

Abstract
Neuraminidase, a key enzyme responsible for influenza virus propagation, has been used as a template for selective synthesis of small subsets of its own inhibitors from theoretically highly diverse dynamic combinatorial libraries. We show that the library building blocks, aldehydes and amines, form significant amounts of the library components resulting from their coupling by reductive amination only in the presence of the enzyme. The target amplifies the best hits at least 120-fold. The dynamic libraries synthesized and screened in such an in vitro virtual mode form the components that possess high inhibitory activity, as confirmed by enzyme assays with independently synthesized individual compounds.
The search for novel small-molecular ligands of biological targets remains a continuing challenge with major implications for drug discovery and fundamental studies of biochemical pathways (1, 2). Despite the increasing success of structure-based design and combinatorial technologies for potential ligand synthesis and screening, the problem still has no general solution. Multiple examples have been reported over the years of using the biological targets as templates for formation of ligands from smaller building blocks. This formation is accomplished either by acceleration of a chemical reaction between the blocks (3–6) or by binding of effective building block combinations by the template, thus shifting the equilibrium between multiple possible combinations to the preferred route (7). Dynamic combinatorial chemistry (DCC) has emerged recently as a coherent approach to self-organization of molecular libraries, thermodynamically driven by the target (8–13). A concept of virtual libraries was proposed (14) and further explored in one of the first applications of DCC to biological targets (15). We report here an example of virtual dynamic libraries in which significant quantities of effective ligands (hits) are formed only in the presence of the target. Notably, the hits result from potentially very diverse libraries that give access to thousands of compounds.
Materials and Methods
Protein Expression and Purification.
The neuraminidase cDNA of the Influenza A/FPV/Rostock/34 virus strain (16) was amplified and modified by PCR (forward primer, GGGGACAAGTTTGTACAAAAAAGCAGGCTGCCACCATGAATCCAAATCAGAAAATATAACC; reverse primer, GGGGACCACTTTGTACAAGAAAGCTGGGTTT ACTAGTGATGGTGATGGTGATGCGATCCCTTGTCAATGGTGAATGGCAACTCAGC) to give pDEST8-tNA-His, which encodes for a neuraminidase with six histidines fused to the C terminus (tNA-His). Spodoptera frugiperda Sf-9 insect cells were cultivated at 27°C in the serum-free medium ExCell400 (JRH Biosciences, Lenexa, KS). Exponentially growing cells (2 × 106 cells/ml) were infected with baculovirus at a multiplicity of infection (moi) of 10. After 72 h of expression the cells were harvested and the neuraminidase (tNA-His) was either released from the plasma membrane by detergent lysis (20 mM Tris, pH 8/150 mM NaCl/2 mM CaCl2/1% Triton X-100) or the extracellular domain (sol-tNA-His) was released by treatment with pronase (17). Briefly, cells were treated for 2 h at 37°C with pronase (1 mg/ml; Calbiochem) and DNaseI (50 μg/ml) in 100 mM sodium acetate (pH 5.5), 2 mM CaCl2, and 10 mM MgCl2. After separation of cellular debris and inactivation of pronase, tNA-His and sol-tNA-His were purified by metal chelate affinity chromatography using Ni-NTA superflow beads (Qiagen). The purification yielded an average of 3 mg of sol-tNA-His and 5 mg of tNA-His out of 1 liter of culture, with a purity of >90% and a specific activity of ≈11 units/mg.
Synthesis.
Scaffolds 2 and 15, as well as individual library components 11-14, 17, and 18, were synthesized according to Schemes 4–8, which are published as supporting information on the PNAS web site, www.pnas.org, and showed analytical parameters (1H and 13C NMR, MS, TLC, and HPLC) consistent with the expected structures. Details of the synthesis will be reported elsewhere.
Diversity Test.
The sample library prepared to test the potential diversity level was prepared by incubation of 0.47 mM 2 with 5 aldehydes, A4, A5, A8, A15, and A22 (4.7 mM each) with 2.36 mM tetrabutylammonium cyanoborohydride (TBC) in 10 mM aqueous imidazole buffer (pH 7.8) at 25°C. The library composition was analyzed within 24, 72, and 120 h.
Library Analysis.
HPLC-MS analyses were performed with electrospray ionization (positive mode) on a Bruker Esquire 3000 ion trap mass spectrometer connected to an Agilent 1100 HPLC. A gradient of 0.1% formic acid in H2O (A) and acetonitrile (B) was applied using a Phenomenex (Belmont, CA) LUNA C18 (2) 5μ reversed-phase HPLC column (250 × 3.00 mm, flow rate 0.5 ml/min). Eluent composition was kept isocratic at 0% B for 5 min. Subsequently, B was linearly increased in two steps to 20% (t = 7 min) and to 50% (t = 15 min) and then kept isocratic at 50% B for 5 min. MS data were acquired in the full scan mode and scanned for protonated molecular ions [M+H]+ of potential products in extracted ion chromatograms. The peak areas (Fig. 1–5) represent concentrations of single components, but generally cannot be used to compare concentrations of two or more different components.
Figure 1.

Mono- and disubstituted components (structures 7 and 8, respectively) detected in a static library of potential neuraminidase inhibitors (see Materials and Methods for synthesis protocol) after 24 h of incubation. Heights of the bars correspond to peak areas of the compound masses in the extracted ion chromatograms.
Figure 5.

Library components of general structure 16 formed from the reactions of 15 in the presence of neuraminidase with the following aldehyde mixtures: Mix I, as in Fig. 4A; Mix II, as in Fig. 3; Mix III, as in Fig. 4B; Mix IV, as in Fig. 2A. Bar colors correspond to coupling products with different aldehydes: A8, blue; A13, red; A22, yellow. For experimental conditions see Fig. 2A. Only the components detected in significant amounts (above signal-to-noise ratio 3:1) are shown. No components were detected in any control experiments in the absence of the enzyme.
Dynamic Combinatorial Chemistry Protocol.
A typical DCC experiment was carried out as follows. Aldehyde stock solutions (1 M) were prepared by dissolving in DMSO and dilution with a 10-mM aqueous imidazole/HCl buffer (pH 7.8) to 10 mM. To a 10-μM solution of the neuraminidase extracellular domain (50 μl) in the imidazole buffer was added the scaffold (2 μl of 250 μM solution in the imidazole buffer, pH 7.8) and premixed stock solutions of aldehydes, 1 μl each. The reaction mixture was incubated for 10 min and then tetrabutylammonium cyanoborhydride (TBC) (2 μl of 2.5 mM solution in acetonitrile) was added. After incubation for 12 h at 25°C the reaction mixture was analyzed by HPLC-MS.
Neuraminidase Assay.
The neuraminidase activity was determined using the synthetic substrate 2′-(4-methylumbelliferyl)-a-D-N-acetylneuraminic acid (MU-NANA) (18, 19). The enzyme was incubated for 30 min at 37°C with 50 μM MU-NANA in 100 mM sodium acetate (pH 5.5) and 2 mM CaCl2, and subsequently the reaction was stopped with 400 mM glycine (pH 10.5) and 250 mM NaCl for measuring the fluorescence of released 4-methylumbelliferone (4-MU) at an excitation wavelength of 355 nm and an emission of 460 nm. The inhibition curves were fitted with the aid of SIGMAPLOT software, using a competitive inhibition model to determine the Ki values. For neuraminidase activity testing we either used purified tNA-His protein originating of the avian influenza A/FPV/Rostock/34 (H7N1) strain (see above) or virus preparations of different human Influenza A [A/Panama/2007/99 (H3N2); A/Johannesburg/33/94 (H3N2)] and Influenza B (B/Harbin/7/94; B/Victoria/504/2000) strains.
Results and Discussion
We used as a target the influenza A virus neuraminidase, a viral surface enzyme that catalyses the cleavage of sialic acid residues terminally linked to glycoproteins and glycolipids and plays an important role in the propagation of the virus (20). For solubility reasons, we expressed neuraminidase and purified the extracellular domain containing the active site.
Neuraminidase is a major target for drug action on influenza (21). The enzyme structure is well characterized and multiple SAR data on the active site ligands are available in the literature (22). It has been shown that the presence and particular spatial arrangement of a number of functional groups in the neuraminidase ligands are crucial and conserved in a majority of known inhibitors. Among the most important functionalities are the carboxylate and acetamido groups, as shown in the example of compound 1, the active component of the commercial influenza drug tamiflu (23). 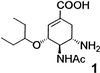 A number of studies have shown the existence of a sizeable hydrophobic pocket next to the neuraminidase active site (21), which is partially occupied by the alkyl chains of 1. We designed the library of potential inhibitors in such a way that the components would be formed from a central scaffold 2 (Scheme S1) that contained a moiety similar to 1 and therefore possessed a basic affinity for the target active site, and a variety of aldehyde substituents. The substituents, all of hydrophobic nature (Scheme S2) were directed toward the hydrophobic pocket of neuraminidase. In the dynamic mixture, each of the scaffold amino groups is in principle capable of reacting with any of the aldehydes to form a set of transient species 3-6 (Scheme S1) existing in a rapid dynamic equilibrium with each other and with the building blocks. 1H NMR studies performed with several single aldehydes‡ showed primarily the presence of hemiaminals 3 and 5, but the formation of corresponding imines 4 and 6, particularly in the presence of the enzyme target, cannot be ruled out. Subsequent chemical reduction of the transient hemiaminals and imines with an externally added reagent (see Materials and Methods) irreversibly converts them into the set of amines 7-10 giving rise to a large array of stable library components, each containing up to four substituents attached to the scaffold. Although the diversity of the transient library is difficult to assess, we have prepared and analyzed a sample library of the reduced products at high concentrations of the building blocks (Fig. 1). The HPLC-MS analysis of the resulting mixture showed the protonated molecular ions corresponding to all mono-, di-, tri-, and most of the tetrasubstituted components, whose ratios changed over the course of the reaction.§ This finding confirmed that the general reaction scheme is in principle capable of generating molecular arrays of high diversity, which rapidly increases with the number of building blocks (see Scheme S1) and where most of the expected components are represented.
A number of studies have shown the existence of a sizeable hydrophobic pocket next to the neuraminidase active site (21), which is partially occupied by the alkyl chains of 1. We designed the library of potential inhibitors in such a way that the components would be formed from a central scaffold 2 (Scheme S1) that contained a moiety similar to 1 and therefore possessed a basic affinity for the target active site, and a variety of aldehyde substituents. The substituents, all of hydrophobic nature (Scheme S2) were directed toward the hydrophobic pocket of neuraminidase. In the dynamic mixture, each of the scaffold amino groups is in principle capable of reacting with any of the aldehydes to form a set of transient species 3-6 (Scheme S1) existing in a rapid dynamic equilibrium with each other and with the building blocks. 1H NMR studies performed with several single aldehydes‡ showed primarily the presence of hemiaminals 3 and 5, but the formation of corresponding imines 4 and 6, particularly in the presence of the enzyme target, cannot be ruled out. Subsequent chemical reduction of the transient hemiaminals and imines with an externally added reagent (see Materials and Methods) irreversibly converts them into the set of amines 7-10 giving rise to a large array of stable library components, each containing up to four substituents attached to the scaffold. Although the diversity of the transient library is difficult to assess, we have prepared and analyzed a sample library of the reduced products at high concentrations of the building blocks (Fig. 1). The HPLC-MS analysis of the resulting mixture showed the protonated molecular ions corresponding to all mono-, di-, tri-, and most of the tetrasubstituted components, whose ratios changed over the course of the reaction.§ This finding confirmed that the general reaction scheme is in principle capable of generating molecular arrays of high diversity, which rapidly increases with the number of building blocks (see Scheme S1) and where most of the expected components are represented.
Scheme 1.

Structures of the scaffold and library composition. a, Hemiaminal species are restricted to those detected by NMR. b, Numbers of compounds 3 and 5 account for diastereomers.
Scheme 2.

Aldehydes used as building blocks for combinatorial libraries. (Only the structures explicitly mentioned in the text are listed. The full list of 41 compounds is given in Schemes 4–8.)
The addition of the target enzyme to the dynamic library is expected to shift the equilibrium between the transient species toward the components with the highest affinity for the target. Chemical reduction of the biased (i.e., re-equilibrated with the target) library should result in the distribution of stable components reflecting the enrichment in the transient best binders. However, the analysis of such a mixture, even of enriched composition, and identification of the active components becomes extremely difficult with increasing size of the library. We therefore explored the conditions where the transient components were present only in trace amounts and the reduction reaction was sufficiently slow so as not to produce significant amounts of any reduced species within a certain time lapse. In such a virtual library, components cannot be detected, unless the presence of a target promotes the formation of preferred structures, due to the equilibrium shift and/or to the acceleration of irreversible coupling.
The substituents were initially grouped in two sets, based on the preliminary activity screens performed with scaffolds mixed with individual aldehydes. The first set contained ten aldehydes, some of which showed statistically significant inhibition of neuraminidase activity in the mixture with the scaffold (Fig. 2B), therefore being potentially promising substituents. The second set included the aldehydes A4, A11, A14, A17, and A18 that did not show inhibitory activity in the preliminary screen.
Figure 2.

(A) Library components of general structure 7 formed in the presence (red bars) and in the absence (blue bars, where applicable) of 10 μM neuraminidase on incubation of the mixture of 2 (10 μM) with A2-A4, A8, A9, A11, A13, A15, A20, and A22 (200 μM each) and 100 μM tetrabutylammonium cyanoborohydride (TBC) for 12 h. See Materials and Methods for analytical protocol. (B) Inhibition of neuraminidase by mixtures of 2 (10 μM) with individual aldehydes, as marked on the axis (2 mM each). Control bars correspond to the enzyme activity without inhibitors (−S) and with the scaffold 2 alone (+S).
Brief incubation of the first scaffold-substituent mixtures with the enzyme, followed by reduction resulted in a striking amplification effect of selected library components, as seen from the HPLC-MS analysis, Fig. 2A. The main type of species detected in the library was the monosubstituted scaffold. This observation is consistent with the properties of known neuraminidase inhibitors, in which one free amino group is engaged in the recognition of amino acids in the enzyme pocket (21).¶ Because the concentrations of most library components in the absence of the enzyme were below detectable level, we could only reliably quantify the amplification of the adduct with aldehyde A22, where trace amounts did form without the target. In this case, the amplification effect amounted to factor of 120. No products could be detected in the experiment with the second set of aldehydes. Notably, the substituents identified in the target-promoted subset of components also showed up in the preliminary activity screen (Fig. 2B). This correlation indicates that the DCC experiment generally reveals the most effective inhibitory species present in the transient library, as discussed in more detail below. In both sets, the progress of the reaction was also controlled by the consumption of the scaffold, which in the first set was completely converted to the selected products, and in the second set was not consumed.
To verify that the amplification effects were due to interactions of library components with the neuraminidase active site and not caused by any nonspecific effects of the protein, two control experiments were performed. In one of them, the incubation was done under the conditions of Fig. 2A, but with neuraminidase replaced by identical concentration of BSA. In the other one, the mixture described in Fig. 2A was incubated with neuraminidase in the presence of 10 μM commercial drug zanamivir, a potent active site inhibitor of neuraminidase [Ki = 1 nM (24)]. Both experiments gave the results identical to that in the absence of the target—i.e., no stable components could be detected within the incubation time.
Kinetic studies of the product evolution in the presence of the target were conducted. Under the conditions of Fig. 2A, the scaffold was consumed and the concentration of products leveled off within 10 h of reduction. No significant change in the mixture composition was observed for the following 48 h of incubation in the presence of the target. A control experiment, without the target, showed slow accumulation of multiple coupling products over prolonged periods of time.
To see whether removal of the most effective substituent will change the library bias, we performed an experiment in which A22 in the first set was replaced by a randomly chosen aldehyde A26. The resulting analytical profile (Fig. 3) showed both secondary hits observed previously (A8 and A13), as well as another minor component formed from A4.
Figure 3.

Library components of general structure 7 formed in the presence (red bars) and in the absence (blue bars, where applicable) of neuraminidase. Experimental conditions are identical to Fig. 2A, with A26 substituted for A22.
We then increased the diversity of the libraries and screened two larger sets of substituents, one that included 20 aldehydes of purely aliphatic nature (Fig. 4A), and another with 17 aldehydes containing aromatic groups (Fig. 4B). Both sets yielded the hits found in the smaller libraries and a new component formed from A39.
Figure 4.

Library components of general structure 7 formed in the presence (red bars) and in the absence (blue bars, where applicable) of neuraminidase from the reactions of 2 with aldehydes containing (A) aliphatic (A4-A11, A14-A19, A23-A26, A28, A29) and (B) aromatic groups (A13, A20-A22, A30-A32, A34, A37, A39, A41, A44-A49). See Fig. 2A for experimental conditions.
It should be noted that the potential diversity level of the virtual libraries is very high, for example the set of 20 aldehydes potentially yields over 40,000 stable components, as shown in Scheme S1. Because the products in the larger libraries cannot be detected in the absence of neuraminidase, their amplification factors by the target can only be conservatively estimated, as the signal-to-noise ratio of the HPLC-MS profile, which for the best hits is close to 100. The actual amplification may, of course, be higher.
To verify that the library components formed in the presence of the target indeed display better inhibition properties, we synthesized several representative library members as individual compounds and tested them in independent enzyme assays. The Ki values summarized in Scheme S3 show that two of three hits possess inhibitory activity considerably higher than the scaffold. Interestingly, one of the hits (13) showed only minor improvement compared with the scaffold, whereas a potential library member that did not emerge in the presence of the target (14) was a somewhat better inhibitor. These data emphasizes one of the key properties of the in vitro virtual libraries: amplification of components in the final stabilized library reflects the inhibitory properties of the corresponding transient components (hemiaminals or imines, Scheme S1). The amplification profile can thus be considered a developed photograph of the transient component's recognition properties, yielding the information that is difficult or impossible to obtain otherwise. With a good probability, these preferred binding properties also show up in the inhibitory activity of the stabilized components, as seen in compounds 11 and 12.
Scheme 3.

Structures of scaffolds and stable library components with corresponding Ki values.
Induction by neuraminidase of the preferred component synthesis may occur both under thermodynamic and kinetic control. In the present case, thermodynamic control seems to be the probable mechanism, because the enzyme was always used at a concentration equal to that of the scaffold. This amount was sufficient to convert the scaffold present in the transient mixture into the best binding components.
We then investigated whether improvement of the scaffold could further increase the efficiency of the inhibitors formed from the virtual libraries. Scaffold 15 was synthesized so as to replace the amino group conserved in the hits with a guanidinium group. Such a replacement typically yields better binding of the known inhibitors, as indeed can be seen from the Ki value of 15 (Scheme S3). The experiments with the virtual libraries were performed similarly to those with 2 and yielded the results summarized in Fig. 5. The subset of substituents selected with scaffold 15 was identical to that with 2, as expected from the high structural similarity of the scaffolds. Two of the preferred components, 17 and 18, were also synthesized individually and showed Ki values of 16 and 52 nM, respectively. The identity of the individually synthesized stable components with the ones formed in the presence of the enzyme was confirmed by the HPLC-MS analysis, as shown in the supporting information.
The library components selected by the target in the DCC experiments and then individually synthesized were also tested as inhibitors of neuraminidases from different virus strains. The resulting Ki values (Table 1) demonstrate that the inhibitory activity of the DCC hits for these enzymes compares well, and in some cases exceeds the values for the extracellular domain, which was used in DCC (cf. Scheme S3).
Table 1.
Ki (nM) of selected inhibitors determined for neuraminidases from different virus subtypes
| Neuraminidase source | 2 | 11 | 12 | 15 | 17 | 18 |
|---|---|---|---|---|---|---|
| B/Victoria/504/2000 | 9,300 ± 500 | 247 ± 64 | 1,210 ± 50 | 665 ± 63 | 18 ± 2 | 114 ± 13 |
| A/Panama/2007/99 | 34,000 ± 4,200 | 145 ± 6 | 130 ± 3 | 209 ± 11 | 5 ± 1 | 4 ± 0.2 |
| B/Harbin/7/94 | 9,400 ± 1,000 | 296 ± 19 | 1,430 ± 110 | 679 ± 66 | 24 ± 4 | 135 ± 19 |
| A/Johannesburg/33/94 | 36,000 ± 3,400 | 303 ± 16 | 1,000 ± 30 | 247 ± 21 | 6 ± 0.3 | 11 ± 1 |
| cHis-tNA full | 11,300 ± 1,800 | 500 ± 50 | 1,090 ± 90 | 104 ± 5 | 19 ± 3 | 16 ± 2 |
Conclusions
The important feature of the libraries described above is the in vitro virtual mode of their formation. A similar term used in in silico combinatorial library screening refers to the arrays of compounds that are screened computationally for predicted affinity to the target and are not physically synthesized until their most effective components are identified (25). The in vitro virtual screening described here is accomplished in a conceptually similar way with the key difference, however, that all possible library components are potentially presented to the target, which promotes a pseudoevolutionary organization of the library into a subset of most effective binders. Although similar processes have been reported with supramolecular assemblies (14), the study described herein represents to our knowledge the first example of new molecular entities created in true virtual libraries under the selection pressure of a biological target.
As a practical advantage, the combined synthesis-screening of a virtual library offers a dramatic informational signal-to-noise increase. Complete synthesis of a library similar to one shown in Fig. 1 in one pot would lead to a mixture with a large number of components and low concentrations of each of them, which would be extremely difficult or impossible to deconvolute. Organization of the virtual library driven by the enzyme not only increases the concentration of strong binders, but also minimizes the nonbinding background. The simplicity of the synthetic and analytical procedure allows for a rapid and straightforward customization of the library composition.
Supplementary Material
Acknowledgments
We thank Prof. H.-D. Klenk (Marburg, Germany), who kindly provided us with the neuraminidase cDNA of the Influenza A/FPV/Rostock/34 strain, and R. Mischler (Berna Biotech AG, Schwitzerland) for A/Panama/2007/99, A/Johannesburg/33/94, B/Harbin/7/94, and B/Victoria/504/2000 virus preparations. We are grateful to Prof. J.-M. Lehn (Strasbourg, France) for helpful comments and continued encouragement of this work. This work was financed in part by Grant BEO 0312391 from Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie, Germany.
Abbreviation
- DCC
dynamic combinatorial chemistry
Footnotes
Species 9 and 10 (Scheme S1) form as products of further reductive amination from 7 and 8 through the hemiaminal intermediates similar to 3 and 5.
It is generally expected that higher substitution degree should be also promoted by a target, if corresponding species possess higher binding affinity. However, adjustment of conditions, such as building block concentrations, is required for detection of the multisubstituted ligands. Formation of some of the multisubstituted species—e.g., 9 and 10—requires an irreversible reduction step. Therefore the expected target effect on the formation of such compounds may be less pronounced.
The reversible interaction between 2 and selected aldehydes was studied by 1H NMR (250 MHz) by titration of 1–5 mM 2 with a 2–10 mM aldehyde in 10 mM aqueous imidazole buffer at pH 7.8–10.0. A new spectral pattern corresponding to monosubstituted species 3 was observed at the aldehyde concentration above 5 mM, and the second pattern corresponding to species 5 at a higher pH/aldehyde concentration. The amino group adjacent to the double bond appears to be more reactive, so that is unlikely that the monosubstitution can occur on the opposite side of the scaffold.
References
- 1.Hung D T, Jamison T F, Schreiber S L. Chem Biol. 1996;3:623–639. doi: 10.1016/s1074-5521(96)90129-5. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber S L. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 3.Rideout D. Science. 1986;233:561–563. doi: 10.1126/science.3523757. [DOI] [PubMed] [Google Scholar]
- 4.Rideout D, Calogeropoulou T, Jaworski J, McCarthy M. Biopolymers. 1990;29:247–262. doi: 10.1002/bip.360290129. [DOI] [PubMed] [Google Scholar]
- 5.Rotenberg S A, Calogeropoulou T, Jaworski J S, Weinstein I B, Rideout D. Proc Natl Acad Sci USA. 1991;88:2490–2494. doi: 10.1073/pnas.88.6.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen R, Huc I. Angew Chem Int Ed Engl. 2001;40:1774–1776. [PubMed] [Google Scholar]
- 7.Swann P G, Casanova R A, Desai A, Frauenhoff M M, Urbancic M, Slomczynska U, Hopfinger A J, Lebreton G C, Venton D L. Biopolymers. 1996;40:617–625. doi: 10.1002/(sici)1097-0282(1996)40:6<617::aid-bip3>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 8.Ganesan A. Angew Chem Int Ed Engl. 1998;37:2828–2831. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2828::AID-ANIE2828>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 9.Eliseev A V, Lehn J M. Combinatorial Chemistry in Biology. 1999;243:159–172. doi: 10.1007/978-3-642-60142-2_9. [DOI] [PubMed] [Google Scholar]
- 10.Lehn J M. Chem Eur J. 1999;5:2455–2463. [Google Scholar]
- 11.Cousins G R L, Poulsen S A, Sanders J K M. Curr Opin Chem Biol. 2000;4:270–279. doi: 10.1016/s1367-5931(00)00088-0. [DOI] [PubMed] [Google Scholar]
- 12.Lehn J M, Eliseev A V. Science. 2001;291:2331–2332. doi: 10.1126/science.1060066. [DOI] [PubMed] [Google Scholar]
- 13.Huc I, Nguyen R. Comb Chem High Throughput Screen. 2001;4:53–74. doi: 10.2174/1386207013331273. [DOI] [PubMed] [Google Scholar]
- 14.Hasenknopf B, Lehn J M, Kneisel B O, Baum G, Fenske D. Angew Chem Int Ed Engl. 1996;35:1838–1840. [Google Scholar]
- 15.Huc I, Lehn J M. Proc Natl Acad Sci USA. 1997;94:2106–2110. doi: 10.1073/pnas.94.6.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hausmann J, Kretzschmar E, Garten W, Klenk H D. J Gen Virol. 1995;76:1719–1728. doi: 10.1099/0022-1317-76-7-1719. [DOI] [PubMed] [Google Scholar]
- 17.McKimm-Breschkin J L, Caldwell J B, Guthrie R E, Kortt A A. J Virol Methods. 1991;32:121–124. doi: 10.1016/0166-0934(91)90192-3. [DOI] [PubMed] [Google Scholar]
- 18.Myers R W, Lee R T, Lee Y C, Thomas G H, Reynolds L W, Uchida Y. Anal Biochem. 1980;101:166–174. doi: 10.1016/0003-2697(80)90056-1. [DOI] [PubMed] [Google Scholar]
- 19.Potier M, Mameli L, Belisle M, Dallaire L, Melancon S B. Anal Biochem. 1979;94:287–296. doi: 10.1016/0003-2697(79)90362-2. [DOI] [PubMed] [Google Scholar]
- 20.Air G M, Laver W G. Proteins. 1989;6:341–356. doi: 10.1002/prot.340060402. [DOI] [PubMed] [Google Scholar]
- 21.Kim C U, Chen X W, Mendel D B. Antivir Chem Chemother. 1999;10:141–154. doi: 10.1177/095632029901000401. [DOI] [PubMed] [Google Scholar]
- 22.Kim C U, Lew W, Williams M A, Wu H W, Zhang L J, Chen X W, Escarpe P A, Mendel D B, Laver W G, Stevens R C. J Med Chem. 1998;41:2451–2460. doi: 10.1021/jm980162u. [DOI] [PubMed] [Google Scholar]
- 23.Lew W, Chen X W, Kim C U. Curr Med Chem. 2000;7:663–672. doi: 10.2174/0929867003374886. [DOI] [PubMed] [Google Scholar]
- 24.Colman P M. J Antimicrob Chemother. 1999;44, Suppl B:17–22. doi: 10.1093/jac/44.suppl_2.17. [DOI] [PubMed] [Google Scholar]
- 25.Waszkowycz B, Perkins T D J, Sykes R A, Li J. IBM Syst J. 2001;40:360–376. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.