

Abstract
Stimulated by the recent surprising results from Wentworth et al. [Wentworth, A. D., Jones, L. H., Wentworth, P., Janda, K. D. & Lerner, R. A. (2000) Proc. Natl. Acad. Sci. USA 97, 10930–10935] that Abs efficiently catalyze the conversion of molecular singlet oxygen (1O2) plus water to hydrogen peroxide (HOOH), we used quantum chemical methods (B3LYP density functional theory) to delineate the most plausible mechanisms for the observed efficient conversion of water to HOOH. We find two reasonable pathways. In Pathway I, (i) H2O catalyzes the reaction of 1O2 with a second water to form HOOOH; (ii) two HOOOH form a dimer, which rearranges to form the HOO-HOOO + H2O complex; (iii) HOO-HOOO rearranges to HOOH-OOO, which subsequently reacts with H2O to form H2O4 + HOOH; and (iv) H2O4 rearranges to the cyclic dimer (HO2)2, which in turn forms HOOH plus 1O2 or 3O2. Pathway II differs in that step ii is replaced with the reaction between HOOOH and 1O2, leading to the formation of HOO-HOOO. This then proceeds to similar products. For a system with 18O H2O, Pathway I leads to a 2.2:1 ratio of 16O:18O in the product HOOH, whereas Pathway II leads to 3:1. These ratios are in good agreement with the 2.2:1 ratio observed in isotope experiments by Wentworth et al. These mechanisms lead to two HOOH per initial 1O2 or one, depending on whether the product of step iv is 1O2 or 3O2, in good agreement with the experimental result of 2.0. In addition to the Ab-induced reactions, the hydrogen polyoxides (H2O3 and H2O4) formed in these mechanisms and their decomposition product polyoxide radicals (HO2, HO3) may play a role in combustion, explosions, atmospheric chemistry, and the radiation chemistry in aqueous systems.
Recently, Wentworth et al. (1, 2) from the Scripps Research Institute reported the surprising result that Abs, regardless of source or antigenic specificity, have an ability to catalyze the generation of H2O2 in a highly efficient manner and by a mechanism that involves the oxidation of H2O by singlet oxygen molecules, O2 (1Δg). This potentially aligns recognition and killing within the same Ab molecule. To understand how Abs can carry out this remarkable and unexpected chemistry, we need to understand how 1O2 can interact with H2O to produce H2O2. Preliminary mechanistic investigation (2)§ suggested that 1O2 might be able to convert H2O to H2O2 via the formation of hydrogen polyoxides, H2O3 and H2O4.
In this article, we use the quantum chemical (QC) methods described in Computational Details to consider the most likely chemical reaction mechanisms for the oxidation of water by singlet molecular oxygen to generate hydrogen peroxide. Quantum Mechanical (QM) Calculations and Plausible Mechanisms reports investigations of the formation mechanisms for hydrogen polyoxides, H2O3 and H2O4, and the related polyoxide radicals, HO2 and HO3. The results lead to plausible mechanisms in good agreement with the Scripps isotope experiments. The Discussion talks about these results, suggesting that these mechanisms account for the Scripps Ab results and they may also be significant for understanding related processes in biochemistry, combustion, explosions, atmospheric chemistry, and radiation chemistry of aqueous systems.
Computational Details
All QM calculations use the Becke three-parameter hybrid functional with Lee–Yang–Parr correlation functional (B3LYP) flavor of density functional theory (3–7), which includes a generalized gradient approximation and some exact exchange. The 6-31G** basis set (8, 9) was used on all atoms for a full geometry optimization. Vibrational frequencies (from the analytic Hessian) were calculated to ensure that each minimum is a true local minimum (containing only positive frequencies) and that each transition state has only a single imaginary frequency (negative eigenvalue of the Hessian). All QM calculations were carried out with jaguar (10–12). Some of these energetics were included in ref. 2.
To obtain more accurate energetics, we carried out calculations with the cc-pVTZ basis set (13) by using the optimized geometries from the 6-31G** basis. Such QM calculations lead to an accuracy of around 3 kcal/mol for simple organic molecules (14).
For molecules such as 1O2 and O3, which have significant open shell character, standard density functional theory methods often lead to much larger errors. Thus, with B3LYP, the singlet and triplet gap for O2 is ΔE (1Δg − 3Σ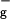 ) = 10.4 kcal/mol, in poor agreement with the experimental value of 22.5 kcal/mol (15). Consequently, we used spin projection techniques (16) to ensure a proper description of the complexes involving 1O2. This leads to ΔE (1Δg − 3Σ
) = 10.4 kcal/mol, in poor agreement with the experimental value of 22.5 kcal/mol (15). Consequently, we used spin projection techniques (16) to ensure a proper description of the complexes involving 1O2. This leads to ΔE (1Δg − 3Σ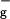 ) = 20.5 kcal/mol, in good agreement with experiment.
) = 20.5 kcal/mol, in good agreement with experiment.
The calculated vibrational frequencies (with no empirical scaling) were used to calculate the zero point energy and temperature corrections so that all energetics are reported for ΔH (298K), in kcal/mol.
QM Calculations and Plausible Mechanisms
Formation of H2O2 from the Reaction Between Two H2O3.
First, we examined the direct reaction of 1O2 with H2O leading to Eq. 0 (See Fig. 1):
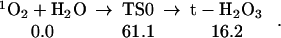 |
0 |
This barrier of 61 kcal/mol is too high for this reaction to play an important role in the observed processes. Indeed, this barrier is above the reaction heat of ΔHr = 46.0 kcal/mol for hydrogen abstraction to form of HO and HO2, which should also have a barrier of ∼46 kcal/mol.
Figure 1.

The reaction of 1O2 with H2O to form H2O3. The top pathway (TS0 at 61.1) is uncatalyzed. The middle pathway is H2O-catalyzed (TS1 at 28.9), whereas the lowest pathway (TS2 at 16.4) is (H2O)2-catalyzed. Either reaction (1) or (2) can be part of Pathways I and II.
Thus, we considered the possible role of a second H2O, as in Eq. 1:
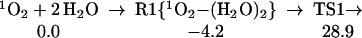 |
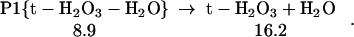 |
1 |
The structures for the reactant cluster (R1), transition state (TS1), and product cluster (P1) are shown in Fig. 1. TS1 shows that the H2O on the left plays the role of a catalyst with one H (pointing down) moving onto the left end of the 1O2 simultaneous with extracting an H from the right H2O, leaving an OH that simultaneously attaches to the other end of the 1O2. This TS1 is 28.9 kcal/mol above the limits of the free reactants and 33.1 kcal/mol above the energy of the 1O2 plus H2O dimer complex (R1). Although high, this activation barrier is small compared to the first HO bond energy of H2O (119 kcal/mol), and the bond energy in 1O2 (96 kcal/mol). Thus, this is a concerted reaction. The second water acts as a catalyst to dramatically reduce the reaction barrier.
We also find that water dimer can serve as a catalyst (See Fig. 1), with a transition state energy only 16.4 kcal/mol above the energy of the separated reactants (31.2 kcal/mol above the stable complex):
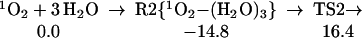 |
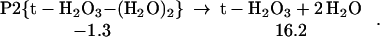 |
2 |
The ground state configuration of the HOOOH product in these reactions is trans (t-H2O3 shown in Table 1), but cis (c-H2O3 shown in Table 1) is only 2.4 kcal/mol higher in energy (17–21, ¶), see Eq. 3.
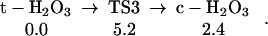 |
3 |
Considering the reverse reactions, Eq. 0 indicates that the unimolecular decomposition of t-H2O3 has a barrier of 44.9 kcal/mol, whereas Eq. 1 indicates that H2O can catalyze decomposition of t-H2O3 with a barrier of 20.0 kcal/mol (from the complex) (22, 23, ‖, ‡‡).
Table 1.
Geometries for some important reaction complexes.
 |
Reaction of HOOOH with HOOOH (Pathway I).
Direct conversion to HOOH.
Starting with two t-H2O3, we find (Eq. 4) that the transition state to form directly two HOOH is 57.8 kcal/mol (TS4 is chair-like), a value we consider too high for this to play a plausible role in the observed chemistry.
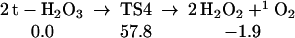 |
4 |
Conversion of the HOOOH dimer to form the HOO-HOOO intermediate plus H2O.
Starting with the dimer of c-H2O3, we find the low barrier process (16.9 kcal/mol from the complex) to form linear HOO-HOOO, as in Eq. 5.
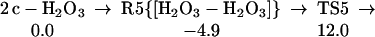 |
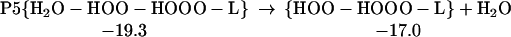 |
5 |
The structures for R5, TS5, and P5 are shown in Table 1. As indicated in Eq. 6, HOO-HOOO-L can rearrange to form the more active complex, HOOH-OOO.
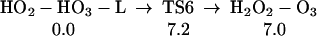 |
6 |
Next we will follow the subsequent reactions involving H2O2-O3.
Formation of H2O4.
Starting with the product of Eq. 5, and allowing the rearrangement in Eq. 6, leads to the complex R7 (Eq. 7), with two hydrogen bonds: one hydrogen of water points to an OH in H2O2, whereas this same OH hydrogen bonds with O3. Fig. 2 shows the reaction profile for Eq. 7. In TS7 H2O2 acts both as a hydrogen donor and a hydrogen acceptor. As the H bonded to the left O of H2O2 is transferred to the right O of O3, to start forming HO3, the right hydrogen of H2O is transferred to the left O of H2O2 to reproduce H2O2. Simultaneously, the remaining HO part of the H2O combines with the newly forming HO3, to form H2O4. Thus, the H2O2 in Eq. 7 catalyzes the formation of H2O4 with a barrier of only 20.1 kcal/mol from the complex (14.4 kcal/mol from the separated reactants). This can be compared with the barrier of 28.5 kcal/mol for O3 to react directly with H2O to form H2O4 (Eq. 8). Eq. 9 shows that a second water reduces the barrier to 11.6 kcal/mol (from separated reactants). This catalytic role of the second water in Eq. 9 is very similar to that in Eq. 1.
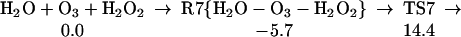 |
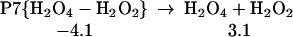 |
7 |
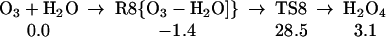 |
8 |
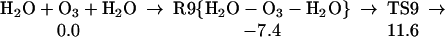 |
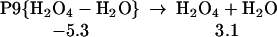 |
9 |
Figure 2.

The reaction of O3 and H2O to form HOOOOH. The top pathway (TS8 at 28.5) is uncatalyzed. The lowest pathway is H2O catalyzed (TS9 at 11.6), whereas the middle pathway (TS7 at 14.4) is H2O2 catalyzed. This latter reaction (7) is part of Pathways I and II.
Conversion of H2O4 to H2O2 and 1O2 or 3O2.
The strength of the central OO bond of the H2O4 produced in Eq. 7 is only 10.2 kcal/mol (Eq. 10a), producing two HO2 radicals. These can in turn associate to form the cyclic dimer, c-[HO2]2, either as a spin singlet or triplet (Eqs. 10b or 10c). In addition there can be interconversion of these spin singlet and triplet as in Eq. 10d. Then, hydrogen transfer from one HO2 to the other in c-[HO2]2 leads to a second H2O2 plus 1O2 or 3O2 (Eqs. 10b or 10c). The optimized structures for 10c are shown in Table 1.
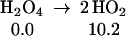 |
10a |
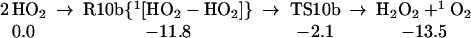 |
10b |
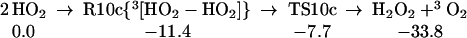 |
10c |
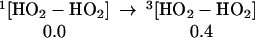 |
10d |
Summary of the HOOOH Dimer Mechanism (Pathway I).
The above reactions lead to two simple reaction mechanisms.
Pathway IA.
[Eq. (1 or 2) + (3)] twice, followed by (5) + (6) + (7) + (10ab) (with the 1O2 product).
Pathway IB.
[Eq. (1 or 2) + (3)] twice, followed by (5) + (6) + (7) + (10adc) (with the 3O2 product). Pathway IA leads to a net reaction of
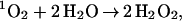 |
A |
whereas Pathway IB leads to a net reaction of
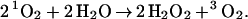 |
B |
Both Pathways IA and IB provide plausible mechanisms by which 1O2 can react with H2O to generate H2O2. Eqs. A and B indicate that this mechanism can lead to two H2O2 per initial 1O2 or one, depending on whether step 10b or 10c is used (leading to 1O2 or 3O2, respectively). This is in good agreement with the experimental result (1, 2) of 2.0.
Wentworth et al. (2) carried out studies by using H2O with 18O but 1O2 with 16O (and also the reverse case). These are difficult experiments requiring removal of all traces of 16O H2O. They found ratios of 16O:18O in the product HOOH ranging from 2.0 to 3.8, with the best experiments leading to ∼2.2:1.
In our reaction mechanism, (denoting 18O as O*):
Eq. 1 leads to HO*OOH and HOOO*H (equal probability).
Eq. 5 starts with four cases of the form (HO*OOH)2.
The product of Eq. 5 after transferring the H as in Eq. 6 leads to four cases, where: HO*H, O*OO-HOOH HO*H, OOO*-HOOH HOH, O*OO-HOO*H HOH, O*OO-HOO*H
Eq. 7 (assuming that the reactant H2O here is the same as the product H2O in Eq. 5) then leads to: HO*O*OOH + HOOH HO*OOO*H + HOOH HOO*OOH + HOO*H HOO*OOH + HOO*H, leading to a 16O:18O ratio of 5:3 in HOOOOH and a 16O:18O ratio of 6:2 in HOOH.
The cyclic (HOO)2 dimer formed in Eq. 10 has the same 16O:18O ratio as in H2O4, leading to the product distribution HO*O*-HOO + HOOH → 0.5 (HO*O*H) + 1.5 HOOH HO*O-HO*O + HOOH → 1.0 (HO*OH) + 1.0 HOOH HOO*-HOO + HOO*H → 1.5 (HOO*H) + 0.5 HOOH HOO*-HOO + HOO*H → 1.5 (HOO*H) + 0.5 HOOH.
The net result is 3.5 HOOH + 4.0 HOO*H + 0.5 (HO*O*H), leading to an overall 16O:18O ratio of 11:5 = 2.2:1 in the two HOOH products. This is in excellent agreement with the experimental value of 2.2:1, giving strong support to this mechanism. Of course, we have neglected isotope-dependence of the rate constants.
Alternatively, assuming that the H2O produced in Eq. 5 is lost and replaced with 18O H2O in Eq. 7, would lead to
HO*O*OOH + HOOH → HO*O*-HOO + HOOH → 0.5 (HO*O*H) + 1.5 HOOH
HO*OOO*H + HOOH → HO*O-HO*O + HOOH → 1.0 (HO*OH) + 1.0 HOOH
HO*O*OOH + HOO*H → HO*O*-HOO + HOO*H → 1.0 (HOO*H) + 0.5 HOOH + 0.5 (HO*O*H)
HO*O*OOH + HOO*H → HO*O*-HOO + HOO*H → 1.0 (HOO*H) + 0.5 HOOH + 0.5 (HO*O*H).
The net result is 3.5 HOOH + 3.0 HOO*H + 1.5 (HO*O*H), giving rise to a 16O:18O ratio of 10:6 or 1.67:1 in the final product of HOOH. This seems outside the range of the experiments. This suggests that the environment in the Ab responsible for this chemistry is isolated such that the product H2O remains in this complex.
Pathway II: Formation of H2O3 Monomer from 1O2.
We also considered an alternative to Pathway I in which the H2O3 molecule reacts with 1O2 to form a sequence of reaction intermediates leading to H2O2.
Reaction of H2O3 with 1O2.
We find (Eq. 11) that 1O2 can react with H2O3, extracting one hydrogen to form HOO-HOOO-7r, a cyclic hydrogen-bonded ring with seven atoms. The linear HOO-HOOO formed here can transfer an H to form HOOH-OOO (similar to Eq. 6, but the barrier is larger because HOO-HOOO-7r is 4.8 kcal/mol more stable than HOO-HOOO-L. The structures for HO2-HO3-7r, TS12, and H2O2-O3 are shown in Table 1.
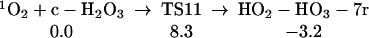 |
11 |
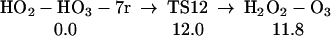 |
12 |
Adding H2O to the HOOH-OOO in Eq. 12 leads to in steps (7) + (10) to form HOOH products just as in Pathway I.
Summary of the H2O3 Plus 1O2 Mechanism (Pathway II).
Pathway IIA.
[Eq. (1 or 2) + (3)] once, followed by (11) + (12) + (7) + (10ab) (with the 1O2 product).
Pathway IIB.
[Eq. (1 or 2) + (3)] once, followed by (11) + (12) + (7) + (10adc) (with the 3O2 product).
These pathways lead to the net reactions in A and B.
Because Eq. 1 leads to one 18O among the outer two O of H2O3, then Eqs. 11 and 12 lead to a product H2O2-O3 in which both O of the H2O2 are 16O, whereas one of the outer two O of the OOO is 18O. Then, Eq. 7 leads to HOOH with only 16O, whereas the HOOOOH has one 18O among the central pair of O and one in the outer pair. The cyclic (HOO)2 dimer formed in Eq. 10 has one 18O next to an H and one away from it so that the product HOOH has one 18O. Thus, the net result is that the two HOOH products have a total of three 16O and one 18O, leading to a 3:1 ratio. This is in the range of the experiments, but probably on the high side. Some decrease from 3:1 might result from the 1O2 reaction product in Eq. 10b (which involves one 18O), reacting subsequently with H2O to form HOOH. It could also result from isotope-dependent rate constants.
Pathway III: Direct Conversion of Singlet Dioxygen to Hydrogen Peroxide.
We also considered the direct conversion of singlet dioxygen to hydrogen peroxide, without formation of the various intermediates:
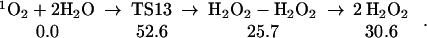 |
13 |
The structures are shown in Table 1. One hydrogen from each H2O moves toward the 1O2 to form one HOOH while the remaining two HO come towards each other to form the other HOOH. The negative charges on O of these two HO repel each other, leading to a high barrier. Spin recoupling (the necessity to flip the lone pairs and radical orbitals on the two hydroxyls simultaneously to make the O–O bond of the departing peroxide) will further increase this barrier. This pathway is unlikely. The isotope ratio would be 16O:18O = 1:1, which is well outside the experimental range.
Discussion
In summary, we find two reasonable pathways by which 1O2 plus water can produce HOOH, providing strong support for the experimental observations by Wentworth et al. (1, 2) that Abs and T cell receptors can catalyst the conversion of 1O2 plus water to HOOH. In Pathway I, (i) H2O catalyzes the reaction of 1O2 with a second water to form HOOOH; (ii) HOOOH forms dimer, which rearranges to form HOO-HOOO + H2O; (iii) HOO-HOOO rearranges to HOOH-OOO that subsequently reacts with H2O to form H2O4; and (iv) H2O4 rearranges to (HO2)2 to form HOOH plus 1O2 or 3O2. Pathway II differs in that step ii is replaced with the reaction between HOOOH and 1O2, leading to the formation of HOO-HOOO. This then proceeds to similar products.
For a system with 18O H2O, these mechanisms lead to a 2.2:1 ratio of 16O:18O in the product HOOH for Pathway I and 3:1 for Pathway II. This is in good agreement with the ratio 2.2:1 observed in isotope experiments by Wentworth et al. These mechanisms lead to two HOOH per initial 1O2 or one, depending on whether the product of step iii is 1O2 or 3O2. This is in good agreement with the experimental result of 2.0.
Relevance for Experiments on Abs.
Datta et al. (24) find that the QM structures reported here for the relevant complexes and transition states involved in forming H2O3 and H2O2 are stabilized at sites unique to Abs and T-cell receptors (TCRs) but not in other structures such as β2-microglobulin. This finding is consistent with the observations from Wentworth et al. (1, 2) that all Abs and TCRs catalyze the oxidation of water, but that other systems, including β2-microglobulin, do not. The predicted sites (24) are at the barrel-like interface of two Greek key domains formed by the light and heavy chains of the Ab (and of the TCR). This structure, referred to as the inter-Greek key domain interface (IGKD) is unique to these systems (24). At these IGKD sites there are several well-ordered crystallographic water molecules spaced just as in water dimers or trimers, providing an environment appropriate for reactions such as Eqs. 1 and 2. In addition, this IGKD region is sufficiently hydrophobic to stabilize both H2O3 and 1O2 (24). These IGKD sites also stabilize HOOOH and its dimer, providing an environment for the balance of Pathways I and II. Indeed, although these IGKD sites have water dimers and trimers, the lack of exposure to bulk water at these sites may be essential to the chemistry. In particular, this environment is consistent with the product H2O in Eq. 5 remaining to be the same as the reactant H2O in Eq. 7, which in turn led to the 2.2:1 isotope ratio, which agrees with experiment (for Pathway I).
Although the only intermediate detected experimentally is HOOH, our mechanism suggests the production of other reactive intermediates (H2O3, HO2-HO3, HOOH-O3, O3, c-(HOO)2, and H2O4), any of which could be responsible for subsequent chemical processes. All these intermediates are expected to form at IGKD sites near the VH-VL region that binds the antigen. In this protected region, these active intermediates might live long enough to travel to the antigen-binding site (indeed, some might be detected by the peroxidase assay used to detect the H2O2). These intermediates might react with antigen and do other chemistry in addition to the formation of H2O2. The function of this chemistry might be to form reactive intermediates next to the antigen that would react with it to nick the protein recognized by the Ab, making it more susceptible to attack by other enzymes in the macrophage. Alternatively, the function of the Ab might be as a 1O2 scavenger, converting 1O2 to H2O2 or other reactive intermediates (like H2O3, O3, or H2O4) that are less damaging or eliminated by other enzymes.
Role of H2O3 in Other Chemistries.
Reaction of 1O2 with H2O.
There are two distinct pathways for the reaction of 1O2 and H2O.
Hydrogen abstraction: leading to the formation of HO and HO2. We calculate that the reaction heat for this process is ΔHr = 46.0 kcal/mol, which should also be the barrier.
O2 addition (Eq. 0): giving rise to H2O3. We calculate that this leads to ΔHr = 16.2 kcal/mol. Thus, losing the π bond in 1O2 is nearly compensated by the formation of σ bond in H2O3. However, the barrier for this reaction is 61.1 kcal/mol, making less favorable than hydrogen abstraction, despite the high endothermicity. On the other hand, including a second H2O catalyzes the O2 addition reaction, reducing the activation barrier from 61.1 to 28.9 kcal/mol (Eq. 1), while a water dimer lowers the effective reaction barrier further to 16.4 kcal/mol (Eq. 2).
If the reaction of 1O2 is so favorable, one might wonder why there have been so few observations of this chemistry. Most significant is that although the barrier for unimolecular decomposition of gas phase H2O3 is 44.9 kcal/mol (the reverse of Eq. 0), water catalyzes the decomposition reducing the barrier to 12.7 kcal/mol (the reverse of Eq. 1). [First pointed out by Plesnicar and Koller (22).]
Production of H2O3.
The existence of higher oxides of hydrogen, such as H2O3 and H2O4, has been postulated repeatedly since the original suggestion of Berthelot in 1880 (25–34). In electrically dissociated water vapor, H2O3 and H2O4 have been postulated on the basis of such experimental observations as oxygen evolution on warming, thermal analysis, or phase changes (25, 26). Infrared absorption of the products from electrically dissociated H2O vapor trapped at liquid nitrogen temperature (26) or photolyzed samples of H2CO in Ar and glyoxal in Ar (28) also provided spectroscopic support for the existence of H2O4. Hydrogen trioxide, H2O3, was proposed as a transient intermediate in the oxygenation of alkanes with ozone in superacid media (32) and in the pulse radiolysis of air-saturated perchloric acid solution (25). UV irradiation of H2O2 in methyl acetate produced an oxygen-rich intermediate, characterized by 1H NMR absorption at about δ14.5 ± 1.0 ppm, downfield from Me4Si. This polyoxide was assigned to H2O3 (30–33). More recently, by means of 17O NMR spectroscopy, Plesnicar et al. (34) concluded that the low-temperature ozonation of isopropyl alcohol and isopropyl methyl ether yields, in addition to the hydrotrioxides of these compounds, hydrogen trioxide H2O3.
Production of H2O2.
The production of H2O2 from H2O3 has been postulated (25–27), but no detailed studies have previously been reported. We find three plausible mechanisms.
H2O3 decomposes to form 1O2 (catalyzed by the presence of H2O as discussed above).
H2O3 reacts with 1O2 in a hydrophobic region to form HOOH and other intermdidats as in Pathway II, where the activation barrier for 1O2 to subtract an H from H2O3 is only 8.3 kcal/mol (Eq. 11). The resultant cyclic hydrogen-bonded complex, HO2-HO3-7r (BE = 8.1 kcal/mol), retards the decomposition of HO3.
H2O3 reacts with another H2O3 in a hydrophobic region to form HOOH and other intermdidats as in Pathway I. This involves the formation of {H2O + HO2-HO3-L} (Eq. 5, ΔHr = −17.0 kcal/mol and ΔEa = 12.0 kcal/mol).
Production of H2O4.
The existence of H2O4 was confirmed spectroscopically by Giguere and Herman (26) and Diem et al. (28). The first theoretical studies of H2O4 were reported by Plesnicar et al. (35), but the most accurate and detailed theoretical studies were performed by Schaefer's group (36, ††). Our calculations show that formation of H2O4 from H2O + O3 is uphill by 3.1 kcal/mol with an effective barrier of 28.5 kcal/mol (Eq. 7). However, H2O or H2O2 can catalyze this reaction, reducing the effective barrier to 11.6 or 14.4 kcal/mol (Eqs. 9 and 7).
Detailed kinetic studies have been reported for the disproportionation of HO2 to H2O2 + O2 (37–42). This reaction possesses a negative activation barrier (40–42), but no molecular level mechenistic study has been reported. Our results show that two HO2 form a planar cyclic hydrogen complex with BE = 11.8 kcal/mol for singlet (HO2)2 and 11.4 kcal/mol for triplet (HO2)2. As one hydrogen in an HOO bends out of the plane, the other hydrogen in the other HOO is pulled over to form HOOH.
The energy surface of singlet (HO2)2 coincides almost exactly with that of the triplet (HO)2, enhancing the rate for singlet to triplet interconversion (Eqs. 10bcd). Because of formation of the cyclic hydrogen-bonded (HO2)2, the entire energy surface is below the potential energy surface of 2 HO2, leading to a negative activation barrier.
Summary
The mechanisms studied here involve the formation of hydrogen polyoxides (H2O3 and H2O4) and the related polyoxide radicals (HO2 and HO3). These mechanisms may be of significance to the understanding of related oxidations in biochemistry, combustion, and explosion chemistry, atmospheric chemistry, and radiation chemistry of aqueous systems. Many of the proposed intermediates and mechanisms might be studied by using spectroscopic and isotope methods.
Acknowledgments
We thank Richard Lerner for suggesting this problem and Albert Eschenmoser, Paul Wentworth, Anita Wentworth, Lyn Jones, and Kim Janda for helpful discussions concerning their experiments. In addition we thank Wely Floriano and Nagarajan Vaidehi for many helpful discussions concerning the possible sites for the reaction intermediates in Abs. We thank Deb Chakraborty for helpful discussions on the QM calculations and Scott Singleton for helpful discussions on the chemistry. This research was funded by the National Institutes of Health. The facilities of the Materials and Process Simulation Center used in these studies were funded by National Science Foundation–Major Research Instrumentation, Defense University Research Instrumentation, and the Beckman Institute. In addition, the Materials and Process Simulation Center is funded by grants from the Department of Energy–Accelerated Strategic Computing Initiative–Academic Strategic Alliances Program, the Army Research Office–Multidisciplinary University Research Initiative, the National Science Foundation, Avery-Dennison, Asahi Chemical, Chevron, 3M, Dow Chemical, Nippon Steel, Seiko-Epson, and Kellogg.
Abbreviations
- QM
quantum mechanical
- IGKD
inter-Greek key domain interface
- TS
transition state
- R
reactant cluster
- P
product cluster
Footnotes
The density functional theory (DFT) results reported here differ by a few kcal/mol from the numbers reported in this article. This is because the calculations in ref. 2 used the UB3LYP/6-31G** level of DFT. For species such as 1O2, this level leads to spin contamination, which can cause errors of several kcal/mol. In the current results, we spin-project to eliminate contamination and use a larger basis set to reduce possible basis-set superposition errors, which can be important for hydrogen-bonded complexes. Experimental heats of formation H2O and H2O2 were used in ref. 2 to calibrate the calculated heat of formation of H2O3. This procedure is not adopted in the present work.
Ref. 17 reported detailed theoretical studies of H2O3.
Ref. 22 studied the decomposition of H2O3 to H2O and O2 (1Δg) with and without the presence of a second water (the reverse reaction of Eqs. 0 and 1). They concluded that HOOOH could be stable in various “basic” solvents like ketones and esters and that a considerable amount of HOOOH might be present in the atmosphere and biological systems.
Ref. 23 concluded that the energy cost to break the O—O bond in H2O3 is 33.9 kcal/mol.
Ref. 35 used high-level ab initio theory to conclude that HOOOOH is 0.1 kcal/mol more stable than (HOO)2.
References
- 1.Wentworth A D, Jones L H, Wentworth P, Janda K D, Lerner R A. Proc Natl Acad Sci USA. 2000;97:10930–10935. doi: 10.1073/pnas.97.20.10930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wentworth P, Jones L H, Wentworth A D, Zhu X Y, Larsen N A, Wilson I A, Xu X, Goddard W A, Janda K D, Eschenmoser A, Lerner R A. Science. 2001;293:1806–1811. doi: 10.1126/science.1062722. [DOI] [PubMed] [Google Scholar]
- 3.Slater J C. Quantum Theory of Molecules and Solids. New York: McGraw–Hill; 1974. [Google Scholar]
- 4.Vosko S H, Wilk L, Nusair M. Can J Phys. 1980;58:1200–1211. [Google Scholar]
- 5.Becke A D. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 6.Becke A D. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 7.Lee C T, Yang W T, Parr R G. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 8.Hehre W J, Ditchfield R, Pople J A. J Chem Phys. 1972;56:2257–2261. [Google Scholar]
- 9.Hariharan P C, Pople J A. Theor Chim Acta. 1973;28:213–222. [Google Scholar]
- 10.Schrodinger, Inc. JAGUAR. Portland, OR: Schrodinger; 1998. , Version 3.5. [Google Scholar]
- 11.Greeley B H, Russo T V, Mainz D T, Friesner R A, Langlois J-M, Goddard W A, III, Donnelly R E, Ringnalda M N. J Chem Phys. 1994;101:4028–4041. [Google Scholar]
- 12.Tannor D J, Marten B, Murphy R, Friesner R A, Sitkoff D, Nicholls A, Ringnalda M, Goddard III W A, Honig B. J Am Chem Soc. 1994;116:11875–11882. [Google Scholar]
- 13.Dunning T H., Jr J Chem Phys. 1989;90:1007–1023. [Google Scholar]
- 14.Curtiss L A, Raghavachari K, Redfern P C, Pople J A. J Chem Phys. 1997;106:1063–1079. [Google Scholar]
- 15.Huber K P, Herzberg G. Molecular Spectra and Molecular Structure: Constants of Diatomic Molecules. New York: Van Nostrand Reinhold; 1979. [Google Scholar]
- 16.Wittbrodt J M, Schlegel H B. J Chem Phys. 1996;105:6574–6577. [Google Scholar]
- 17.Cremer D. J Chem Phys. 1978;69:4456–4471. [Google Scholar]
- 18.Gonzalez C, Theisen J, Zhu L, Schlegel H B, Hase W L, Kaiser E W. J Phys Chem. 1991;95:6784–6792. [Google Scholar]
- 19.Jackels C F. J Chem Phys. 1993;99:5768–5779. [Google Scholar]
- 20.Blint R J, Newton M D. J Chem Phys. 1973;59:6220–6228. [Google Scholar]
- 21.Lay T H, Bozzelli J W. J Phys Chem A. 1997;101:9505–9510. [Google Scholar]
- 22.Koller J, Plesnicar B. J Am Chem Soc. 1996;118:2470–2472. [Google Scholar]
- 23.McKay D J, Wright J S. J Am Chem Soc. 1998;120:1003–1013. [Google Scholar]
- 24. Datta, D., Vaidehi, N., Xu, X. & Goddard, W. A., III (2001) Proc. Natl. Acad. Sci. USA 99, in press. [DOI] [PMC free article] [PubMed]
- 25.Czapski G, Bielski B H J. J Phys Chem. 1963;67:2180–2184. [Google Scholar]
- 26.Giguere P A, Herman K. Can J Chem. 1970;48:3473–3482. [Google Scholar]
- 27.Nangia P S, Benson S W. J Phys Chem. 1979;83:1138–1142. [Google Scholar]
- 28.Diem M, Tso T-L, Lee E K C. J Chem Phys. 1982;76:6452–6454. [Google Scholar]
- 29.Sugimoto H, Sawyer D T. J Am Chem Soc. 1988;110:8707–8708. [Google Scholar]
- 30.Plesnicar B, Kovac F, Schara M. J Am Chem Soc. 1988;110:214–222. [Google Scholar]
- 31.Plesnicar B, Cerkovnik J, Koller J, Kovac F. J Am Chem Soc. 1991;113:4946–4953. [Google Scholar]
- 32.Cerkovnik J, Plesnicar B. J Am Chem Soc. 1993;115:12169–12170. [Google Scholar]
- 33.Speranza M. Inorg Chem. 1996;35:6140–6151. [Google Scholar]
- 34.Plesnicar B, Cerkovnik J, Tekavec T, Koller J. Chem Eur J. 2000;6:809–819. doi: 10.1002/(sici)1521-3765(20000303)6:5<809::aid-chem809>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 35.Plesnicar B, Kaiser S, Azman A. J Am Soc Chem. 1973;95:5476–5477. [Google Scholar]
- 36.Fermann J T, Hoffman B C, Tschumper G S, Schaefer H F., III J Chem Phys. 1997;106:5102–5108. [Google Scholar]
- 37.Simonaitis R, Helcklen J. J Phys Chem. 1982;86:3416–3418. [Google Scholar]
- 38.Takacs G, Howard C J. J Phys Chem. 1984;88:2110–2116. [Google Scholar]
- 39.Kircher C, Sander S P. J Phys Chem. 1984;88:2082–2091. [Google Scholar]
- 40.Lii R-R, Gorse R A, Jr, Saur M C, Gordon S. J Phys Chem. 1979;83:1803–1804. [Google Scholar]
- 41.Mozurkewich M, Benson S W. Int J Chem Kinet. 1985;17:787–807. [Google Scholar]
- 42.Benson S W, Dobis O. J Phys Chem A. 1998;102:5175–5181. [Google Scholar]