

Abstract
Cell walls of the Arabidopsis mutant mur2 contain less than 2% of the wild-type amount of fucosylated xyloglucan because of a point mutation in the fucosyltransferase AtFUT1. The mur2 mutation eliminates xyloglucan fucosylation in all major plant organs, indicating that Arabidopsis thaliana fucosyltransferase 1 (AtFUT1) accounts for all of the xyloglucan fucosyltransferase activity in Arabidopsis. Despite this alteration in structure, mur2 plants show a normal growth habit and wall strength. In contrast, Arabidopsis mur1 mutants that are defective in the de novo synthesis of l-fucose exhibit a dwarfed growth habit and decreased wall strength [Reiter, W. D., Chapple, C. & Somerville, C. R. (1993) Science 261, 1032–1035]. Because the mur1 mutation affects several cell wall polysaccharides, whereas the mur2 mutation is specific to xyloglucan, the phenotypes of mur1 plants appear to be caused by structural changes in fucosylated pectic components such as rhamnogalacturonan-II. The normal growth habit and wall strength of mur2 plants casts doubt on hypotheses regarding roles of xyloglucan fucosylation in facilitating xyloglucan–cellulose interactions or in modulating growth regulator activity.
The cell walls of higher plants determine the boundaries of plant form, represent the main sink for photoassimilated carbon, protect against pathogen invasion, and provide mechanical strength to the plant body (1, 2). Cell walls comprise primarily cellulose microfibrils, cross-linking glycans (also referred to as hemicelluloses), pectic polysaccharides, and small amounts of structural proteins (2, 3). Compared with other biological macromolecules, little is known about the synthesis and assembly of plant cell walls. The main components of pectic material are homogalacturonans, and the highly branched rhamnogalacturonans I and II (RG-I and RG-II). The major cross-linking glycan in the primary cell wall of most higher plants is xyloglucan, a polysaccharide composed of a (1 → 4)-β-D-glucan backbone substituted at regular intervals by α-D-xylosyl(1 → 6) residues. In most plant families, a subset of the xylose residues bear β-D-galactosyl(1 → 2) residues, some of which are in turn decorated by α-L-fucosyl(1 → 2) units to form a trisaccharide side chain (3, 4); however, other types of xyloglucan structures have been found in some species (5). Cellulose and xyloglucan interact to form a three-dimensional network hypothesized to be a major load-bearing element within the cell wall (3, 6). Previous studies have suggested that a key component in the association between cellulose and xyloglucan is the presence of the L-fucose-containing trisaccharide side-chain. Computer modeling of xyloglucan structure predicts that fucose-containing xyloglucans adopt spatial conformations more favorable for cellulose binding than nonfucosylated xyloglucan (7). Fucosylated xyloglucans bind cellulose in vitro at a 2-fold higher rate than do nonfucosylated xyloglucans (8); however, xyloglucan fucosylation is not absolutely required for the formation of cellulose–xyloglucan networks (9).
Fucose-containing xyloglucan is also thought to play a role in the regulation of plant growth. Several studies have shown that xyloglucan-derived oligosaccharides, called oligosaccharins, act as inhibitors of auxin-stimulated elongation of pea epicotyls (reviewed in ref. 10). The biological activity of these oligosaccharins depends on the presence of terminal L-fucose (11, 12).
Recently a (1 → 2)-α-L-fucosyltransferase was purified from pea epicotyls, and an Arabidopsis coding region with high sequence similarity to this protein was identified and termed Arabidopsis thaliana fucosyltransferase 1 (AtFUT1) (13, 14). Using nonfucosylated tamarind seed xyloglucan as a substrate AtFUT1 catalyzes the addition of L-fucose at the appropriate D-galactose residue. The Arabidopsis genome contains nine AtFUT1-like sequences (14), the functions of which are currently unknown.
Although the majority of work on the determinants of cell wall tensile properties has focused on the xyloglucan/cellulose network, the pectic polysaccharide RG-II may play a significant role in cell wall integrity. Recent work on RG-II indicates that it forms a homodimer cross-linked by borate esters (15–17). Boron is essential for normal plant growth (18), and the covalent bridging of RG-II via borate esters may be a major determinant of cell wall porosity (19).
To determine the functions of specific cell wall polysaccharides and clone cell wall-related genes, Arabidopsis mutants with altered monosaccharide composition of their cell wall material have been isolated (20, 21). Three of these mutant lines (mur1, mur2, and mur3) exhibit significant decreases in the fucose content of cell wall polymers (20). The mur1 plants are defective in the interconversion of GDP-D-mannose to GDP-L-fucose (22), which leads to the absence of fucose in glycoproteins, pectins, and xyloglucan (23, 24). The mur1 mutants are slightly dwarfed and have a more than 50% reduction in wall strength (21). Interestingly, approximately one-third of the missing L-fucose residues in mur1 xyloglucan are replaced by L-galactose, which is structurally similar to L-fucose (24).
Whereas mur1 plants are completely deficient in cell wall L-fucose levels, mur2 and mur3 plants are both approximately 50% reduced in total cell wall L-fucose content (20). This study reports the genetic and biochemical characterization of mur2 plants, indicating a reduction in xyloglucan fucosylation to about 1% of wild type, due to a mutation in the AtFUT1 gene.
Materials and Methods
Plant Lines and Growth Conditions.
Unless otherwise stated, all plants were grown in environmental chambers at 23°C and 70% relative humidity under continuous fluorescent light (60–70 μmol⋅m−2⋅s−1). Seeds were planted on either ProMix BX soil, or on nutrient agar plates (25).
Construction of a mur1, mur2 Double Mutant.
To obtain mur1, mur2 double mutants, mur2 plants were crossed to plants carrying the mur1–2 allele, and several mur1 plants were identified in the F2 generation based on their complete lack of fucose. Progeny of these mur1 plants were screened for partial fucose deficiency in the presence of 20 mM L-fucose, which rescues the mur1 phenotype.
Genetic Mapping and Sequence Determination.
To map the mur2 locus at high resolution, a mapping population of approximately 300 plants was generated. F1 plants from a mur2/mur2 (Columbia) X MUR2/MUR2 (Ler) cross were allowed to self-pollinate, and the F2 progeny were screened by means of gas-liquid chromatography of alditol acetates for the mur2 phenotype (21). Individual F2 plants homozygous for the mur2 mutation were allowed to self-pollinate, and DNA was isolated (26) from bulked F3 plant material. A rough map position was established using PCR-amplifiable markers as described (27). Fine mapping was done by means of Southern analysis using the 32P-labeled restriction fragment length polymorphism (RFLP) markers mi320 and m246 and continued with the use of 32P-labeled bacterial artificial chromosomes (BACs) that mapped between mi320 and m246. Nucleic acid hybridizations (Southern analysis and BAC library screening) were done as described (22). Nucleotide sequences were obtained from both strands by using the ABI Prism Big Dye Terminator Cycle Sequence Reaction Kit (Perkin–Elmer Applied Biosystems) and an ABI Prism 377 DNA sequencer.
Genetic Complementation.
A 7-kb SacI fragment containing the AtFUT1 gene was cloned into the pCAMBIA 1301 plant transformation vector (CAMBIA, Canberra, Australia), and transformed into Escherichia coli XL1 Blue MRF′ supercompetent cells following the manufacturer's protocol (Stratagene). Plasmid DNA was isolated as described (22), and used to transform Agrobacterium tumefaciens GV3101/pMP90 (28), employing a Bio-Rad Genepulser. Recombinant A. tumefaciens cells were then used to transform wild-type Columbia and mur2 plants as described (29). T1 plants were selected on plates containing MS salts and the appropriate antibiotics, then transferred to soil and allowed to self-pollinate. The resulting T2 plants were selected on plates as described above, and T3 plants homozygous for the transgene were analyzed for cell wall fucose content as described (21). As a control mur2 plants were transformed as described above with the pCAMBIA 1301 vector alone.
Cell Wall Fractionation.
Plants were grown in pots for approximately 20 days and placed in the dark for 72 h to deplete starch reserves. Approximately 15 g of leaf material were harvested and ground into a fine powder with a combination of liquid N2 and dry ice. The ground material was resuspended in 50 ml of cold 1× extraction buffer (0.1 M Mops/NaOH, pH 7.0, containing 1.5% SDS and 5 mM sodium bisulfite), and cell disruption was verified by light microscopy. The homogenate was centrifuged for 10 min at 15,000 rpm in a Sorvall SS34 rotor at 4°C. The resulting supernatants (S1 fraction) were used to investigate carbohydrate content of glycoproteins including N-linked glycans and arabinogalactan proteins. The pellet was washed twice with 50 ml of 0.25× extraction buffer, centrifuging between each wash. The pellet was incubated for 1 h at 40°C in 25 ml of PAW (phenol/acetic acid/water, 2:1:1, w/vol/vol), a solvent for proteins and phenolics (30). Following centrifugation the supernatant was discarded and the residue was kept for further fractionation. Four subsequent extractions were used to isolate pectic material: (i) 15 ml of 50 mM CDTA, pH 7.5, containing 0.02% thimerosal, incubated overnight at 23°C; (ii) 15 ml of ice-cold 50 mM sodium carbonate, with stirring at 4°C overnight; (iii) 15 ml of 50 mM sodium carbonate containing 20 mg ml−1 NaBH4, with stirring at 23°C overnight; and (iv) 160 units of endopolygalacturonase (EPG) in 10 ml of 0.1 M sodium acetate, pH 5.2, 0.02% thimerosal, with constant agitation at 35°C overnight. Following centrifugation the pellet was washed once with 30 ml of water. The sodium carbonate extracts were neutralized with acetic acid, and all fractions were dialyzed against water overnight at 4°C. Isolation of cross-linking glycans was done by sequential extraction with 1 M KOH and 4 M KOH, which disturbs hydrogen-bond interactions (31, 32). After neutralization with acetic acid these xyloglucan-enriched fractions were dialyzed against water, concentrated by lyophilization, and stored at −20°C. Glycoproteins were precipitated from the S1 fraction as described (33), resuspended in 4 M guanidinium chloride, and dialyzed against water.
Purification of Xyloglucan Oligosaccharides.
Equal proportions of the dialyzed 1 M KOH and 4 M KOH extracts were combined and samples containing 50–200 mg of total carbohydrate were adjusted to 50 mM sodium acetate (pH 5.5). Five microliters of a 1:100 dilution of Trichoderma viride cellulase (Megazyme, Bray, Ireland; 500 units⋅ml−1 in 3.2 M ammonium sulfate, pH 7) was added and samples were incubated at 37°C overnight. Boiled digestion products (20–100 μl) were separated on a Dionex HPLC system with a CarboPac PA-1 anion exchange column equilibrated in 200 mM NaOH. Oligosaccharides were eluted in 200 mM NaOH, using a 20-min linear gradient of sodium acetate from 0–80 mM, followed by a 10-min linear gradient of sodium acetate from 80–200 mM. Xyloglucan-derived oligosaccharides were detected by pulsed amperometric detection (Dionex), and pooled fractions were desalted by passage through a proton exchange column (On-Guard H-columns, Dionex), followed by lyophilization to remove residual acetic acid.
Monosaccharide and Linkage Composition Analysis.
Purified xyloglucan oligosaccharides were converted into partially methylated alditol acetates (PMAAs) as described (34), and separated by means of GC-MS on a 30-m SP2330 capillary column (35). Monosaccharides were quantified as alditol acetates by using standard procedures (21).
Electrospray MS and MS/MS.
Samples obtained from Trichoderma cellulase digestion were analyzed by electrospray MS on a Finnigan MAT LCQ (Thermoquest, San Jose, CA) mass spectrometer system. Freeze-dried samples containing 0.1–1 mg of carbohydrate were dissolved in 100 μl of water, and 10 μl of glacial acetic acid and 10 μl of methanol were added. The source voltage was set at 3.5 kV, the capillary voltage was varied between 20 and 30 V, depending on sample abundance, and the capillary temperature was 225°C. Typical background source pressures were 1.5 × 10−5 torr (1 torr = 133 Pa). The sample flow rate was 10 μl/min, and the drying gas was nitrogen. The liquid chromatography quadrupole (LCQ) was scanned to m/z 2,000. Helium was introduced into the system to an estimated pressure of 1 × 10−5 torr to improve trapping efficiency, and it also acted as the collision gas during collisionally activated decomposition (MS/MS) experiments. The stereoisomers XXLG and XLXG were differentiated by their unique MS/MS fragmentation patterns to produce diagnostic ions of m/z 659 and m/z 773, respectively. Quantitation was based on response factors determined by the relative abundance of these fragments recovered after MS/MS of XLLG (m/z 1409) obtained from tamarind xyloglucan.
Scanning Electron Microscopy.
Rosette leaves from wild-type, mur2, and complemented mur2 plants were excised and immediately placed in 4% glutaraldehyde in 0.1 M phosphate buffer (sodium phosphate, pH 7.4). Samples were vacuum infiltrated for 5 min and fixed overnight at 4°C. The following day samples were washed three times for 15 min each in phosphate buffer, and incubated overnight in 2% osmium tetroxide diluted in phosphate buffer. Samples were washed as before and dehydrated through an ethanol series (30%, 50%, 70%, 90%, and 100% twice). Tissues were dried in a Polaron (Watford, U.K.) E3000 critical point dryer for 3 h, using CO2 as the transition fluid. Carbon tape was applied to precleaned aluminum stubs and the dried tissue was attached to the tape. Stubs were left for one day under vacuum, then gold coated for 1 min with a Polaron E5100 Series II sputter coater. Tissue was examined in a Zeiss (Leo) DSM 982 field emission scanning electron microscope at an acceleration voltage of 2 kV.
Heterologous Expression in Mammalian Cells.
Full-length cDNAs corresponding to the wild-type and mur2 genes were cloned into the mammalian expression vector pcDNA3 (Invitrogen). The plasmid was transformed into E. coli XL1 Blue MRF′ following the manufacturer's instructions (Stratagene). Plasmid DNA was used to transfect 293T cells by calcium phosphate transfection following the manufacturer's instructions (Invitrogen). Approximately 8 × 105 cells were plated in 100-mm-diameter plates and grown in DMEM containing 10% FBS, 4.5 μg⋅ml−1 glucose, and 1 μg⋅ml−1 fungizone (amphotericin B). Cells were grown overnight in a humidified 5% CO2 incubation chamber at 37°C. Four hours before transfection the growth media were removed, and 10 ml of fresh growth medium was added to each plate. Approximately 20 μg of DNA was used to transfect each plate. Twenty-four hours after transfection cells were suspended in 50 mM Hepes/KOH, pH 7.4, 0.25 M sucrose, 0.4% CHAPS, and 1% mammalian protease inhibitor mixture (Sigma). After 10 min of incubation at 4°C the cells were disrupted by nitrogen cavitation (750 psi for 5 min; 1 psi = 6.89 kPa). Cell debris was spun down at 1000 × g for 5 min, and the supernatant was immediately used for fucosyltransferase assays or frozen at −80°C in small aliquots.
Fucosyltransferase Assays.
Assays were performed essentially as described (13). Each reaction contained approximately 100,000 dpm of GDP-[U-14C]-L-fucose at 10 GBq/mmol, 33 μl of tamarind xyloglucan (3 mg/ml stock), 33 μl of cell free extract (≈66 μg of total protein), and 100 μl of FT assay buffer (50 mM Mops/KOH, pH 6.8, with 0.05% Triton X-100 and 10 mM MnCl2). Assays were conducted at room temperature for 20 min, stopped by the addition of 800 μl of cold absolute ethanol, and incubated at 4°C for at least 2 h. Samples were then applied to glass fiber filters in a vacuum manifold and washed three times with 15 ml of cold 70% ethanol. Radioactivity was determined by liquid scintillation counting.
Generation of AtFUT1-Specific Antibodies.
Approximately 0.1 mg of the N-terminal peptide MDQNSYRRRSSPIRTTT was conjugated to keyhole limpet hemocyanin and used to immunize New Zealand White rabbits. After 10 weeks the animals were bled and the serum was collected by centrifugation. Before use the antibody was affinity-purified using an affinity column with the antigenic peptide conjugated to it. Peptide synthesis and antibody preparation was carried out by Research Genetics (Huntsville, AL).
Immunoblot Analysis.
Approximately 66 μg of protein isolated from transfected 293T cells were loaded on an SDS–8% polyacrylamide gel (36). After electrophoretic separation proteins were transferred to a PVDF membrane (Millipore) by using the Panther SemiDry Electroblotter following the manufacturer's protocol (Owl Scientific, Woburn, MA). Immunological detection was carried out as described (37), using the anti-AtFUT1 primary antibody diluted 1:1000 in PBS for 2 h at room temperature, followed by the secondary antibody (alkaline phosphate conjugated goat anti-rabbit-IgG; Sigma) diluted 1:50,000 for 45 min. Visualization was carried out by chemiluminescence using CDP-Star (Boehringer Mannheim) following the manufacturer's protocol. Protein molecular weights were estimated using a wide range multicolored protein marker (NEN Life Science).
Results
Positional Cloning of the MUR2 Gene.
To clone the gene carrying the mur2 mutation, a mapped-based approach was undertaken (Fig. 1A). Extensive mapping identified markers mi320 and m246 [Chromosome II, 9.5 and 11 cM on the Lister and Dean recombinant inbred genetic map (38), respectively] to be flanking the gene carrying the mur2 mutation. Because we suspected that mur2 plants may be defective in a glycosyltransferase, a BAC library was screened with a cDNA encoding the AtFUT1 fucosyltransferase (13). Of 14 BACs hybridizing to AtFUT1, one clone (T18E12) mapped to the region known to contain the gene affected by the mur2 mutation (Fig. 1A). Sequencing the AtFUT1 gene in the wild-type and mur2 plants identified a G to A transition in mur2 plants changing aspartic acid residue 550 close to the carboxy terminus of the protein to an asparagine. Interestingly, the alteration in amino acid sequence creates a potential N-glycosylation site (N-X-S/T) by changing the amino acid sequence from DIS to NIS; however, it is not known whether N-glycosylation occurs at this position in the mutant protein. To verify that the MUR2 gene had been cloned, mur2 plants were transformed with the wild-type AtFUT1 gene by using an Agrobacterium-based transformation system. Analysis of the transgenic plants for cell wall monosaccharide content revealed L-fucose levels similar to wild-type Arabidopsis plants, indicating that the complementation was successful (Fig. 1B).
Figure 1.

(A) Positional cloning of the mur2 gene. A map position was established between markers mi320 (9.5 cM) and m246 (11 cM) on chromosome 2, using previously characterized restriction fragment length polymorphisms (RFLPs). The closest recombinants were found using BAC T8K22 and m246 (one recombinant per marker), thus this region spanning approximately 350 kbp was identified as containing the MUR2 gene. A gene corresponding to the AtFUT1 cDNA (13) was identified on BAC T18E12 and sequenced in wild-type and mur2 plants revealing a single-point mutation. (B) The AtFUT1 gene complements the mur2 mutation. Six independent mur2 plants transformed with the AtFUT1 gene have fucose levels similar to wild type, whereas mur2 plants and mur2 plants transformed with vector alone have reduced fucose levels. Fucose was quantitated from total leaf cell wall material as described (21). Fucose levels from pooled leaf material were compared relative to rhamnose, arabinose, xylose, mannose, and galactose. Complementation was verified by electrospray MS on xyloglucan oligosaccharides from AtFUT1-transformed mur2 plants (data not shown).
Enzymatic Activity of MUR2 and mur2 Proteins.
Using a mammalian expression system, wild-type and mutant MUR2 proteins were produced and assayed for xyloglucan fucosyltransferase activity by using the nonfucosylated tamarind seed xyloglucan as the fucose acceptor. Both proteins were approximately 65 kDa in size when separated by SDS/PAGE, which is in reasonable agreement with the predicted molecular weight of 63 kDa (Fig. 2A). When assayed for activity the wild-type MUR2 protein showed substantial incorporation of radiolabeled L-fucose into the acceptor substrate, whereas an equal amount of the mutant mur2 protein had an activity similar to nontransfected control protein (Fig. 2B). Equal expression of wild-type and mur2 proteins was verified by immunoblot analysis (Fig. 2A).
Figure 2.
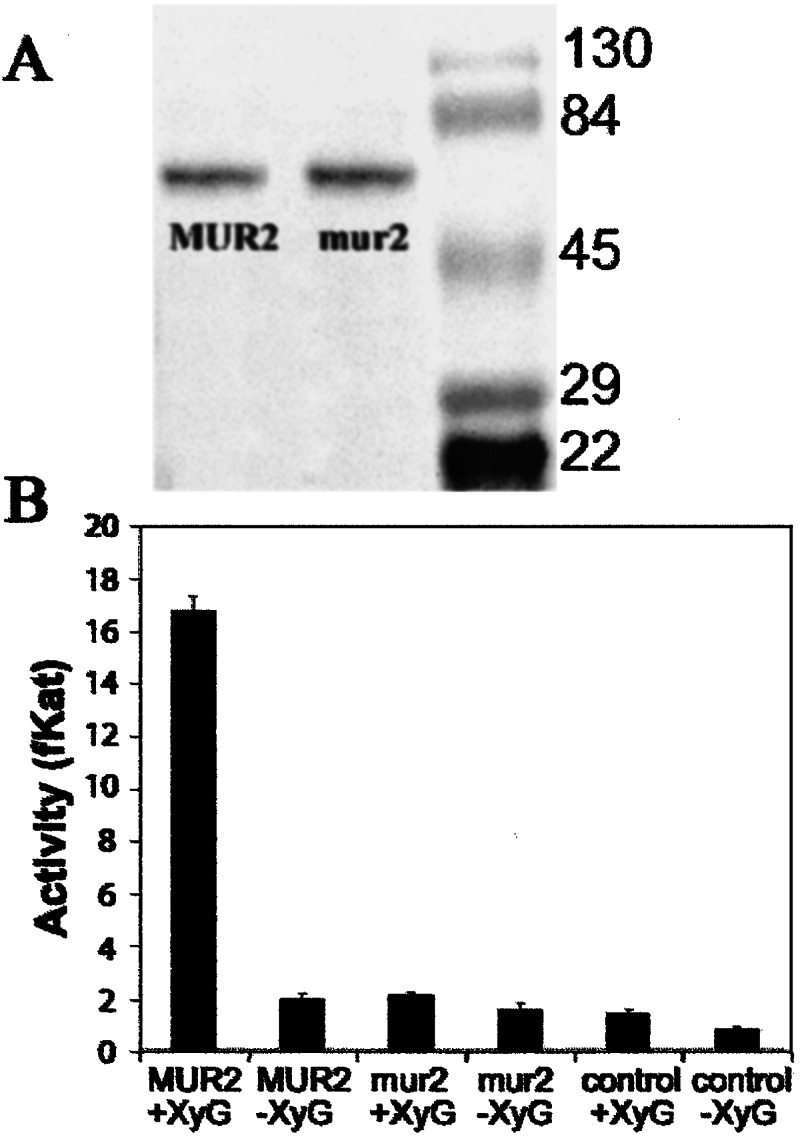
Immunoblot and enzymatic activity of wild-type and mutant MUR2 proteins expressed in mammalian cells. (A) Both wild-type MUR2 and mutant mur2 proteins are approximately 65 kDa in size when separated by SDS/PAGE, which is close to the molecular mass predicted from the amino acid sequence (63 kDa). The third lane represents a size marker. (B) Fucosyltransferase assays were performed with tamarind xyloglucan, which is nonfucosylated, and radiolabeled GDP-l-fucose. Xyloglucan fucosylation was measured by determining the incorporation of radiolabeled fucose into ethanol-insoluble material by using liquid scintillation counting. One fKat is defined as incorporation of one fmol of fucose into ethanol-insoluble material per second. Equal amounts of expressed protein were assayed as determined by immunoblots. Control protein was isolated from nontransfected cells, and the control protein concentration was similar to the protein concentration in the experimental samples. Assays were conducted in the presence (+XyG) or absence (−XyG) of tamarind xyloglucan as an acceptor substrate.
The Only Polysaccharide Affected in the mur2 Mutant Is Xyloglucan, with a More than 98% Reduction in l-Fucose Levels.
A cell wall fractionation experiment was undertaken to determine the effect of the mur2 mutation on each L-fucose containing cell wall polymer (xyloglucan, RG-I, and RG-II) as well as the L-fucose content of glycoproteins (i.e., N-linked glycans plus arabinogalactan proteins). Upon initial separation of glycoproteins, pectins, and cross-linking glycans, only the latter class of polymers was affected by the mur2 mutation (Fig. 3A). Because the initial fractionation was not sufficient to purify specific polysaccharides to homogeneity, xyloglucan oligosaccharides were purified from the alkali-soluble fractions via endoglucanase digestion followed by HPLC. The monosaccharide compositions and linkage types were determined by gas chromatography–mass spectrometry (GC-MS). Wild-type xyloglucan contained an abundance of fucose, whereas only trace amounts (less than 2% of wild type) were present in mur2 xyloglucan (Fig. 3A). The relative amounts of 2-linked galactose were 3.6% in wild type and 0.04% in mur2 xyloglucan, representing a loss of 99% of the substitution at the normally fucosylated galactose residue. These data also indicated that the missing fucose residues were not replaced by other monosaccharides.
Figure 3.

Fucose content of glycoproteins and cell wall components in wild-type and mur2 plants. Glycoproteins and cell wall polymers were purified as described in Materials and Methods. (A) Relative fucose content of leaf-derived glycoproteins, pectins, and xyloglucan from wild-type and mur2 plants as measured by gas-liquid chromatography of alditol acetates (21). Error bars represent SE with a sample number of four. Pectic fractions were combined before GLC analysis. Xyloglucan oligosaccharides were purified by high pH anion-exchange chromatography before GLC analysis. (B) Electrospray MS analysis of wild-type and mur2 xyloglucan. Xyloglucan oligosaccharides were isolated as described in Materials and Methods, and their nomenclature is illustrated in Table 1. Samples were analyzed using a Finnigan MAT liquid chromatograph quadrupole mass spectrometer system (Thermoquest). The m/z values of ionized xyloglucan fragments are indicated.
Further analysis of wild-type and mur2 xyloglucan by electrospray MS-MS supported the results from the monosaccharide and linkage composition analysis. Endoglucanase-generated xyloglucan fragments were subjected to mass determination and compared with known values for xyloglucan oligosaccharides. Table 1 illustrates the relative mol % of each xyloglucan oligosaccharide. The major difference between wild type and mur2 was the nondetectability of m/z 1,393 and m/z 1,555 ions in mur2, corresponding to the fucosylated xyloglucan oligosaccharides XXFG and XLFG, respectively (Fig. 3B). The mur2 samples also contained large quantities of the galactosylated oligosaccharides XXLG (m/z 1,247) and XLLG (m/z 1,409) because of the lack of conversion to their fucosylated forms (XXFG and XLFG, respectively).
Table 1.
Oligosaccharide content of wild type and mur2 xyloglucan (mol %) based on electrospray MS
 |
Although initially performed on leaf tissue, the absence of mur2 xyloglucan fucosylation was confirmed using material isolated from stems, flowers, and roots. The mur2 plants from various growth stages were also shown to be lacking fucosylated xyloglucan (data not shown). Lastly, electrospray MS-MS analysis of xyloglucan from mur2 plants or a mur1, mur2 double mutant did not detect the L-galactosylated oligosaccharides XXJG and XLJG, which are present uniquely in mur1 xyloglucan (24). This result indicates that the replacement of L-galactose for L-fucose that occurs in mur1 xyloglucan does not occur in the mur1, mur2 double mutant (data not shown).
Cell Wall Tensile Strength and Growth Habit of mur1, mur2 and a mur1, mur2 Double Mutant.
Previous work concerning the cell wall tensile strength of the L-fucose-deficient mur1 mutant indicated that when compared with wild-type plants, mur1 plants require about one-half the force to break elongating inflorescence stems (21). Using an extensiometer (21), the cell wall tensile strengths of wild-type, mur1, mur2, and a mur1, mur2 double mutants were compared. The wall strength of mur2 plants was indistinguishable from wild type, whereas mur1, mur2 plants had a decrease in wall strength similar to mur1 plants (Table 2).
Table 2.
Comparison of the effects of the mur1 and mur2 mutations on growth and cell wall strength
| WT Columbia | mur1 | mur2 | mur1, mur2 | |
|---|---|---|---|---|
| Growth habit | Normal | Dwarfed | Normal | Dwarfed |
| Wall strength* | 20.2 ± 5.3 | 9.1 ± 2.5 | 20.8 ± 6.4 | 10.9 ± 1.5 |
| Xyloglucan fucosylation | + | − | − | − |
| Xyloglucan l-galactosylation | − | + | − | − |
N/mg cell wall material, as measured in an extensiometer (21).
Along with a decrease in cell wall tensile strength, mur1 plants are slightly dwarfed (21). Whereas this phenotype persisted in the mur1, mur2 double mutant, mur2 plants had a growth habit indistinguishable from wild type. Furthermore, wild-type and mur2 plants responded similarly to cold stress, heat stress, and conditions of high salinity (data not shown).
The mur2 Plants Contain Altered Trichome Papillae.
Although macroscopic inspection of mur2 plants showed no visible phenotype, ultrastructural analysis by scanning electron microscopy (SEM) revealed an alteration in trichome structure (Fig. 4). Trichomes are unicellular, nonglandular hairs that protrude from the surfaces of many plant species including Arabidopsis. Trichomes are nonessential to Arabidopsis plants (39), and although they normally contain three branches, different Arabidopsis ecotypes contain a variety of branching patterns (40). In later stages of development wild-type trichomes form papillae, which are small protrusions from the cell wall. In mur2 plants these papillae appear wrinkled and collapsed when compared with wild type plants. Trichome papillae appear wild type in mur2 plants transformed with the AtFUT1 gene, indicating that the alteration in papillae structure is due to the mur2 mutation (data not shown).
Figure 4.
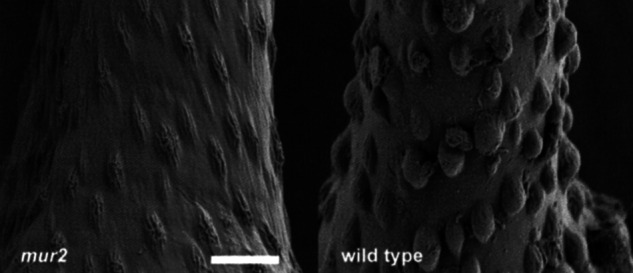
Surface structures of wild-type and mur2 trichomes. Axenically grown plants were fixed, stained, and dehydrated as described in Materials and Methods. (Scale bar, 5 μm.)
Discussion
To identify plants defective in the fucosylation of specific cell wall polymers, we studied the mur2 mutant of Arabidopsis that shows a 50% reduction in the fucose content of cell wall material. To determine the molecular basis of the mur2 mutation the MUR2 gene was positionally cloned and shown to be identical to AtFUT1, a recently characterized xyloglucan fucosyltransferase (13).
In most higher plants, including Arabidopsis, L-fucose is found in glycoproteins, the pectic polysaccharides RG-I and RG-II, and in xyloglucan. Fractionation studies indicated that mur2-derived glycoproteins and pectins contained wild-type levels of L-fucose, whereas mur2-derived xyloglucan isolated from all major plant organs was essentially nonfucosylated. A detailed analysis of xyloglucan structure in mur2 plants, using electrospray MS-MS and linkage composition analysis, revealed that the missing fucose residues were not replaced by any other monosaccharide, resulting in a polysaccharide structure unlike any known primary cell wall xyloglucan. However, mur2 xyloglucan was structurally similar to the seed storage xyloglucans of nasturtium and tamarind. In this sense, the change of xyloglucan structure is more severe in mur2 plants than in the fucose-deficient mur1 mutant, where one-third of the missing L-fucose residues are replaced by L-galactose (24). The mur2 and mur1, mur2 plants had xyloglucan structures missing terminal L-fucose and L-galactose residues, whereas both mur1 and mur1, mur2 plants displayed dwarfism and reduced wall strength (Table 2). These results indicate that the altered growth habit and wall strength of mur1 plants is neither caused by a lack of xyloglucan fucosylation nor the replacement of L-fucose with L-galactose in mur1 xyloglucan. Because lack of glycoprotein fucosylation does not lead to an obvious visible phenotype or reduction in wall strength (21), alterations in fucosylated cell wall components such as RG-I and RG-II appear to be responsible for the altered growth and wall properties of mur1 plants. This result is supported by recent studies indicating that mur1 plants are partially deficient in the cross-linking of RG-II molecules by borate esters, and that the visible phenotypes of mur1 plants are rescued by growth in the presence of millimolar concentrations of borate (41).
The virtual absence of fucosylated xyloglucan in all organs of mur2 plants, the ability of MUR2 protein to fucosylate xyloglucan in vitro, and the normal fucosylation of mur2 glycoproteins and pectic components indicate that the MUR2 protein is both necessary and sufficient for xyloglucan fucosylation, and does not appear to be involved in the fucosylation of cell wall components other than xyloglucan. Although the Arabidopsis genome contains at least nine MUR2 paralogs (14), our genetic and biochemical data suggest that none of them is functionally redundant with MUR2 under laboratory conditions. MUR2-like proteins may be involved in the fucosylation of pectic components or arabinogalactan proteins. Alternatively, one or more of the hypothetical proteins may represent xyloglucan fucosyltransferases expressed under specific environmental conditions.
L-Fucose- or L-galactose-containing xyloglucan fragments are believed to serve as signal molecules regulating extension growth (10, 24). Furthermore, fucosylation of xyloglucan is thought to facilitate binding of xyloglucan to cellulose by straightening the glucan backbone, thereby contributing to wall strength and integrity (7, 8). However, mur2 plants have a growth habit and wall strength indistinguishable from wild type under a variety of growth conditions, speaking against a role of fucosylated xyloglucans in establishing a strong cellulose–xyloglucan network or as a source for growth-regulating signal molecules in vivo.
Acknowledgments
We thank James Murphy and Debra Kendall for assistance in expressing the MUR2 protein, and James Romanow and Marie Cantino for their electron microscopy advice. Supported by National Science Foundation Grant MCB-9728779 (to W.-D.R.) and a grant from the U.S. Department of Agriculture National Research Initiative Competitive Grants Program (to N.C.C.).
Abbreviations
- AtFUT1
Arabidopsis thaliana fucosyltransferase 1
- BAC
bacterial artificial chromosome
- RG
rhamnogalacturonans
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Bacic A, Harris P J, Stone B A. In: The Biochemistry of Plants. Stumpf P K, Conn E E, editors. Vol. 14. New York: Academic; 1988. pp. 297–371. [Google Scholar]
- 2.Carpita N C, Gibeaut D M. Plant J. 1993;3:1–30. doi: 10.1111/j.1365-313x.1993.tb00007.x. [DOI] [PubMed] [Google Scholar]
- 3.McNeil M, Darvill A G, Fry S C, Albersheim P. Annu Rev Biochem. 1984;53:625–663. doi: 10.1146/annurev.bi.53.070184.003205. [DOI] [PubMed] [Google Scholar]
- 4.Fry S C. J Exp Bot. 1989;40:1–11. [Google Scholar]
- 5.Vierhuis E, York W S, Kolli V S K, Vincken J-P, Schols H A, Van Alebeek G W, Voragen A G. Carbohydr Res. 2001;332:285–297. doi: 10.1016/s0008-6215(01)00096-9. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi T. Annu Rev Plant Physiol Plant Mol Biol. 1989;40:139–168. [Google Scholar]
- 7.Levy S, York W S, Stuike-Prill R, Meyer B, Staehelin L A. Plant J. 1991;1:195–215. [PubMed] [Google Scholar]
- 8.Levy S, Maclachlan G, Staehelin L A. Plant J. 1997;11:373–386. doi: 10.1046/j.1365-313x.1997.11030373.x. [DOI] [PubMed] [Google Scholar]
- 9.Whitney S E C, Brigham J E, Darke A H, Reid J S G, Gidley M J. Plant J. 1995;8:491–504. [Google Scholar]
- 10.Cote F, Hahn M G. Plant Mol Biol. 1994;26:1379–1411. doi: 10.1007/BF00016481. [DOI] [PubMed] [Google Scholar]
- 11.Augur C, Benhamou N, Darvill A, Albersheim P. Plant J. 1993;3:415–426. doi: 10.1046/j.1365-313x.1993.t01-24-00999.x. [DOI] [PubMed] [Google Scholar]
- 12.McDougall G J, Fry S C. Plant Physiol. 1989;89:883–887. doi: 10.1104/pp.89.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrin R M, DeRocher A E, Bar-Peled M, Zeng W, Norambuena L, Orellana A, Raikhel N V, Keegstra K. Science. 1999;284:1976–1979. doi: 10.1126/science.284.5422.1976. [DOI] [PubMed] [Google Scholar]
- 14.Sarria R, Wagner T A, O'Neill M, Faik A, Wilkerson C, Keegstra K, Raikhel N V. Plant Physiol. 2001;127:1595–1606. [PMC free article] [PubMed] [Google Scholar]
- 15.O'Neill M A, Warrenfeltz D, Kates K, Pellerin P, Doco T, Darvill A G, Albersheim P. J Biol Chem. 1996;271:22923–22930. doi: 10.1074/jbc.271.37.22923. [DOI] [PubMed] [Google Scholar]
- 16.Ishii T, Matsunaga T, Pellerin P, O'Neill M A, Darvill A, Albersheim P. J Biol Chem. 1999;274:13098–13104. doi: 10.1074/jbc.274.19.13098. [DOI] [PubMed] [Google Scholar]
- 17.Matoh T, Takasaki M, Kobayashi M, Takabe K. Plant Cell Physiol. 2000;41:363–366. doi: 10.1093/pcp/41.3.363. [DOI] [PubMed] [Google Scholar]
- 18.Loomis W D, Durst R W. Biofactors. 1992;3:229–239. [PubMed] [Google Scholar]
- 19.Fleischer A, O'Neill M A, Ehwald R. Plant Physiol. 1999;121:829–838. doi: 10.1104/pp.121.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reiter W-D, Chapple C, Somerville C R. Plant J. 1997;12:335–345. doi: 10.1046/j.1365-313x.1997.12020335.x. [DOI] [PubMed] [Google Scholar]
- 21.Reiter W-D, Chapple C, Somerville C R. Science. 1993;261:1032–1035. doi: 10.1126/science.261.5124.1032. [DOI] [PubMed] [Google Scholar]
- 22.Bonin C P, Potter I, Vanzin G F, Reiter W-D. Proc Natl Acad Sci USA. 1997;94:2085–2090. doi: 10.1073/pnas.94.5.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rayon C, Cabanes-Macheteau M, Loutelier-Bourhis C, Salliot-Maire I, Lemoine J, Reiter W-D, Lerouge P, Faye L. Plant Physiol. 1999;119:725–734. doi: 10.1104/pp.119.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zablackis E, York W S, Pauly M, Hantus S, Reiter W-D, Chapple C C S, Albersheim P, Darvill A. Science. 1996;272:1808–1810. doi: 10.1126/science.272.5269.1808. [DOI] [PubMed] [Google Scholar]
- 25.Haughn G W, Somerville C. Mol Gen Genet. 1986;204:430–434. [Google Scholar]
- 26.Dellaporta S L, Wood J, Hicks J B. Plant Mol Biol Rep. 1983;1:19–22. [Google Scholar]
- 27.Konieczny A, Ausubel F M. Plant J. 1993;4:403–410. doi: 10.1046/j.1365-313x.1993.04020403.x. [DOI] [PubMed] [Google Scholar]
- 28.Koncz C, Schell J. Mol Gen Genet. 1986;204:383–396. [Google Scholar]
- 29.Bechtold N, Ellis J, Pelletier G. C R Acad Sci Ser Gen Vie Sci. 1993;316:1194–1199. [Google Scholar]
- 30.Selvendran R R, Stevens B J H, O'Neill M A. In: Biochemistry of Plant Cell Walls. Brett C T, Hillman J R, editors. Cambridge, U.K.: Cambridge Univ. Press; 1985. pp. 39–78. [Google Scholar]
- 31.McCann M C, Roberts K. In: The Cytoskeletal Basis of Plant Growth and Form. Lloyd C W, editor. London: Academic; 1991. pp. 109–129. [Google Scholar]
- 32.Pauly M, Albersheim P, Darvill A, York W S. Plant J. 1999;20:629–639. doi: 10.1046/j.1365-313x.1999.00630.x. [DOI] [PubMed] [Google Scholar]
- 33.Burget E G, Reiter W-D. Plant Physiol. 1999;121:383–389. doi: 10.1104/pp.121.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibeaut D M, Carpita N C. Proc Natl Acad Sci USA. 1993;90:3850–3854. doi: 10.1073/pnas.90.9.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.York W S, Darvill A G, McNeil M, Stevenson T T, Albersheim P. Methods Enzymol. 1985;118:3–40. [Google Scholar]
- 36.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 37.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 38.Lister C, Dean C. Plant J. 1993;4:745–750. [Google Scholar]
- 39.Koornneef M, Dellaert L W, van der Veen J H. Mutat Res. 1982;93:109–123. doi: 10.1016/0027-5107(82)90129-4. [DOI] [PubMed] [Google Scholar]
- 40.Marks M D, Esch J, Herman P, Sivakumaran S, Oppenheimer D. Symp Soc Exp Biol. 1991;45:77–87. [PubMed] [Google Scholar]
- 41.O'Neill M A, Eberhard S, Albersheim P, Darvill A G. Science. 2001;294:846–849. doi: 10.1126/science.1062319. [DOI] [PubMed] [Google Scholar]