

Abstract
The specificity of the reactions of nitric oxide (NO) with its neuronal targets is determined in part by the precise localizations of neuronal NO synthase (nNOS) within the cell. The targeting of nNOS is mediated by adapter proteins that interact with its PDZ domain. Here, we show that the nNOS adapter protein, CAPON, interacts with synapsins I, II, and III through an N-terminal phosphotyrosine-binding domain interaction, which leads to a ternary complex comprising nNOS, CAPON, and synapsin I. The significance of this ternary complex is demonstrated by changes in subcellular localization of nNOS in mice harboring genomic deletions of both synapsin I and synapsin II. These results suggest a mechanism for specific actions of NO at presynaptic sites.
Nitric oxide (NO), a major biological messenger molecule with multiple functions in the immune, cardiovascular, and nervous systems, is generated by three distinct enzymes, inducible NO synthase (NOS), endothelial NOS, and neuronal NOS (nNOS). Neurally generated NO has numerous roles including regulation of neurotransmitter release (1–4) and neuronal process extension (5) consistent with the multiple intracellular localizations of nNOS in the cytoplasm and cytoskeleton (6, 7). Because the cytosol contains compounds that are capable of interacting with NO, such as metal ions, ascorbate, glutathione, and superoxide, covalent modification of proteins by NO is enhanced by the juxtaposition of nNOS with its targets (8, 9).
The delivery of nNOS to discrete sites in neurons is mediated by adapter proteins. For instance, nNOS is linked to postsynaptic densities by the cytoskeletal protein PSD95/93 (9). This interaction places nNOS adjacent to other PSD95 ligands, such as the NR2 subunit of the N-methyl-D-aspartate receptor (10), which accounts for the efficient activation of nNOS by N-methyl-D-aspartate receptor stimulation (11) and may explain the preferential S-nitrosylation of NR2 subunits in vivo (12). Recently, we identified another adapter protein for nNOS, designated CAPON (13). CAPON contains a C-terminal PDZ domain-binding motif (13), which interacts with the N-terminal PDZ domain of nNOS. Evidence from binding studies indicates that CAPON is stoichiometrically associated with soluble nNOS (13), indicating that CAPON may serve as an nNOS-targeting protein in neurons. CAPON has an N-terminal phosphotyrosine-binding (PTB) domain whose ligands include Dexras1, a novel member of the ras family (8). The physiologic activity of Dexras1 is determined by nNOS and CAPON, as Dexras1 activation is selectively diminished in mice harboring a targeted deletion of nNOS (8), and Dexras1 activation by nNOS is enhanced in the presence of CAPON (8).
To seek other proteins that might be associated with CAPON and nNOS, we conducted blot overlay experiments by using the radiolabeled PTB domain of CAPON as a probe. We report the identification of synapsins I, II, and III as binding partners of CAPON. We also find that nNOS, CAPON, and synapsin I can form a ternary complex. The physiologic significance of these interactions is indicated by the changes in subcellular localization of nNOS and CAPON in mice with targeted deletion of both synapsin I and II.
Materials and Methods
Overlay Experiments.
For blot-overlay analysis, pGEX-4T2 (Amersham Pharmacia) was modified such that two phosphorylation sites for protein kinase A (PKA) encoded on complementary synthetic oligonucleotides were ligated into the EcoRI–SalI sites in the multiple cloning site to generate plasmid pGEX4T-2K (14). Kinase reactions and blot overlays were performed as described (15). Samples for overlay analysis were prepared by homogenizing the indicated tissues in homogenization buffer (50 mM Tris·HCl, pH 7.0/100 mM NaCl/2 mM EDTA/1 mM phenylmethylsulfonyl fluoride/20 μg/ml leupeptin), and 50 μg were loaded on gels and transferred to Immobilon-P membranes (Millipore). Purified bovine synapsin containing both Ia and Ib for overlay assays was prepared as described (16).
Purification of the 80-kDa PTB-Binding Protein.
Thirty rat brains were homogenized in 150 ml of homogenization buffer. The debris was removed by centrifugation at 800 × g for 5 min, and the supernatant was centrifuged at 15,000 × g for 15 min at 4°C. The supernatant was discarded, and the pellet was sonicated in homogenization buffer adjusted to 1% Triton X-100 and then incubated on ice for 30 min. After this solubilization step, the “cytoskeleton” fraction was obtained by centrifugation at 20,000 × g for 30 min. The supernatant (“membranes”) was discarded, and the pellet was washed once in 20 ml of homogenization buffer without Triton X-100.
The cytoskeletal fraction was solubilized by sonication in homogenization buffer adjusted to 1% sodium deoxycholate followed by incubation on ice for 30 min, and the debris was removed by centrifugation. The supernatant, which contained the 80-kDa PTB-binding protein, p80, was adjusted to 5% Triton X-100 and loaded onto 40 ml of Q-Sepharose. Under these conditions, all of the p80 flowed through the column. The flow-through was applied to a CM-Sepharose column and eluted in 100 ml of homogenization buffer in a gradient from 0–800 mM NaCl. Fractions were assayed by overlay assay, and fractions containing p80 were pooled, diluted to a final salt concentration of 50 mM NaCl in homogenization buffer without NaCl, and loaded onto a 10-ml SP-Sepharose column. This column was subjected to a linear NaCl gradient as with the CM-Sepharose column, and fractions that contained p80 were pooled and concentrated by using Amicon filtration devices according to the manufacturer's instructions. p80 was identified by aligning the band identified by overlay assay with the bands on the Coomassie stain of the same gel. p80 was excised from poly(vinylidene difluoride) membranes and sequenced by Edman degradation.
Binding Experiments.
Fusion proteins were prepared in Escherichia coli BL21(DE3) (Novagen) with glutathione-agarose (Sigma) as described (17), except that bacterial pellets were lysed in lysis buffer (50 mM Tris·HCl, pH 7.7/100 mM NaCl/2 mM EDTA), supernatants were adjusted to 1% Triton X-100, and protein was purified by using elution buffer (50 mM Tris·HCl, pH 7.7/100 mM NaCl/10 mM reduced glutathione/2 mM EDTA).
Synapsin constructs were prepared by PCR by using primers containing SalI and NotI sites, and the PCR products were subcloned into pCIS-GST (18). COS7 cells were transfected with plasmids for nNOS (19), CAPON (13), or synapsin by using the calcium phosphate method. Following transfection, cells were sonicated in buffer A (50 mM Tris·HCl, pH 7.7/100 mM NaCl/2 mM EDTA/1% Triton X-100) and cleared by centrifugation. This cellular lysate was incubated with glutathione-agarose for 1 h at 4°C and washed extensively in HNTG buffer (20 mM Hepes, pH 7.4/500 mM NaCl/10% glycerol/0.1% Triton X-100). The material remaining on the resin was eluted with SDS/PAGE sample buffer, and the indicated proteins were detected by immunoblotting by using nNOS- and CAPON-specific antibodies as described (13).
Immunoprecipitations were performed by homogenizing one rat cerebellum in 3 ml lysis buffer followed by centrifugation at 100,000 × g for 30 min at 4°C. Supernatant (0.2 ml) was incubated with 40 μl of protein A/G-agarose (Oncogene Science) and 5 μg of mouse anti-synapsin Ia/b antibody (Chemicon) or mouse anti-PKC-zeta (Santa Cruz Biotechnology) for 60 min at 4°C. The resins were then washed with IP wash buffer (50 mM Tris·HCl, pH 7.7/400 mM NaCl/2 mM EDTA) six times and eluted in 60 μl of 1 × SDS/PAGE sample buffer by boiling.
Equilibrium dialysis experiments were performed by using 1 nmol of purified bovine synapsin Ia/b (16) and 100 fmol of thrombin-cleaved 32P-PTB. 32P-PTB was adjusted to a final specific activity of 31,000 cpm per 100 fmol by using unlabeled protein. Dialysis was performed by using 10 ml of dialysis buffer (20 mM Hepes, pH 7.7/100 mM NaCl) with the synapsin and CAPON loaded in a volume of 1 ml in Spectra/Por MWCO 50,000-Da membranes (Spectrum Laboratories, Houston) for 48 h at 4°C.
Subcellular Fractionations.
Subcellular fractions were prepared as described (20). P3 and S3 represent the pellet and supernatant fractions obtained after high-speed centrifugation of S2. Mice harboring genomic deletions in both synapsin I and synapsin II and littermates have been described (21).
Results
The CAPON PTB Domain Interacts with an 80-kDa Brain-Enriched Protein.
CAPON has an N-terminal PTB domain (amino acids 20–180) (Fig. 1A), which has previously been shown to interact with Dexras1 (8). To identify other interacting proteins, we conducted binding studies by using a protein overlay assay with a radioactively labeled form of the PTB domain of CAPON. A fusion protein comprising the glutathione S-transferase (GST) domain, two cAMP-dependent PKA sites, and the PTB domain of CAPON was prepared in E. coli, immobilized to glutathione agarose, phosphorylated with [32P]ATP and PKA, and purified by elution with glutathione. This radioactive probe, 32P-PTB, was used in an overlay assay to probe blots containing proteins resolved by SDS/PAGE (Fig. 1B). We identified several radiolabeled bands including a prominent 80-kDa protein (p80). Using the overlay assay to probe for the presence of p80 in a variety of central and peripheral tissues, we found that p80, like CAPON, was only detected in brain (Fig. 1C).
Figure 1.

Identification of an 80-kDa CAPON PTB domain-binding protein, p80. (A) Schematic representation of CAPON. CAPON has a C-terminal PDZ domain-binding consensus sequence that binds nNOS. The N terminus contains a PTB domain. A fusion protein comprising GST, two PKA consensus phosphorylation sites, and the CAPON-PTB domain was phosphorylated in vitro with [32P]ATP and PKA. The radiolabeled protein, 32P-PTB, was used in overlay assays. (B) Protein overlays by using 32P-PTB detects a prominent 80-kDa protein in rat brain cortex homogenates (50 μg per lane). (C) p80 is detected in neuronal tissues, whereas it is undetectable in peripheral tissues.
Purification and Identification of p80 as Synapsin I.
We purified the 80-kDa protein by using a series of column fractionations, with the final step being a SP-Sepharose column (Fig. 2A). Through the rounds of chromatography, each collected fraction was subjected to the overlay assay with 32P-PTB to detect the presence of p80, and fractions containing p80 were pooled and diluted to an NaCl concentration of less than 50 mM before use on the subsequent column (Fig. 2B). In the final step, the majority of 32P-PTB binding activity (reflecting p80) was detected in a single fraction eluting at approximately 150 mM NaCl, with a smaller amount seen in the following fraction. Coomassie staining of the same fractions revealed a single predominant band at 80 kDa, which comigrated, and was found with similar intensity as seen in the p80 band seen by overlay, suggesting that this protein was indeed p80.
Figure 2.

Purification and identification of p80 as synapsin I. (A) Schematic of the purification strategy for p80. (B) Final step in p80 purification. Protein overlays of aliquots (50 μl) of the final chromatographic separation reveal that p80 comigrates with an 80-kDa band seen on Coomassie staining, indicating that the Coomassie-stained band is p80. (C) Alignment of p80-derived peptides with synapsin I. Two tryptic peptides derived from p80 align exactly with rat synapsin I a/b. (D) Confirmation that p80 is synapsin I. Synapsin I purified by immunoprecipitation was labeled by 32P-PTB in the overlay assay and exhibited the same electrophoretic mobility as p80 from cortical homogenates.
The region of the poly(vinylidene difluoride) membrane that contained p80 was excised and subjected to trypsinolysis followed by protein sequencing of two HPLC-purified peptides by sequential Edman degradation. Sequencing of two tryptic peptides revealed 100% identity to amino acids 257–269 and 282–299 of rat synapsin I (Fig. 2C). To confirm that synapsin I was p80, we immunoprecipitated authentic synapsin I and subjected it to the overlay assay with 32P-PTB. Immunoprecipitated synapsin I was labeled by the probe and comigrated with p80 from cortex cytoskeletal fractions (Fig. 2D). These findings indicate that p80 is synapsin I.
Amino Acids 200–300 of Synapsin I Bind CAPON.
Synapsin I can be alternatively spliced into synapsin Ia and Ib, which differ by an alternate C terminus (22). The N-terminal 659 aa are identical in both proteins. To determine whether CAPON selectively binds to one isoform, we assayed the ability of a purified synapsin I preparation containing both Ia and Ib to bind to the PTB domain in the overlay assay. To ensure resolution of the two bands, we ran the SDS/PAGE gel slightly longer than usual. 32P-PTB labeled both bands (Fig. 3A), indicating that the CAPON binding site lies within the N-terminal 659 aa.
Figure 3.

Characterization of the CAPON/Synapsin I interaction. (A) 32P-PTB binds both synapsin Ia and synapsin Ib. Purified bovine synapsin I (0.1 μg) containing an approximate molar ratio of 1:2 for the Ia and Ib isoforms, respectively, was assayed for binding by overlay with 32P-PTB. Binding of both isoforms indicates that the CAPON-binding region lies within residues 1–659 of the two alternatively spliced isoforms. (B) A domain consisting of amino acids 200–300 of synapsin I is the common region of interaction with CAPON. The indicated truncations of synapsin I were expressed as GST-fusion proteins in COS-7 cells along with an expression plasmid encoding CAPON. Following incubation of the lysates with glutathione agarose, complexes of synapsin I and CAPON were detected by immunoblot of protein retained on glutathione agarose. (C) Schematic diagram of the results in B. (D) CAPON interacts with synapsins I, II, and III. Each synapsin was expressed as a myc-tagged fusion protein in COS-7 cells and immunoprecipitated with an anti-myc antibody. The purified synapsins were subjected to overlay assay by using the 32P-PTB probe (Upper). An anti-myc blot of the immunoprecipitated synapsins is shown to compare the relative expression of the different synapsin isoforms (Lower). (E) CAPON is detected in synapsin I immunoprecipitates. Cerebellar membranes were solubilized with 1% Triton X-100 and then incubated with anti-synapsin I antibodies or the control antibody against PKC-zeta (Upper). Only immunoprecipitates prepared by using anti-synapsin I antibodies contained appreciable amounts of CAPON. Blots were stained with Coomassie blue to visualize the heavy chain of the immunoprecipitating antibody to confirm that approximately equivalent amounts of antibody were used in immunoprecipitation experiments (Lower).
To further delineate a binding site for CAPON in synapsin I, we expressed fusion proteins of GST and various fragments of synapsin I in COS7 cells along with CAPON. Using a GST pull-down assay, we found that fragments containing amino acids 1–300 or 200–700 pulled down cotransfected CAPON (Fig. 3 B and C). However, fragments of synapsin containing amino acids 400–705 did not interact with synapsin I, indicating that residues 200–300 contain the binding site of CAPON in synapsin I. GST-synapsin I fusions comprising amino acids 200–300 of synapsin also pulled down CAPON but less efficiently than larger fusion proteins, suggesting that this domain might not fold properly when truncated (data not shown).
CAPON Interacts with All Three Synapsin Family Members.
The region of synapsin I containing the CAPON binding site, amino acids 200–300, is a part of the highly conserved C domain common to all isoforms of synapsin (22–24), suggesting that CAPON might bind to all of them. We expressed myc-tagged synapsin Ia, IIa, and IIIa in COS7 cells and purified them by immunoprecipitation. The purified synapsins were then subjected to the overlay assay with 32P-PTB. Each of the three synapsins was capable of interacting with CAPON (Fig. 3D).
To further characterize the interaction of CAPON with synapsin, we performed equilibrium dialysis experiments by using 100 fmol of 32P-PTB and 1 nmol of purified bovine synapsin Ia/b. Following 48 h of dialysis, 70.4% of 32P-PTB remained bound to synapsin Ia/b, giving an approximate Kd of 39 nM.
CAPON and Synapsin I Interact in Vivo.
To determine whether CAPON and synapsin I interact in intact cells, we immunoprecipitated solubilized rat cerebellar membranes with an antibody to synapsin I. CAPON was detected in synapsin I immunoprecipitates but not in control immunoprecipitates (Fig. 3E).
Detection of an nNOS, CAPON, and Synapsin I Ternary Complex.
Because CAPON can bind to nNOS through the C-terminal PDZ-binding domain of CAPON and to synapsin I through the N-terminal PTB module, we speculated that CAPON can link nNOS to synapsin. To explore this possibility, we conducted GST pull-down experiments in COS7 cells transfected with GST-synapsin, CAPON, and nNOS in various combinations (Fig. 4). Glutathione-agarose pulled down nNOS in cells transfected with synapsin, CAPON, and nNOS, but not when CAPON was not transfected, establishing that CAPON is required for interactions between synapsin and nNOS. Neither nNOS nor CAPON bound to the resin directly, as neither was precipitated in the absence of GST-synapsin I. These findings indicate that nNOS, synapsin, and CAPON are capable of forming a ternary complex.
Figure 4.
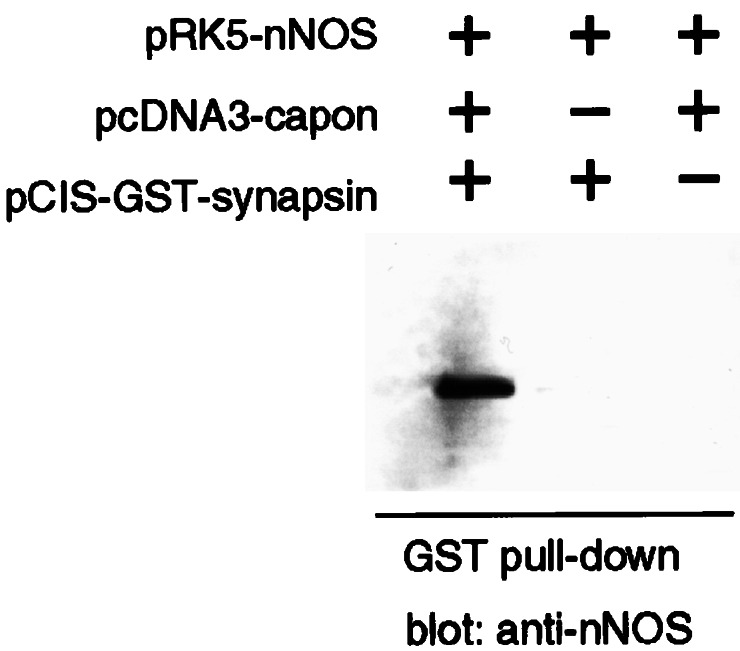
NOS, CAPON, and synapsin I form a ternary complex in transfected cells. COS7 cells were cotransfected with the indicated plasmids, and the lysates were incubated with glutathione-agarose. After extensive washing of the resin, bound nNOS was detected by immunoblot. nNOS adhered to the resin only in the presence of GST-synapsin 200–705 and CAPON, but not in the presence of only one, demonstrating the existence of a ternary complex composed of nNOS, CAPON, and synapsin I.
Altered Subcellular Localization of nNOS and CAPON in Synapsin Double Knockout Mice.
We determined regions of the cell in which nNOS, CAPON, and synapsin I are colocalized by subcellular fractionation of rat and mouse forebrain (Fig. 5). In rat brain, the subcellular localization of synapsin I was similar to that previously reported (20), with high densities in the crude synaptic vesicle fraction (LP2) that further increased after purification of synaptic vesicles through sucrose density centrifugation (SG2 fraction) and CPG chromatography (SV fraction) (Fig. 5A). Dilution of the SV fraction in a high salt buffer (200 mM NaCl) led to about 90% depletion of synapsins from the salt-treated SV fraction. Synapsins II and III showed similar subcellular distributions and sensitivity to ionic strength. CAPON and nNOS localizations were similar to each other, although nNOS was also associated with soluble fractions such as LS2, where CAPON was barely present. Localizations of CAPON, nNOS, and synapsins in rat brain displayed a partial overlap. Thus, CAPON and, to a lesser extent, nNOS were consistently observed in fractions containing synaptic vesicles (e.g., LP2, SG2, SV, and P3), although their pattern of enrichment was not fully correlated with that of synapsins I and II. The pools of CAPON and nNOS associated with synaptic vesicle fractions could not be dissociated by high salt treatment (Fig. 5A) and did not result from contamination of the highly purified synaptic vesicle fractions, as other nerve terminal proteins (e.g., synuclein, Na+/K+-ATPase, or GAP-43) were not significantly detected in the SG2 and SV fractions (F.B., unpublished results). Differences in CAPON and synapsin I levels were also observed, including relatively high levels in SG1 and SG4 fractions, which had very low levels of synapsin I, and the similar enrichment of CAPON in SG1 and SG2 in contrast to the marked concentration of synapsin I in SG2 (Fig. 5).
Figure 5.

Subcellular localization of CAPON and nNOS is altered in synapsin I−/−, synapsin II−/− double knockout mice. (A) Subcellular fractionation of rat brain. Subcellular fractions from rat brain were blotted for synapsin I, nNOS, and CAPON as indicated. (B) Subcellular fractions (50 μg) derived from wild-type (W) or double knockout mice (K) were blotted with synapsin I, nNOS, CAPON, or FAK-specific antibodies. CAPON and nNOS levels were increased in S2, P3, SG1, SG3, and SG4 fractions of knockout mice relative to wild-type mice. Levels of FAK were not changed in wild-type vs. double knockout mice. H, homogenate; S1, postnuclear supernatant; S2, supernatant of P2; P2, crude synaptosomes; LP1, crude synaptic plasma membranes; LS1, supernatant of LP1; LP2, crude synaptic vesicles; LS2, supernatant of LP2 (synaptosol); SG1-SG4, sucrose gradient fractions of LP2 in which SG2 is enriched in synaptic vesicles and SG4 in small synaptic membranes; SV, synaptic vesicles purified from the SG2 fraction by controlled-pore glass chromatography; SSV, salt-treated synaptic vesicles prepared by elution of the SV fraction with 200 mM NaCl; S3, supernatant of P3 after high-speed centrifugation of S2 (cytosol); P3, microsomes. Subcellular fractionations were performed twice with essentially identical results.
If the localizations of CAPON and nNOS in various neuronal compartments are, at least in part, determined by interactions with the synapsins, then deletion of synapsins should alter the subcellular distribution of CAPON and nNOS. To explore this possibility, we compared the subcellular distribution of nNOS and CAPON in wild-type mice and mice with a targeted deletion of both synapsin I and II (21) (Fig. 5B). Comparison of nNOS and CAPON in wild-type and double knockout mice revealed marked alterations in their levels in specific subcellular fractions. Thus, in the knockout mice, levels of CAPON and nNOS were tripled in S2 and P3 fractions containing microsomal membranes as well as some synaptic vesicles and other cytoskeletal constituents. To purify fractions enriched in various synaptic components, the LP2 pellet was fractionated on a sucrose gradient. In the synapsin double knockout mice, CAPON and nNOS levels were markedly increased in the SG1 and, to a lesser extent, in the SG4 fractions, whereas there was no change in CAPON and nNOS in the SG2 fraction containing synaptic vesicles. A control protein, focal-adhesion kinase (FAK), exhibited identical localizations in both wild-type and double knockout mice (Fig. 5B). No significant differences in CAPON and nNOS levels and/or distribution were observed in single synapsin I or synapsin II knockout mice with respect to wild-type littermates (not shown).
Discussion
NO mediates its effects by stimulating guanylyl cyclase and by posttranslational modification of proteins. Because NO is highly reactive, nonspecific interactions of NO are minimized by localizing nNOS in proximity to NO targets. Here, we show that the nNOS adapter protein CAPON interacts with the synapsin family of proteins. This interaction is mediated by the CAPON PTB domain. CAPON is able to assemble a ternary complex comprising nNOS, CAPON, and synapsin. We also find changes in the subcellular localization of nNOS and CAPON in mice lacking both synapsin I and synapsin II.
Several roles of synapsins are similar to those of nNOS. Synapsins regulate neurotransmitter release and synaptic plasticity and are enriched in presynaptic terminals, where they have a role in the assembly of synaptic vesicle clusters (25) and synaptogenesis (21, 26). Synapsins also participate in neurite outgrowth and extension (21, 26). These effects are mediated in part by a diverse set of binding proteins such as src (27, 28), annexin VI (29), fodrin (30), neurofilaments (31), and actin (32). Synapsins may function as scaffolding proteins that target a diverse set of proteins to presynaptic sites. Like synapsins, NO has been localized in presynaptic terminals to the presynaptic cytoskeleton, including actin and microtubules (6), as well as synaptic vesicles (6). NO has multiple functions within neurons, such as influencing release of neurotransmitters from synaptic vesicles (2, 33, 34), through a mechanism that most likely involves S-nitrosylation (35). Additionally, nNOS−/− mice display defects in dendritic arborization (5), which are similar to dendritic defects found in synapsin I knockout mice (26). It is intriguing to speculate that the deficits in dendritic morphology or neurotransmitter release in synapsin-deficient mice reflect decreased access of nNOS to synaptic sites.
Synapsin binds to the PTB domain located at the N terminus of CAPON. The PTB domain of CAPON is most similar to the PTB domain of mouse numb, which, like CAPON, lacks certain residues that are expected to contact the phosphotyrosine based on extrapolation from the crystal structure of the Shc PTB domain (36). In the case of numb, its protein targets lack phosphotyrosine, as exemplified by Numb-associated protein kinase (37) and LNX (38). Similarly, Dexras1, the other identified ligand of CAPON PTB domain, also has no requirement for phosphotyrosine for binding to occur (8). Synapsin I has previously been shown to be phosphorylated on serine and threonine residues, but tyrosine phosphorylation has not been detected, suggesting that the CAPON–synapsin interactions are unlikely to be phosphotyrosine-dependent. The highly conserved calcium- and ATP-binding region C domain of still unknown function (39) contains the CAPON-binding region. The CAPON–synapsin interaction does not require ATP or calcium, as overlay assays were done in the absence of nucleotides and divalent ions.
Because the exact identity of the constituents of various subcellular fractions is never definitive, it is difficult to interpret the physiologic meaning of the increases of CAPON and nNOS in certain subcellular fractions of synapsin double knockout mice. The total amount of CAPON and nNOS in crude homogenates of the brain is not notably altered in the knockout mice, implying that the increases in certain fractions most likely reflect translocation of CAPON and nNOS from other fractions, rather than changes in expression levels. Because no fractions have pronounced decreases of CAPON and nNOS, it is likely that a modest decrease in some fractions would be sufficient to account for the increases in other fractions. Nonetheless, the marked and selective alterations in localization suggest that the association of CAPON and nNOS with synapsins physiologically determines their intracellular localizations. Conceivably, the marked increases of CAPON and nNOS in the S2, P3, SG1, and SG4 fractions may reflect slightly higher expression levels of these proteins in synapsin double knockout mice; however, the specificity of the increases in specific subcellular fractions rather than generalized increases in all subcellular fractions makes this possibility less likely and implies a significant change in their subcellular distribution. Other proteins that bind nNOS, including PIN/LC8/DLC8 (14), heat shock protein 90 (40), and phosphofructokinase (41), may also function as targeting proteins for nNOS. Interestingly, a PIN/LC8-binding consensus sequence has been found in dynamin (42), providing another mechanism by which nNOS may be targeted presynaptically.
In summary, CAPON has now been found to function as an adapter protein linking nNOS to specific targets such as Dexras1 (8) and synapsins. nNOS is also coupled to N-methyl-D-aspartate receptors through PSD95 (11, 43), and this binding is a determinant of nNOS targeting to postsynaptic sites. Synapsins are exclusively presynaptic (22) and may be a determinant of presynaptic targeting of nNOS. Because NO reacts relatively nonspecifically with cysteines to form nitrosothiol adducts, which are capable of modulating protein activity (12, 44), anchoring nNOS to specific proteins may deliver neurally generated NO selectively to its physiologic targets. Indeed, the linkage of nNOS to Dexras1 by CAPON mediates NO-dependent activation of Dexras1 (8). Similarly, targeting of nNOS to synapsin may facilitate selective exposure of various synapsin-associated proteins to NO.
Acknowledgments
We thank H. T. Kao for genotyping synapsin double knockout mice and C. Ferris for valuable suggestions. This work was supported by U.S. Public Health Service Grant MH-18501 and Research Scientist Award DA00074 (to S.H.S.), a Young Investigator Award from the National Alliance for Research on Schizophrenia and Depression (to S.R.J.), and Telethon-Italy Grant 1131, AIRC-Italy, and MURST-Cofin 2000 (to F.B.).
Abbreviations
- GST
glutathione S-transferase
- NOS
nitric-oxide synthase
- nNOS
neuronal nitric-oxide synthase
- PKA
protein kinase A
- PTB
phosphotyrosine-binding domain
References
- 1.Kano T, Shimizu-Sasamata M, Huang P L, Moskowitz M A, Lo E H. Neuroscience. 1998;86:695–699. doi: 10.1016/s0306-4522(98)00179-1. [DOI] [PubMed] [Google Scholar]
- 2.Montague P R, Gancayco C D, Winn M J, Marchase R B, Friedlander M J. Science. 1994;263:973–977. doi: 10.1126/science.7508638. [DOI] [PubMed] [Google Scholar]
- 3.Hanbauer I, Wink D, Osawa Y, Edelman G M, Gally J A. NeuroReport. 1992;3:409–412. doi: 10.1097/00001756-199205000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Sporns O, Jenkinson S. Neuroscience. 1997;80:1057–1073. doi: 10.1016/s0306-4522(97)00152-8. [DOI] [PubMed] [Google Scholar]
- 5.Inglis F M, Furia F, Zuckerman K E, Strittmatter S M, Kalb R G. J Neurosci. 1998;18:10493–10501. doi: 10.1523/JNEUROSCI.18-24-10493.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loesch A, Belai A, Burnstock G. J Neurocytol. 1994;23:49–59. doi: 10.1007/BF01189816. [DOI] [PubMed] [Google Scholar]
- 7.Hecker M, Mulsch A, Busse R. J Neurochem. 1994;62:1524–1529. doi: 10.1046/j.1471-4159.1994.62041524.x. [DOI] [PubMed] [Google Scholar]
- 8.Fang M, Jaffrey S R, Sawa A, Ye K, Luo X, Snyder S H. Neuron. 2000;28:183–193. doi: 10.1016/s0896-6273(00)00095-7. [DOI] [PubMed] [Google Scholar]
- 9.Brenman J E, Chao D S, Gee S H, McGee A W, Craven S E, Santillano D R, Wu Z, Huang F, Xia H, Peters M F, et al. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 10.Kornau H C, Schenker L T, Kennedy M B, Seeburg P H. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- 11.Sattler R, Xiong Z, Lu W Y, Hafner M, MacDonald J F, Tymianski M. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- 12.Jaffrey S R, Erdjument-Bromage H, Ferris C D, Tempst P, Snyder S H. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 13.Jaffrey S R, Snowman A M, Eliasson M J, Cohen N A, Snyder S H. Neuron. 1998;20:115–124. doi: 10.1016/s0896-6273(00)80439-0. [DOI] [PubMed] [Google Scholar]
- 14.Jaffrey S R, Snyder S H. Science. 1996;274:774–777. doi: 10.1126/science.274.5288.774. [DOI] [PubMed] [Google Scholar]
- 15.Kavanaugh W M, Williams L T. Science. 1994;266:1862–1865. doi: 10.1126/science.7527937. [DOI] [PubMed] [Google Scholar]
- 16.Bahler M, Greengard P. Nature (London) 1987;326:704–707. doi: 10.1038/326704a0. [DOI] [PubMed] [Google Scholar]
- 17.Smith D B, Johnson K S. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 18.Tsai R Y, Reed R R. BioTechniques. 1997;23:794–800. doi: 10.2144/97235bm06. [DOI] [PubMed] [Google Scholar]
- 19.Bredt D S, Hwang P M, Glatt C E, Lowenstein C, Reed R R, Snyder S H. Nature (London) 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- 20.Huttner W B, Schiebler W, Greengard P, De Camilli P. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferreira A, Chin L S, Li L, Lanier L M, Kosik K S, Greengard P. Mol Med. 1998;4:22–28. [PMC free article] [PubMed] [Google Scholar]
- 22.Sudhof T C, Czernik A J, Kao H T, Takei K, Johnston P A, Horiuchi A, Kanazir S D, Wagner M A, Perin M S, De Camilli P, et al. Science. 1989;245:1474–1480. doi: 10.1126/science.2506642. [DOI] [PubMed] [Google Scholar]
- 23.Kao H T, Porton B, Czernik A J, Feng J, Yiu G, Haring M, Benfenati F, Greengard P. Proc Natl Acad Sci USA. 1998;95:4667–4672. doi: 10.1073/pnas.95.8.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosaka M, Sudhof T C. J Biol Chem. 1998;273:13371–13374. doi: 10.1074/jbc.273.22.13371. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Chin L S, Shupliakov O, Brodin L, Sihra T S, Hvalby O, Jensen V, Zheng D, McNamara J O, Greengard P, Andersen P. Proc Natl Acad Sci USA. 1995;92:9235–9239. doi: 10.1073/pnas.92.20.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chin L S, Li L, Ferreira A, Kosik K S, Greengard P. Proc Natl Acad Sci USA. 1995;92:9230–9234. doi: 10.1073/pnas.92.20.9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Onofri F, Giovedi S, Vaccaro P, Czernik A J, Valtorta F, De Camilli P, Greengard P, Benfenati F. Proc Natl Acad Sci USA. 1997;94:12168–12173. doi: 10.1073/pnas.94.22.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foster-Barber A, Bishop J M. Proc Natl Acad Sci USA. 1998;95:4673–4677. doi: 10.1073/pnas.95.8.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inui M, Watanabe T, Sobue K. J Neurochem. 1994;63:1917–1923. doi: 10.1046/j.1471-4159.1994.63051917.x. [DOI] [PubMed] [Google Scholar]
- 30.Iga M, Inui M, Sobue K. Biochem Biophys Res Commun. 1997;231:852–855. doi: 10.1006/bbrc.1997.6202. [DOI] [PubMed] [Google Scholar]
- 31.Steiner J P, Ling E, Bennett V. J Biol Chem. 1987;262:905–914. [PubMed] [Google Scholar]
- 32.Greengard P, Benfenati F, Valtorta F. Adv Second Messenger Phosphoprotein Res. 1994;29:31–45. doi: 10.1016/s1040-7952(06)80005-4. [DOI] [PubMed] [Google Scholar]
- 33.Meffert M K, Premack B A, Schulman H. Neuron. 1994;12:1235–1244. doi: 10.1016/0896-6273(94)90440-5. [DOI] [PubMed] [Google Scholar]
- 34.Hirsch D B, Steiner J P, Dawson T M, Mammen A, Hayek E, Snyder S H. Curr Biol. 1993;3:749–754. doi: 10.1016/0960-9822(93)90022-g. [DOI] [PubMed] [Google Scholar]
- 35.Meffert M K, Calakos N C, Scheller R H, Schulman H. Neuron. 1996;16:1229–1236. doi: 10.1016/s0896-6273(00)80149-x. [DOI] [PubMed] [Google Scholar]
- 36.Zhou M M, Ravichandran K S, Olejniczak E F, Petros A M, Meadows R P, Sattler M, Harlan J E, Wade W S, Burakoff S J, Fesik S W. Nature (London) 1995;378:584–592. doi: 10.1038/378584a0. [DOI] [PubMed] [Google Scholar]
- 37.Chien C T, Wang S, Rothenberg M, Jan L Y, Jan Y N. Mol Cell Biol. 1998;18:598–607. doi: 10.1128/mcb.18.1.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dho S E, Jacob S, Wolting C D, French M B, Rohrschneider L R, McGlade C J. J Biol Chem. 1998;273:9179–9187. doi: 10.1074/jbc.273.15.9179. [DOI] [PubMed] [Google Scholar]
- 39.Esser L, Wang C R, Hosaka M, Smagula C S, Sudhof T C, Deisenhofer J. EMBO J. 1998;17:977–984. doi: 10.1093/emboj/17.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bender A T, Silverstein A M, Demady D R, Kanelakis K C, Noguchi S, Pratt W B, Osawa Y. J Biol Chem. 1999;274:1472–1478. doi: 10.1074/jbc.274.3.1472. [DOI] [PubMed] [Google Scholar]
- 41.Firestein B L, Bredt D S. J Biol Chem. 1999;274:10545–10550. doi: 10.1074/jbc.274.15.10545. [DOI] [PubMed] [Google Scholar]
- 42.Liang J, Jaffrey S R, Guo W, Snyder S H, Clardy J. Nat Struct Biol. 1999;6:735–740. doi: 10.1038/11501. [DOI] [PubMed] [Google Scholar]
- 43.Christopherson K S, Hillier B J, Lim W A, Bredt D S. J Biol Chem. 1999;274:27467–27473. doi: 10.1074/jbc.274.39.27467. [DOI] [PubMed] [Google Scholar]
- 44.Stamler J S, Simon D I, Osborne J A, Mullins M E, Jaraki O, Michel T, Singel D J, Loscalzo J. Proc Natl Acad Sci USA. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]