:45–62. doi: 10.1016/s0006-8993(99)01909-5. [DOI] [PubMed] [Google Scholar]
- 25.Mains R E, Eipper B A. Handbook of Physiology. London: Oxford Univ. Press; 2000. pp. 85–101. [Google Scholar]
- 26.Zhou A, Bloomquist B T, Mains R E. J Biol Chem. 1993;268:1763–1769. [PubMed] [Google Scholar]
- 27.Benjannet S, Rondeau N, Day R, Chretien M, Seidah N G. Proc Natl Acad Sci USA. 1991;88:3564–3568. doi: 10.1073/pnas.88.9.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas L, Leduc R, Thorne B A, Smeekens S P, Steiner D, Thomas G. Proc Natl Acad Sci USA. 1991;88:5297–5301. doi: 10.1073/pnas.88.12.5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Devine S E, Boeke J D. Nucleic Acids Res. 1994;22:3765–3772. doi: 10.1093/nar/22.18.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Westphal C H, Leder P. Curr Biol. 1997;7:530–533. doi: 10.1016/s0960-9822(06)00224-7. [DOI] [PubMed] [Google Scholar]
- 31.Day R, Schafer M K, Watson S J, Chretien M, Seidah N G. Mol Endocrinol. 1992;6:485–497. doi: 10.1210/mend.6.3.1316544. [DOI] [PubMed] [Google Scholar]
- 32.Tilders F J, Mulder A H. J Endocrinol. 1975;64:63P–64P. [PubMed] [Google Scholar]
- 33.Gomez F, Lahmame A, de Kloet E R, Armario A. Neuroendocrinology. 1996;63:327–337. doi: 10.1159/000126973. [DOI] [PubMed] [Google Scholar]
- 34.Oyarce A M, Hand T A, Mains R E, Eipper B A. J Neurochem. 1996;67:229–241. doi: 10.1046/j.1471-4159.1996.67010229.x. [DOI] [PubMed] [Google Scholar]
- 35.Anisman H, Lacosta S, Kent P, McIntyre D C, Merali Z. Stress. 1998;2:209–220. doi: 10.3109/10253899809167284. [DOI] [PubMed] [Google Scholar]
- 36.Matthys L, Castello R, Zilz A, Widmaier E P. Neuroendocrinology. 1998;67:403–411. doi: 10.1159/000054339. [DOI] [PubMed] [Google Scholar]
- 37.Autelitano D J, Lundblad J R, Blum M, Roberts J L. Annu Rev Physiol. 1989;51:715–726. doi: 10.1146/annurev.ph.51.030189.003435. [DOI] [PubMed] [Google Scholar]
- 38.Chronwall B M, Hook G R, Millington W R. Endocrinology. 1988;123:1992–2002. doi: 10.1210/endo-123-4-1992. [DOI] [PubMed] [Google Scholar]
- 39.Stack J, Surprenant A. J Physiol. 1991;439:37–58. doi: 10.1113/jphysiol.1991.sp018655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goudreau J L, Lindley S E, Lookingland K J, Moore K E. Neuroendocrinology. 1992;56:100–105. doi: 10.1159/000126214. [DOI] [PubMed] [Google Scholar]
- 41.Cote T E, Eskay R L, Frey E A, Grewe C W, Munemura M, Stoof J C, Tsuruta K, Kerabian J W. Neuroendocrinology. 1982;35:217–224. doi: 10.1159/000123384. [DOI] [PubMed] [Google Scholar]
- 42.Munemura M, Cote T E, Tsuruta K, Eskay R L, Kebabian J W. Endocrinology. 1980;107:1676–1683. doi: 10.1210/endo-107-6-1676. [DOI] [PubMed] [Google Scholar]
- 43.Bower A, Hadley M E, Hruby V J. Science. 1974;184:70–72. doi: 10.1126/science.184.4132.70. [DOI] [PubMed] [Google Scholar]
- 44.Saiardi A, Borelli E. Mol Endocrinol. 1998;12:1133–1139. doi: 10.1210/mend.12.8.0144. [DOI] [PubMed] [Google Scholar]
- 45.Bosse R, Fumagalli F, Jaber M, Giros B, Gainetdinov R R, Wetsel W C, Missale C, Caron M G. Neuron. 1997;19:127–138. doi: 10.1016/s0896-6273(00)80353-0. [DOI] [PubMed] [Google Scholar]
- 46.Pivonello R, Faggiano A, Di Salle F, Filippella M, Lombardi G, Colao A. Endocrinol Invest. 1999;22:860–865. doi: 10.1007/BF03343660. [DOI] [PubMed] [Google Scholar]
- 47.Mercado-Asis L B, Yanovski J A, Tracer H L, Chik C L, Cutler G B., Jr J Clin Endocrinol Metab. 1997;82:514–517. doi: 10.1210/jcem.82.2.3742. [DOI] [PubMed] [Google Scholar]
- 48.Peterson M E. Vet Clin North Am Small Anim Pract. 2001;31:1005–1014. doi: 10.1016/s0195-5616(01)50010-8. [DOI] [PubMed] [Google Scholar]
- 49.Rouge-Pont F, Deroche V, Le Moal M, Piazza P V. Eur J Neurosci. 1998;10:3903–3907. doi: 10.1046/j.1460-9568.1998.00438.x. [DOI] [PubMed] [Google Scholar]
- 50.Ortiz J, De Caprio J L, Kosten T A, Nestler E J. Neuroscience. 1995;67:383–397. doi: 10.1016/0306-4522(95)00018-e. [DOI] [PubMed] [Google Scholar]
- 51.Barrot M, Abrous D N, Marinelli M, Rouge-Pont F, Le Moal M, Piazza V. Eur J Neurosci. 2001;13:812–818. doi: 10.1046/j.1460-9568.2001.01434.x. [DOI] [PubMed] [Google Scholar]
- 52.Huszar D, Lynch C A, Dunmore J H, Fang Q, Berkemeier L R, Gu W, Kesterson R A, Boston B A, Cone R D. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 53.Yaswen L, Diehl N, Brennan M B, Hochgeschwender U. Nat Med. 1999;5:1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- 54.Raffin-Sanson M L, Bertherat J. Eur J Endocrinol. 2001;144:207–208. doi: 10.1530/eje.0.1440207. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Y, Proenca R, Maffel M, Barone M, Leopold L, Friedman J M. Nature (London) 1994;372:425–431. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 56.Muglia L J, Jenkins N A, Gilbert D J, Copeland N G, Mazjoub J A. J Clin Invest. 1994;93:2066–2072. doi: 10.1172/JCI117201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muglia L J, Jacobson L, Dikkes P, Mazjoub J. Nature (London) 1995;373:427–432. doi: 10.1038/373427a0. [DOI] [PubMed] [Google Scholar]
- 58.Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A. Nat Genet. 1998;19:155–157. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 59.Bultman S J, Michaud E J, Woychik R P. Cell. 1992;71:1195–1204. doi: 10.1016/s0092-8674(05)80067-4. [DOI] [PubMed] [Google Scholar]
- 60.Butler A A, Kesterson R A, Khong K, Cullen M J, Pelleymounter M A, Dekoning J, Baetscher M, Cone R D. Endocrinology. 2000;141:3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]