

Abstract
Kaposi sarcoma-associated herpes virus (KSHV) contains a gene that has functional and sequence homology to the apoptotic Bcl-2 family of proteins [Sarid, R., Sato, T., Bohenzky, R. A., Russo, J. J. & Chang, Y. (1997) Nat. Med. 3, 293–298]. The viral Bcl-2 protein promotes survival of infected cells and may contribute to the development of Kaposi sarcoma tumors [Boshoff, C. & Chang, Y. (2001) Annu. Rev. Med. 52, 453–470]. Here we describe the solution structure of the viral Bcl-2 homolog from KSHV. Comparison of the KSHV Bcl-2 structure to that of Bcl-2 and Bcl-xL shows that although the overall fold is the same, there are key differences in the lengths of the helices and loops. Binding studies on peptides derived from the Bcl-2 homology region 3 of proapoptotic family members indicate that the specificity of the viral protein is very different from what was previously observed for Bcl-xL and Bcl-2, suggesting that the viral protein has evolved to have a different mechanism of action than the host proteins.
Kaposi sarcoma-associated herpes virus (KSHV) is directly involved in the etiology of Kaposi sarcoma tumors (1–4) and shows a high incidence among immunocompromised individuals such as patients with HIV and transplant recipients. Like other gammaherpesviruses, KSHV contains genes that mimic those that the host uses to regulate cell growth and apoptosis (3). Dysregulation of these processes helps the virus to survive and successfully replicate. Recent in vivo work has shown that the gammaherpesvirus expresses several cellular protein mimics including a viral-cyclin and a viral-bcl-2 that are critical for viral reactivation from latency and replication in immunocompromised hosts (5). Furthermore, these viral genes likely increase the propensity for oncogenic transformation of virus-infected cells based on their ability to interfere with established tumor-suppressor pathways (2).
KSHV encodes a protein with both sequence and functional homology to human Bcl-2 (6). Members of the Bcl-2 family of proteins play an important role in tissue homeostasis, embryogenesis, and the immune response through their actions as either inhibitors or promoters of apoptosis (7–10). The prosurvival function of cellular Bcl-2 is a result of heterodimerization with proapoptotic family members such as Bak, Bax, and Bad. Based on a sequence alignment (Fig. 1), the KSHV Bcl-2 protein contains the Bcl-2 homology (BH) motifs BH1 and BH2. In addition, the BH1 region of KSHV Bcl-2 contains the signature “NWGR” sequence, which is believed to be essential for the antiapoptotic function of Bcl-2 and for the ability of Bcl-2 to heterodimerize with other family members (11). However, the mechanism by which KSHV Bcl-2 protects against apoptosis is still unclear. Cellular assays suggest that KSHV Bcl-2 does not heterodimerize with proapoptotic Bcl-2 family members such as Bax and Bak (12).
Figure 1.

Sequence alignment based on the observed secondary structure of KSHV Bcl-2 and human full-length Bcl-xL and Bcl-2. α-Helices for KSHV Bcl-2 are shown above the sequence. The BH regions are shown below the sequence. The transmembrane domain (TM) is indicated below the sequence.
KSHV Bcl-2 contains only limited homology to the BH3 and BH4 motifs (12). Structural and mutagenesis studies have shown that the BH3 region is important for the antiapoptotic activity of Bcl-2 family members, and that the BH4 region may be important for the regulation of Bcl-2 activity. Caspase cleavage of human Bcl-2 at Asp-34 separates the BH4 region from the rest of the protein and produces a truncated protein, which is proapoptotic vs. antiapoptotic. The viral KSHV homolog of Bcl-2 does not contain this caspase cleavage site, and therefore, escapes this extra level of regulation (13, 14).
The viral KSHV homolog of Bcl-2 may also protect virus-infected cells from apoptosis induced by the expression of KSHV viral cyclin (v-cyclin) (15, 16). Expression of v-cyclin in cells with increased levels of CDK6 induces apoptosis by a v-cyclin-CDK6-mediated inactivating phosphorylation of cellular Bcl-2. As with caspase cleavage, the phosphorylation of cellular Bcl-2 occurs in the unstructured loop of the proteins (amino acids 32–80) (15, 16). Because viral Bcl-2 lacks this loop it is able to retain its antiapoptotic activity even in the presence of v-cyclin-CDK6 (16). The viral protein thus exhibits different antiapoptotic properties than cellular Bcl-2.
Suppression of apoptosis seems to be important to enhance the survival of KSHV-infected cells so that the virus may replicate, spread, and persist in its host. A better understanding of how the virus mimics the host proteins may aid in the development of therapies for the dysregulated pathway found in virus-infected cells. Here we describe the solution structure of the Bcl-2 homolog found in KSHV and compare it to the previously determined structures of Bcl-2 (17), Bcl-xL (18, 19), and Bax (20). In addition, we investigated the ability of KSHV Bcl-2 to form a complex with peptides derived from the BH3 region of the proapoptotic proteins Bak, Bax, and Bad. Finally, the importance of various residues of the viral protein for binding to the BH3 peptides was explored through site-directed mutagenesis.
Materials and Methods
Plasmid Construction.
Several different KSHV Bcl-2 constructs were prepared and evaluated for their suitability for NMR structural studies. A fragment coding amino acids 1–146 of KSHV Bcl-2 (Fig. 1) was obtained from full-length KSHV Bcl-2. The fragment was inserted into the NcoI and XhoI sites of the pET21d(+) plasmid (Novagen) for expression. An additional 12 residues were added at the C terminus (DDDDLEHHHHHH), and mutations at N67D and V117A were included to optimize the expression and solubility of the protein in Escherichia coli. Mutants of KSHV Bcl-2 were prepared by using the QuickChange site-directed mutagenesis kit (Stratagene) with the double mutant KSHV Bcl-2 as the template. Coding sequences were confirmed by DNA sequencing.
Expression and Purification.
In all cases, the KSHV Bcl-2 protein was prepared by expressing the protein in E. coli BL21(DE3) grown on M9 media. Uniformly 15N-labeled, uniformly 15N,13C-labeled, and uniformly 15N,13C-labeled 75% 2H samples were prepared with media containing either 15NH4Cl, 15NH4Cl plus [U-13C]glucose, or 15NH4Cl, [U-13C]glucose and 75% 2H2O, respectively. In addition, a sample was prepared that was uniformly 15N,2H-labeled; 13C-labeled at the methyl groups of leucine, valine, and the δ1 methyl of isoleucine; and protonated at phenylalanine, tyrosine, and tryptophan with M9 media containing 15NH4Cl, [3-13C]-α-ketobutyrate, [3,3′-13C]-α-ketoisovalerate, protonated phenylalanine, protonated tyrosine, protonated tryptophan, and 100% 2H2O (21). Soluble protein was purified by Ni2+ affinity chromatography. NMR samples contained 0.5–1.0 mM protein in either 90% H2O/10% 2H2O or 100% 2H2O, 20 mM 2H-Tris (pH 7.8), and 5 mM 2H-DTT.
NMR Spectroscopy.
All NMR experiments were acquired at 298 K on a Bruker (Billerica, MA) DRX500, DRX600, or DRX800 NMR spectrometer. Backbone 1H, 13C, and 15N resonance assignments were achieved with [15N,13C(75%)2H] KSHV Bcl-2 by using a suite of deuterium-decoupled triple-resonance experiments [HNCA, HN(CO)CA, HN(CA)CB, HN(COCA)CB, HNCO, and HN(CA)CO] (22). The side-chain 1H and 13C NMR signals were assigned from HCCH-total correlation spectroscopy experiments (23), and stereospecific assignments of the valine and leucine methyl groups were obtained from an analysis of the 13C-13C coupling patterns observed for biosynthetically directed, fractionally 13C-labeled KSHV Bcl-2 (24). Nuclear Overhauser effect (NOE) distance restraints were obtained from three-dimensional 15N- and 13C-edited NOE spectroscopy spectra (25, 26) acquired with a mixing time of 80 ms. Slowly exchanging amide protons were identified in an 15N heteronuclear single quantum coherence spectrum recorded immediately after exchanging the protein into a buffer prepared with 2H2O.
Structure Calculations.
KSHV Bcl-2 structures were calculated by using a simulated annealing protocol (27) with the program cnx (Molecular Simulations, Waltham, MA). A square-well potential (FNOE = 50 kcal mol−1) was used to constrain NOE-derived distances. Based on the cross-peak intensities, NOE-derived distance restraints were given upper bounds of 3.5, 4.5, or 6.0 Å. In the refinement stage, additional ambiguous constraints were added, with an upper bound of 6.0 Å, for unassigned cross peaks that were consistent with the chemical shift table (i.e., error bars of 0.07 ppm for protons, 0.7 ppm for hetero atoms) and the structure. Torsion angle restraints, φ and ψ, were generated from analysis of N, C′, Cα, and Hα chemical shifts by using the talos program (28). A force constant of 200 kcal mol−1 rad−2 was applied to all torsional restraints. Explicit hydrogen bonds were included in α-helices only for residues observed to have slowly exchanging amide protons. The program procheck was used to analyze the geometric quality of the calculated structures in the ensemble (29).
Peptide Binding.
A fluorescence polarization-based competition assay was used to determine the relative affinity of BH3 peptides (SynPep, Dublin, CA) to the Bcl-2 proteins. Fluorescence polarization measurements were carried out as described (30) by using an SLM8000 fluorimeter and a fluorescein-labeled Bak peptide with the sequence GQVGRQLAIIGDK(FITC)INR as the probe. The dissociation constant of this peptide for KSHV Bcl-2 and human Bcl-2 was found to be 144 and 350 nM, respectively. Titrations were carried out in a buffer containing 120 mM sodium phosphate (pH 7.55), 0.01% bovine gamma globulin, and 0.1% sodium azide. For KSHV BCL-2, the protein concentration was 290 nM with the probe concentration at 2.9 nM, whereas for cellular Bcl-2, the protein concentration was 550 nM with the probe concentration at 2.9 nM. Dissociation constants were determined from titration curves with in-house-written software by using the analytical expressions of Wang (31).
Results and Discussion
A portion of the KSHV Bcl-2 protein (residues 1–146) lacking the putative C-terminal transmembrane helix (Fig. 1) was expressed in E. coli, isotopically labeled, and purified. This protein was found to aggregate at concentrations above 0.1 mM over a wide pH range and was therefore unsuitable for NMR structural studies. To improve the solubility and expression of the protein in E. coli, a double mutant protein (N67D, V117A) was prepared. The 15N heteronuclear single quantum coherence spectrum of this mutant was essentially identical to that of the wild-type (wt) protein, indicating that the fold of the protein is preserved. Furthermore, the mutant and wt protein bind with similar affinities to a fluoresceinated peptide from Bak with dissociation constants of 144 and 139 nM, respectively.
The backbone and side-chain resonances of the protein were assigned from standard heteronuclear three-dimensional NMR experiments (see Materials and Methods) recorded on a protein sample that was uniformly labeled with 15N and 13C and 75% deuterated. To rapidly obtain assignments and unambiguous structural constraints, experiments were also recorded on a sample labeled uniformly with 15N and 2H and selectively enriched with 13C at the methyl groups of leucine, valine, and the δ1 methyl group of isoleucine. In addition, the phenylalanine, tyrosine, and tryptophan residues of this sample were protonated (21).
The structure of the KSHV Bcl-2 protein was determined from a total of 1,953 unambiguous NMR-derived distance and torsion angle restraints along with 1,023 ambiguous distance restraints (Table 1). Fig. 2A depicts a backbone (N,Cα,C′) superposition of 10 low-energy structures that were derived from the NMR data by using the program cnx. Excluding residues 1–5, 23–31, and 146–152, the atomic rms deviation about the mean position is 0.39 ± 0.05 Å for the backbone atoms and 0.75 ± 0.06 Å for all heavy atoms. The structural statistics for the ensemble and energy minimized structure are given in Table 1. There are no dihedral-angle violations greater than 5o and no NOE violations greater than 0.4 Å. Only covalent geometry, NOE, torsion, and repulsive terms were included in the structure refinement. Even so, the Lennard–Jones energy is large and negative, indicating that the structures have favorable nonbonded contacts. Analysis of the average-minimized structures with the program procheck (29) showed that 70.4% of the residues for KSHV Bcl-2 lie in the most favored region of the Ramachandran plot, whereas an additional 25.2% lie in allowed regions.
Table 1.
Structural statistics for KSHV Bcl-2
| 〈SA〉* | 〈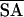 〉r 〉r
|
|
|---|---|---|
| rmsd from experimental distance restraints, Å | ||
| Intraresidue (397) | 0.023 ± 0.002 | 0.014 |
| Sequential (407) | 0.017 ± 0.003 | 0.035 |
| Short range (366) | 0.026 ± 0.003 | 0.026 |
| Long range (452) | 0.028 ± 0.002 | 0.035 |
| Hydrogen bonds (39) | 0.085 ± 0.002 | 0.092 |
| Talos (204) | ||
| cnx potential energies (kcal mol−1)† | ||
| Etot | 94.6 ± 0.8 | 114.6 |
| Ebond | 1.8 ± 0.1 | 3.6 |
| Eang | 54.8 ± 0.9 | 68.3 |
| Eimp | 5.2 ± 0.4 | 6.7 |
| Erepel | 17.7 ± 0.8 | 13.5 |
| Enoe | 14.1 ± 1.1 | 21.4 |
| EL-J | −724.4 ± 12.2 | −739.1 |
| Cartesian coordinate rmsd (Å)‡ | Backbone | All heavy |
〈SA〉 vs. 〈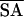 〉 〉 |
0.39 ± 0.05 | 0.76 ± 0.06 |
〈SA〉 is the ensemble of the 10 lowest-energy structures, 〈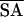 〉 is the mean structure, and 〈SA〉r is the energy-minimized mean structure. rmsd, rms deviation.
〉 is the mean structure, and 〈SA〉r is the energy-minimized mean structure. rmsd, rms deviation.
EL-J was not used in the refinement but is included as an independent assessment of nonbonded geometry.
Excluding residues 1–5, 23–31, and 146–152.
Figure 2.

(A) Backbone (N,Cα,C′) superposition of 10 low-energy NMR-derived structures for viral KSHV Bcl-2. Helices are numbered with respect to those observed in the structure of Bcl-xL. (B) Ribbon (36) depiction of the average-minimized structure for viral KSHV Bcl-2. The central helix, α5, is colored yellow. (C) Solvent-accessible surface showing hydrophobic groove for viral KSHV Bcl-2. Leucine, isoleucine, valine, methionine, tyrosine, phenylalanine, and tryptophan residues are colored yellow. Aspartate and glutamate are colored red. Lysine, arginine, and histidine are colored blue. All other residue types are colored gray.
Description of the Structure.
KSHV Bcl-2 is an all α-helical protein (Fig. 2B). A central hydrophobic α-helix (α5) forms the core of the protein and is sandwiched between the amphipathic helices α3 and α4 on one side and by amphipathic helices α1, α2, and α6 on the other side. The N-terminal helix, α1 (residues 10–21), is connected to α2 (residues 36–46) by a 13-residue loop (residues 22–35). A prominent bend at Lys-47 separates α2 from α3 (residues 48–56) and orients these helices in a nearly orthogonal fashion. Helices α4 (residues 63–77), α5 (residues 84–100), and α6 (residues 105–131) are oriented in a nearly antiparallel fashion with a kink in helix 6 at Gly-124. A turn composed of two glycine residues (residues 132 and 133) separates α6 from α7 (residues 134–141) and orients α7 nearly perpendicular to helices 4–6. A hydrophobic groove is formed on the surface of the protein from residues of α2, α3, α4, and the N-terminal portion of α5 (Fig. 2C).
Structural Comparison of KSHV Bcl-2 to Other Bcl-2 Family Members.
KSHV Bcl-2 shows substantial structural homology to the related human proteins Bcl-2 (17), Bcl-xL (18, 19), and Bax (20). They all contain the same number of α-helices and share the same overall fold. In addition, the hydrophobic groove formed by the BH1, BH2, and BH3 regions is maintained in the viral protein. This groove is lined with hydrophobic residues that are highly conserved among Bcl-2 family members. A backbone superposition of the average minimized structure of KSHV Bcl-2, excluding the turns and loops, with Bcl-xL and Bcl-2 gives an rms deviation of 2.7 and 2.5 Å, respectively.
As in other Bcl-2 family members, the signature NWGR sequence is conserved. From the structure it is clear that these residues serve the same purpose in the viral protein as they do in Bcl-xL. In both Bcl-xL and KSHV Bcl-2, the tryptophan residue (W84) sits at the N terminus of α5 and contacts hydrophobic residues on α6 and α7. The structural data suggests that it is important for maintaining the fold of the protein. Indeed, no soluble protein was obtained after expression in E. coli when we mutated this tryptophan to an alanine or leucine (i.e., W84A or W84L). The glycine and arginine residues of the signature sequence sit at the top of the hydrophobic cleft. Mutation of either of these residues (G85A, and R86Q) produced a soluble, well folded protein, suggesting that these residues are not critical for maintaining the fold of KSHV Bcl-2. However, in Bcl-xL these residues were shown to be important for the binding of a BH3 peptide from the proapoptotic protein Bak (18).
The sequences of the BH3 and BH4 domains in KSHV Bcl-2 are very poorly conserved (13). However, the structure of the viral protein in these regions is homologous to other Bcl-2 family members. The first helix of viral Bcl-2 helps to bury the hydrophobic residues from α5 and α6 and is thus structurally similar to the helix contained in the BH4 region of Bcl-2 and Bcl-xL. When buried residues in the first helix of Bcl-2 are mutated (e.g., V15E), it effects the antiapoptotic activity of the protein and its ability to heterodimerize with Bax (32). The second helix of viral Bcl-2 corresponds to the BH3 regions of Bcl-2 and Bcl-xL and as in these human proteins, forms part of the hydrophobic groove of KSHV Bcl-2.
Despite the overall similarity of KSHV Bcl-2 to Bcl-2 and Bcl-xL, there are key differences in the length of the helices, turns, and loops between the viral and human proteins (Fig. 3). The first helix of the viral protein is two turns shorter than the corresponding helix in Bcl-2 and Bcl-xL. In addition, the loop connecting α1 to α2 is much shorter in the viral protein than in the wt human proteins (Fig. 1). In Bcl-xL and Bcl-2, this loop contains a caspase cleavage site that converts these antiapoptotic proteins to proapoptotic proteins. The shorter loop of the viral homolog does not contain this caspase cleavage (13, 14) or the inactivating phosphorylation sites present in cellular Bcl-2 (16). Additional differences between the viral protein and its cellular homologs are the lengths of the loops connecting α3 to α4 and α4 to α5. In KSHV Bcl-2, the loop between α3 and α4 is four residues shorter than that in Bcl-2 and Bcl-xL, whereas the loop connecting α4 to α5 is three residues longer. Because both of these regions are on the periphery of the binding groove, they would not be expected to make a major contribution to the binding of KSHV Bcl-2 to other apoptotic proteins.
Figure 3.

Comparison of KSHV Bcl-2 (A) to Bcl-xL (B) and to Bcl-2 (C). Residues of the hydrophobic groove that are homologous to those that contact the Bak and Bad peptides when complexed to Bcl-xL are shown along with the tryptophan residue of the NWGR sequence. To emphasize the differences in the structured regions of the proteins, we have made our comparisons to truncated forms of Bcl-2 and Bcl-xL (17, 18).
A significant structural difference that was observed between Bcl-xL and Bcl-2 involves the region around α3 (17). In Bcl-2 a compact bend separates α2 from α3, whereas in Bcl-xL a short loop separates these two helices. In addition, α3 is translated toward the core of the protein in Bcl-xL compared with Bcl-2. In the viral protein, the region around α3 resembles Bcl-2 more than Bcl-xL (Fig. 3). The residues in the viral protein are, however, different and this has an effect on the chemical character of the bottom of the binding groove.
Recognition of Proapoptotic BH3 Peptides.
The binding interactions between Bcl-2 family members show a defined selectivity (17, 18). To investigate the selectivity of KSHV Bcl-2, we used a fluorescence polarization-based assay to measure the affinity of a series of BH3-containing peptides from the proapoptotic proteins Bak, Bax, and Bad (Table 2). Earlier studies on Bcl-xL have shown a good correlation between BH3 peptide binding and binding to full-length protein (33, 34). In the present study we found that the Bak peptide binds tightly (Kd < 50 nM) to viral KSHV Bcl-2. These results are inconsistent with an earlier report that indicated that KSHV Bcl-2 does not bind to full-length Bak in cellular assays (12). The Bax peptide also binds to KSHV Bcl-2, although with reduced affinity compared with Bak, and binding of the Bad 25-mer to viral Bcl-2 is weaker still.
Table 2.
BH3 peptide binding to Bcl family members
The observed specificity of the viral protein for the Bak, Bax, and Bad peptide is very different from what is observed for either Bcl-2 or Bcl-xL (17, 18). As shown in Table 2, Bcl-xL binds tightly to both the Bak and Bad peptides but only weakly to the Bax peptide, whereas Bcl-2 binds tightly to the Bad peptide but only weakly to the Bak and Bax peptides. The greater affinity of the viral protein for the Bak and Bax peptides vs. the Bad peptide is interesting based on their postulated mechanism for promoting apoptosis. Bak and Bax are directly involved in the mitochondrial dysfunction required for cell death whereas Bad and other BH3-only family members are tissue-specific triggers for the homooligomerization of Bak and Bax (35).
Mutation of the Bak peptide to alanine at V74, L78, and I85 causes a large decrease in the affinity of the peptide for KSHV Bcl-2 (Table 3). In the structure of the Bcl-xL/Bak peptide complex, these residues were observed to make hydrophobic contacts to residues in the groove of Bcl-xL (18) and in fact, these mutations have a similar affect on the affinity of the Bak peptide for Bcl-xL. In addition, mutation of Asp-83 to alanine decreases the affinity of the Bak peptide for KSHV Bcl-2, although not to as great an extent as observed for Bcl-xL.
Table 3.
Binding of Bak peptide mutants to KSHV Bcl-2 and Bcl-xL
To investigate where the Bak and Bax BH3 peptide binds to KSHV Bcl-2, we monitored spectral changes for both amide and methyl resonances after titration with the peptides. Amide- and methyl-group resonances that shift significantly in the titration are plotted on the surface of the protein in Fig. 4. As expected, residues that are significantly affected are in the groove formed by the BH1, BH2, and BH3 regions of KSHV Bcl-2. The amino acid side chains of Bcl-xL, which make intimate contact with the bound Bak and Bad peptides, are shown in Fig. 3. Some of these residues are identical in KSHV Bcl-2 (F105, F146, R138, and A142), some are conservatively substituted (L108M, V130I, and V141L), whereas the rest differ greatly in character between the two proteins (A93Y, F97L, A104L, and L130G). Thus, although the binding groove of KSHV Bcl-2 is clearly hydrophobic as it is in Bcl-xL and Bcl-2, the contact surface between the viral protein and the peptides is likely quite different.
Figure 4.

Solvent-accessible surface showing the hydrophobic groove of KSHV Bcl-2. The orientation of A is the same as in Fig. 2C whereas B is the opposite face. Residues that shift significantly when the BH3 peptide of Bak is titrated into a solution of the protein are colored magenta.
The importance of the conserved glycine and arginine residues of the signature NWGR sequence was investigated by site-directed mutagenesis. Mutation of these residues (G85A, G85V, and R86Q) greatly reduces the affinity of viral Bcl-2 for the fluorescein-labeled Bak peptide, with dissociation constants of 4,800, >10,000, and 1,900 nM, respectively. The wt protein shows a dissociation constant for the peptide of 144 nM. Because the 15N heteronuclear single quantum coherence spectra of these mutated proteins are essentially identical to that of the wt protein, the observed changes in affinity are most likely because of changes in the contacts that the mutated proteins make with the peptide. For the glycine mutants, the reduced affinity is consistent with what would be expected based on the structure of the complex of Bcl-xL with the Bak and Bad peptides (18, 30). In both complexes, the peptide packs tightly against this glycine, suggesting that a larger residue, like alanine or valine, would lead to an unfavorable steric contact.
In the Bcl-xL/Bak complex, the side chain of the signature arginine residue is positioned to make favorable electrostatic contact with Asp-83 of the Bak peptide and in fact, mutation of this arginine to glutamine in Bcl-xL inhibits its binding to the Bax protein. Similarly, the same mutation in the KSHV Bcl-2 protein, R86Q, decreases its affinity for the Bak peptide, although the effect is not as dramatic as observed for Bcl-xL. These data, although surprising, are consistent with the observation that the D83A Bak peptide mutant reduces, but does not abolish, peptide binding (Table 3). These data, therefore, suggest that the interaction between D83 of the peptide and R86 of the protein is not as important in KSHV Bcl-2 as it is for Bcl-xL.
Conclusions
The solution structure of the KSHV Bcl-2 reveals how the virus mimics the host's own antiapoptotic proteins. Although the viral protein has the same overall fold as the larger human homologs, the lengths of certain helices and loops are different. The structural homology that we have found between the viral and human proteins is not surprising based on their functional similarities. We have also shown that the KSHV Bcl-2 has its own distinct selectivity for binding BH3 peptides from the proapoptotic proteins Bak, Bax, and Bad. The functional significance of this observation needs to be studied further. The importance of the antiapoptotic proteins in the life cycle of gammaherpesviruses suggests that they could be intriguing targets for drug discovery. Recent data have shown the importance of viral Bcl-2 in maintaining the persistence of the virus (5). The role of viral Bcl-2 in latent and persistent infection, the stages of infection associated with human disease, makes the viral Bcl-2 a particularly attractive target for pharmacologic manipulation. The structural data provided here point to subtle differences between the viral and human proteins that may make it possible to specifically target these proteins for antiviral therapy.
Acknowledgments
We thank Dr. Rob Meadows for help with the structure calculations and for useful discussions.
Abbreviations
- NOE
nuclear Overhauser effect
- KSHV
Kaposi sarcoma-associated herpes virus
- BH
Bcl-2 homology
- wt
wild type
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1k3k).
References
- 1.Blattner W A. Proc Assoc Am Phys. 1999;111:563–572. doi: 10.1046/j.1525-1381.1999.99210.x. [DOI] [PubMed] [Google Scholar]
- 2.Boshoff C, Chang Y. Annu Rev Med. 2001;52:453–470. doi: 10.1146/annurev.med.52.1.453. [DOI] [PubMed] [Google Scholar]
- 3.Hardwick J M. Semin Cell Dev Biol. 1998;9:339–349. doi: 10.1006/scdb.1998.0243. [DOI] [PubMed] [Google Scholar]
- 4.Muralidhar S, Veytsmann G, Chandran B, Ablashi D, Doniger J, Rosenthal L J. J Clin Virol. 2000;16:203–213. doi: 10.1016/s1386-6532(99)00081-5. [DOI] [PubMed] [Google Scholar]
- 5. Gangappa, S., Van Dyk, L. F., Jewett, T. J., Speck, S. H. & Virgin, H. W. (2002) J. Exp. Med., in press. [DOI] [PMC free article] [PubMed]
- 6.Sarid R, Sato T, Bohenzky R A, Russo J J, Chang Y. Nat Med. 1997;3:293–298. doi: 10.1038/nm0397-293. [DOI] [PubMed] [Google Scholar]
- 7.Adams J M, Cory S. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 8.Chao D T, Korsmeyer S J. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 9.Tsujimoto Y, Shimizu S. FEBS Lett. 2000;466:6–10. doi: 10.1016/s0014-5793(99)01761-5. [DOI] [PubMed] [Google Scholar]
- 10.Thompson C B. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 11.Yin X M, Oltvai Z N, Korsmeyer S J. Nature (London) 1994;369:321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- 12.Cheng E H, Nicholas J, Bellows D S, Hayward G S, Guo H G, Reitz M S, Hardwick J M. Proc Natl Acad Sci USA. 1997;94:690–694. doi: 10.1073/pnas.94.2.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bellows D S, Chau B N, Lee P, Lazebnik Y, Burns W H, Hardwick J M. J Virol. 2000;74:5024–5031. doi: 10.1128/jvi.74.11.5024-5031.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng E H, Kirsch D G, Clem R J, Ravi R, Kastan M B, Bedi A, Ueno K, Hardwick J M. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- 15.Ojala P M, Tiainen M, Salven P, Veikkola T, Castanos-Velez E, Sarid R, Biberfeld P, Makela T P. Cancer Res. 1999;59:4984–4989. [PubMed] [Google Scholar]
- 16.Ojala P M, Yamamoto K, Castanos-Velez E, Biberfeld P, Korsmeyer S J, Makela T P. Nat Cell Biol. 2000;2:819–825. doi: 10.1038/35041064. [DOI] [PubMed] [Google Scholar]
- 17.Petros A M, Medek A, Nettesheim D G, Kim D H, Yoon H S, Swift K, Matayoshi E D, Oltersdorf T, Fesik S W. Proc Natl Acad Sci USA. 2001;98:3012–3017. doi: 10.1073/pnas.041619798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sattler M, Liang H, Nettesheim D, Meadows R P, Harlan J E, Eberstadt M, Yoon H S, Shuker S B, Chang B S, Minn A J, Thompson C B, Fesik S W. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 19.Muchmore S W, Sattler M, Liang H, Meadows R P, Harlan J E, Yoon H S, Nettesheim D, Chang B S, Thompson C B, Wong S L, et al. Nature (London) 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki M, Youle R J, Tjandra N. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 21.Medek A, Olejniczak E T, Meadows R P, Fesik S W. J Biomol NMR. 2000;18:229–238. doi: 10.1023/a:1026544801001. [DOI] [PubMed] [Google Scholar]
- 22.Yamazaki T, Lee W, Arrowsmith C H, Muhandiram D R, Kay L E. J Am Chem Soc. 1994;116:11655–11666. [Google Scholar]
- 23.Clore G M, Gronenborn A M. Methods Enzymol. 1994;239:349–363. doi: 10.1016/s0076-6879(94)39013-4. [DOI] [PubMed] [Google Scholar]
- 24.Neri D, Szyperski T, Otting G, Senn H, Wüthrich K. Biochemistry. 1989;28:7510–7516. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 25.Fesik S W, Zuiderweg E R P. J Magn Reson. 1988;78:588–593. [Google Scholar]
- 26.Marion D, Driscoll P C, Kay L E, Wingfield P T, Bax A, Gronenborn A M, Clore G M. Biochemistry. 1989;29:6150–6156. doi: 10.1021/bi00441a004. [DOI] [PubMed] [Google Scholar]
- 27.Brunger A T. X-PLOR. New Haven, CT: Yale Univ. Press; 1992. , Version 3.1. [Google Scholar]
- 28.Cornilescu G, Delaglio F, Bax A. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 29.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 30.Petros A M, Nettesheim D G, Wang Y, Olejniczak E T, Meadows R P, Mack J, Swift K, Matayoshi E D, Zhang H, Thompson C B, Fesik S W. Protein Sci. 2000;9:2528–2534. doi: 10.1110/ps.9.12.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z-X. FEBS Lett. 1995;360:111–114. doi: 10.1016/0014-5793(95)00062-e. [DOI] [PubMed] [Google Scholar]
- 32.Hirotani M, Zhang Y, Fujita N, Naito M, Tsuruo T. J Biol Chem. 1999;274:20415–20420. doi: 10.1074/jbc.274.29.20415. [DOI] [PubMed] [Google Scholar]
- 33.Kelekar A, Chang B S, Harlan J E, Fesik S W, Thompson C B. Mol Cell Biol. 1997;17:7040–7046. doi: 10.1128/mcb.17.12.7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ottilie S, Diaz J L, Horne W, Chang J, Wang Y, Wilson G, Chang S, Weeks S, Fritz L C, Oltersdorf T. J Biol Chem. 1997;272:30866–30872. doi: 10.1074/jbc.272.49.30866. [DOI] [PubMed] [Google Scholar]
- 35.Wei M C, Zong W X, Cheng E H, Lindsten T, Panoutsakopoulou V, Ross A J, Roth K A, MacGregor G R, Thompson C B, Korsmeyer S J. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carson M. J Mol Graphics. 1987;5:103–106. [Google Scholar]