

Abstract
Cell cycle checkpoints are evolutionarily conserved surveillance systems that protect genomic stability and prevent oncogenesis in mammals. One important target of checkpoint control is ribonucleotide reductase (RNR), which catalyzes the rate-limiting step in dNTP and DNA synthesis. In both yeast and humans, RNR is transcriptionally induced after DNA damage via Mec1/Rad53 (yeast) and ATM/CHK2 (human) checkpoint pathways. In addition, yeast checkpoint proteins Mec1 and Rad53 also regulate the RNR inhibitor Sml1. After DNA damage or at S phase, Mec1 and Rad53 control the phosphorylation and concomitant degradation of Sml1 protein. This new layer of control contributes to the increased dNTP production likely necessary for DNA repair and replication; however, the molecular mechanism is unclear. Here we show that Dun1, a downstream kinase of Mec1/Rad53, genetically and physically interacts with Sml1 in vivo. The absence of Dun1 activity leads to the accumulation of Sml1 protein at S phase and after DNA damage. As a result, dun1Δ strains need more time to finish DNA replication, are defective in mitochondrial DNA propagation, and are sensitive to DNA-damaging agents. Moreover, phospho-Sml1 is absent or dramatically reduced in dun1Δ cells. Finally, Dun1 can phosphorylate Sml1 in vitro. These results suggest that Dun1 kinase function is the last step required in the Mec1/Rad53 cascade to remove Sml1 during S phase and after DNA damage.
Ribonucleotide reductase (RNR) catalyzes the conversion of NDP to dNDP, the rate-limiting step of dNTP synthesis. RNR activity directly affects the levels and balances of the dNTP pools and subsequently both genetic fidelity and cell viability (1). As an important enzyme in the early stages of DNA synthesis, RNR activity is tightly regulated during DNA replication and repair. Besides its intrinsic allosteric regulation, RNR activity is regulated at the transcriptional level (2) and also by its inhibitor, Sml1 (3, 4). In yeast, these last two types of control both require the evolutionarily conserved checkpoint kinases, Mec1 and Rad53 (5).
In response to DNA damage, Mec1 and Rad53 activate the downstream checkpoint kinase Dun1, which leads to the transcriptional induction of the RNR genes (6–8). Such induction is achieved by the relief of transcriptional repression by the Crt1 protein (9). However, the direct downstream substrate(s) of the Dun1 kinase remain unknown. Similar transcriptional induction of the RNR genes also exists in mammals. For example, a human RNR small subunit (p53R2) is induced by p53 after DNA damage, and this induction is crucial for DNA repair (10). Given that the p53 is activated after DNA damage by the Mec1 and Rad53 homologs, ATM/ATR and CHK2 (11), it is likely that the RNR transcriptional induction circuitry is largely conserved between yeast and mammals.
In addition to transcriptional induction of the RNR genes, the Mec1/Rad53 pathway also increases RNR activity by regulating the removal of the RNR inhibitor, Sml1 (3, 4). Sml1 is a 104-residue peptide that binds the large subunit of RNR through its C-terminal half and inhibits RNR enzymatic activity efficiently. After DNA damage, the activation of Mec1 and Rad53 via the bifurcated checkpoint pathways, Rad9 and Rad17/Rad24/Mec3, leads to rapid phosphorylation and concomitant degradation of Sml1 protein (5). Such precipitous removal of this RNR inhibitor likely complements the slower mode of transcriptional induction, providing increased dNTP pools necessary for DNA repair.
However, unlike the transcriptional induction of RNR genes, the Mec1/Rad53-dependent phosphorylation and removal of Sml1 also occurs at S phase (5). In fact, the S phase-specific decrease of Sml1 levels is essential for cell survival as the failure to reduce Sml1 protein levels at S phase in mec1Δ strains leads to incomplete DNA replication and cell death (5). Moreover, hypomorphic mec1 and rad53 cells contain lower dNTP levels and subsequently exhibit increased levels of petite (mitochondrial DNA-deficient) cells (5). Deletion of Sml1 suppresses all these defects, suggesting that removal of the RNR inhibitor is an important regulatory step during normal cell growth.
Although this regulation requires the Mec1/Rad53 checkpoint proteins, it is unclear whether another kinase(s) actually phosphorylates Sml1. Three lines of genetic evidence hint that Dun1 might be the kinase involved in regulating Sml1. First, sml1Δ suppresses the DNA damage sensitivity of dun1Δ strains without affecting RNR transcription (3). Second, sml1Δ suppresses the loss of telomere position effect in dun1Δ strains (12). Third, overexpression of Dun1, like a deletion of SML1, suppresses mec1Δ and rad53Δ lethality (13). These results support the idea that Dun1 functions after Mec1/Rad53 in Sml1 regulation during normal growth and after DNA damage. In this work, we further explore the relationship between SML1 and DUN1. We find that dun1 in combination with hypomorphic alleles of mec1 and rad53 exhibits synthetic growth defects or lethality. In addition, dun1Δ mec1 strains exhibit increased levels of spontaneous petite formation. All of the growth defects as well as the increased petite formation are suppressed by sml1Δ, suggesting that Dun1 functions in the same pathway as Mec1/Rad53 to control dNTP levels. We next demonstrate that the phosphorylation and removal of Sml1 at S phase is partially defective in dun1Δ strains leading to a longer S phase. This regulation of Sml1 depends even more on Dun1 after DNA damage, because it is completely absent in dun1Δ strains. Finally, we show that Dun1 interacts with Sml1 by two-hybrid analysis and that it phosphorylates Sml1 in vitro, strongly supporting the notion that Dun1 is the last step in the Mec1/Rad53 cascade that regulates Sml1 stability.
Materials and Methods
Yeast Strains.
Strain PJ69-4A was used as a two-hybrid host (14). Other yeast strains used in this study are isogenic or congenic (more than 6 backcrosses) to W303 (15) and are RAD5. Therefore, only the genotype of a wild-type strain (W1588-4C) is described below. For the rest, only alleles that differ from W303 are given. We used the strains W1588-4C (MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1), W1763-8B (dun1Δ∷URA3), U1195 [(mec1Δ∷TRP1 (pWJ841: 2 μm-RNR1)], W1830-3A (dun1Δ∷URA3 sml1Δ∷HIS3), W1967 (MATa/α mec1-3/+ dun1Δ∷URA3/+ sml1Δ∷HIS3/+), W1968 (MATa/α mec1-8/+ dun1Δ∷URA3/+ sml1Δ∷HIS3/+), W1973 (MATa/α rad53-1/+ dun1Δ∷URA3/+ sml1Δ∷HIS3/+), and W1974 (MATa/α rad53-17/+ dun1Δ∷URA3/+ sml1Δ∷HIS3/+).
Plasmids.
To construct pWJ736 (GBD-RAD53), the RAD53 gene was PCR-amplified by using primer pGBD-C1MEC2 5′ (5′- atccatcgatatggaaaatattacacaacccac-3′) and primer MEC2 3′ (5′-aactgcaggttgtaccacatcaagcagg-3′). The PCR product was cloned into pGBD-C1 (14) between ClaI and PstI sites. To construct pWJ772 [glutathione S-transferase (GST)-Dun1] and pWJ996 (GST-Dun1-D328A), DUN1 and dun1-D328A were PCR-amplified by using primer pair DUN1 5′ + 0 BI (5′-ggggatccatgagtttgtccacgaaaag-3′) and DUN13′SalI (5′-acgcgtcgacagagctgcaggagaaagtag-3′). The PCR products were cloned into a 2 μm plasmid, pEG(KT) (16), between BamHI and SalI sites. All of the constructs were sequenced and confirmed for the correct insertions. GBD-Rad53 and GBD-Dun1 fusion proteins are functional as they fully rescue the synthetic lethality of rad53-1 dun1Δ strains. The construction and complementation tests of other plasmids have been described (3).
Dun1 Kinase Assay.
The expression of GST-Dun1 and GST-Dun1-D328A fusion proteins was induced by 2% galactose. The preparation of these fusion proteins was carried out as described (3), except during extraction, the Nonidet P-40 buffer was complemented with 5 mM MgCl2, 50 mM KCl, 10% glycerol, 0.1 M DTT, 0.1 M Na3VO4, and 30 mM NaF. In vitro kinase assays were performed under conditions that have been used for screening peptide substrates for Dun1 kinase (17). In brief, GST-Dun1 and GST-Dun1-D328A fusion proteins were incubated in a 30-μl kinase reaction buffer containing 50 mM Tris (pH 7.5), 10 mM MgCl2, 1 mM DTT, 60 μM ATP, 3 μCi (1 Ci = 37 GBq) of [γ-32P]ATP, and 0.1 μg/μl of Sml1 protein purified from Escherichia coli extracts (4). The mixtures were incubated at 30°C for 30 min and separated on denaturing polyacrylamide gels.
Other Techniques.
Standard yeast manipulations and media preparation were carried out as described (18). To analyze sml1Δ suppression of the synthetic growth defect of rad53-1 dun1Δ and mec1-3 dun1Δ strains, we dissected 16 tetrads for each of the two crosses shown in Fig. 1 a and b. Six rad53-1 dun1Δ sml1Δ spores and eight mec1-3 dun1Δ sml1Δ were recovered, respectively, but no rad53 dun1Δ or mec1-3 dun1Δ spores were recovered. The synthetic lethality of dun1Δ rad53-1 or dun1Δ rad53-17 could be rescued by either DUN1 or RAD53 plasmids (data not shown). Measurement of the frequency of petite formation and the two-hybrid assay method has been described (3). Cell cycle arrest, fluorescence-activated cell sorting, genotoxin treatment, extraction of yeast proteins, and protein blot analysis have been described (5, 19). The imagequant program (Molecular Dynamics) was applied to measure the density of bands on protein blots.
Figure 1.

sml1Δ suppresses synthetic growth defects of dun1Δ mec1 and dun1Δ rad53 strains and the increased petite formation in dun1Δ mec1 and dun1Δ strains. (a) Three tetrads (displayed horizontally) are shown for each diploid. The genotype of inviable spores is deduced from those of the sister spore clones and are shown dun1Δ rad53-1 Left and dun1Δ rad53-17 Right. The arrows indicate dun1Δ rad53-1 sml1Δ and dun1Δ rad53-17 sml1Δ triple mutants. Spore clones containing other genotypes grow equally well. (b) The slow-growing spore clones indicated by the arrows are dun1Δ mec1-3 and dun1Δ mec1-8. The faster growing triple mutants (dun1Δ mec1-3 or -8 sml1Δ) are indistinguishable from wild-type or the single mutants. (c) The averages and SDs of the frequency of petite formation are plotted for the four strains indicated. (d) Spore clones from b were streaked on yeast extract/peptone/dextrose (YPD) medium and then replica-plated onto YPGlycerol medium. Petite cells grow only on YPD but not on YPGlycerol medium. The strains depicted are (1) sml1Δ, (2) dun1Δ mec1-8 sml1Δ, (3) dun1Δ mec1-8, (4) dun1Δ mec1-3, and (5) dun1Δ mec1-3 sml1Δ. Cells in sectors 3 and 4 are mostly petite.
Results
Deletion of SML1 Rescues the Synthetic Growth Defect and Synthetic Lethality Between dun1Δ and Hypomorphic mec1 and rad53 Alleles.
Unlike MEC1 and RAD53, deletion of DUN1 does not cause any obvious growth defect (6). However, when DUN1 is deleted in strains containing hypomorphic alleles of mec1 and rad53, cells exhibit growth defects or lethality. As shown in Fig. 1, single mutants of dun1Δ, hypomorphic mec1 (mec1-3 and mec1-8), and rad53 (rad53-1 and rad53-17), grow normally on their own. However, dun1Δ rad53-1 and dun1Δ rad53-17 double mutants are inviable, and dun1Δ mec1-3 and dun1Δ mec1-8 cells grow very slowly (Fig. 1 a and b and Materials and Methods). These growth defects are completely suppressed by removal of Sml1 (Fig. 1 a and b).
sml1Δ Suppresses Increased Petite Formation in dun1Δ and dun1Δ mec1 Strains.
We next examined the formation of mitochondrial DNA-deficient cells (petite cells) in dun1Δ and dun1Δ mec1 strains. Spontaneous petite cells, deficient in mitochondrial DNA, arise more frequently when dNTP levels are low (such as in mec1, rad53, or rnr strains) and less frequently when dNTPs are abundant (such as in sml1Δ strains) (3, 5, 8, 20). Here, we find that a dun1 deletion alone leads to a 4-fold increase in the frequency of petite formation (Fig. 1c). Deletion of SML1 in dun1Δ strains reduces the frequency of petite formation to that of sml1Δ strains (Fig. 1c), suggesting that SML1 acts downstream of DUN1. In addition, dun1Δ mec1-3 and dun1Δ mec1-8 mutant strains exhibit extremely high levels of petites, even higher than either of the single mutants. Again, sml1Δ suppresses this defect (Fig. 1d). The suppression of defects in growth and in mitochondrial DNA propagation by sml1Δ strongly supports the notion that Dun1 shares functions with Mec1/Rad53 in the removal of Sml1 during normal growth.
Dun1 Strains Are Defective in Phosphorylation and Removal of Sml1 at S Phase.
We have shown that the S phase-specific phosphorylation of Sml1 and the concomitant decrease of its protein levels depend on Mec1 and Rad53 (5). We next examined whether Dun1 was also required for this regulation. In wild-type strains, Sml1 levels decrease 6-fold in S phase compared with G1 (Fig. 2 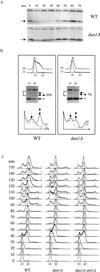 a). In dun1Δ strains, Sml1 levels in S phase fall only 2-fold (Fig. 2a). Moreover, the fraction of phosphorylated Sml1 found during S phase is reduced 5-fold in dun1Δ strains (7% vs. 35% in wild-type strains; Fig. 2b Middle). Thus, phosphorylation of Sml1 and the decreased levels at S phase largely depend on Dun1.
a). In dun1Δ strains, Sml1 levels in S phase fall only 2-fold (Fig. 2a). Moreover, the fraction of phosphorylated Sml1 found during S phase is reduced 5-fold in dun1Δ strains (7% vs. 35% in wild-type strains; Fig. 2b Middle). Thus, phosphorylation of Sml1 and the decreased levels at S phase largely depend on Dun1.
Figure 2.
dun1Δ strains are defective in Sml1 regulation and exhibit a longer S phase. (a) Wild-type and dun1Δ strains were arrested in G1 phase with α-mating factor and then were released into the cell cycle. Samples were taken every 10 min, and proteins from the 0–70-min samples were examined by protein blot with anti-Sml1 Ab. Samples from these time points were examined by fluorescence-activated cell sorting (FACS) analysis and are shown in c. The arrowhead indicates Sml1 protein. The band above Sml1 crossreacts to anti-Sml1 serum and is used as a loading control. (b) In wild-type and dun1Δ strains, the phosphorylated form of Sml1 appears only briefly during S phase and is not abundant (5). We therefore optimized the sampling time after release from cell cycle arrest (12 and 24 min). DNA content (Top) and Sml1 protein levels (Middle) were examined after release from a G1 block. The protein blots were overexposed to visualize the phosphorylated Sml1 band (marked by ←•). Unphosphorylated Sml1 bands (marked by ←○) and phospho-Sml1 bands were quantitated. The percentage of phospho-Sml1 protein is shown and was calculated by dividing the amount of phospho-Sml1 by the sum of both forms of Sml1. The quantitation of the bracketed region on each blot is depicted (Bottom). This region includes the phospho-Sml1 band and an adjacent weak crossreacting band, which serves as a loading control (marked with an arrow). Dotted and solid lines depict the profiles of 12- and 24-min samples, respectively. (c) As in a, wild-type, dun1Δ, and dun1Δ sml1Δ strains were arrested in G1 phase and then released into the cell cycle. Samples were taken every 10 min and their DNA content was examined by FACS analysis. Note that both wild-type and dun1Δ strains start the second cell cycle at 90 min.
dun1Δ Strains Exhibit a Prolonged S Phase That Can Be Suppressed by sml1Δ.
To determine whether the partially defective regulation of Sml1 in dun1Δ strains leads to any cell cycle abnormality, we carefully examined cell cycle progression. We found that dun1Δ cells exhibit a longer S phase and a shorter G2/M phase. As shown in Fig. 2c, both wild-type and dun1Δ cells enter S phase at the same time (20 min); however, dun1Δ cells finish S phase 10 min later than wild-type cells (50 vs. 40 min). The longer S phase in dun1Δ cells is also apparent in the following cell cycle (Fig. 2c). In contrast to S phase, the G2/M phase in dun1Δ cells is 10 min shorter than in wild-type (Fig. 2c). Thus, the overall length of the cell cycle in dun1Δ cells is normal, consistent with its normal growth rate (6). As is the case for each aspect of the dun1Δ phenotype examined thus far, the longer S phase in dun1Δ cells is completely suppressed by removal of Sml1 (Fig. 2c). These observations suggest that the reduction of Sml1 levels at S phase is important for successful DNA replication.
Sml1 Protein Is Not Phosphorylated or Degraded After DNA Damage in dun1Δ Strains.
Another cellular process that is sensitive to dNTP levels is DNA repair. Accordingly, Sml1 protein is phosphorylated and disappears after DNA damage in a Mec1/Rad53-dependent fashion to provide sufficient dNTPs for repair (5). To determine whether Dun1 plays a role in this process, we examined the levels and phosphorylation state of Sml1 protein in dun1Δ strains after DNA damage. In wild-type strains, Sml1 protein rapidly diminishes to undetectable levels after treatment with several DNA-damaging agents, including methyl methanesulfonate, UV, and γ-ray (Fig. 3a). In contrast, Sml1 protein levels in dun1Δ strains remain unaltered after these treatments (Fig. 3a).
Figure 3.

After DNA damage, Sml1 protein accumulates in dun1Δ strains whereas the phosphorylated forms of Sml1 are absent. (a) Wild-type, dun1Δ, and mec1Δ strains were treated with 0.05% methyl methanesulfonate for 1 h or were irradiated by UV light (120 J/m2) or γ-rays (30 krads). Sml1 protein levels were examined by protein blot with anti-Sml1 Ab. Arrowheads indicate the position of Sml1 protein. The band above Sml1 crossreacts with anti-Sml1 serum and serves as a loading control. The mec1Δ strain, which is also defective in Sml1 regulation, is shown for comparison. Because the mec1Δ strain is unable to grow in the presence of Sml1, its viability was maintained by a 2 μm-RNR1 plasmid (24). (b) dun1Δ strains containing either GST-Dun1 or GST-Dun1-D328A were irradiated by γ-rays (30 krads). The levels of the GST-fusion proteins as well as Sml1 protein were examined by protein blot with anti-GST Ab and anti-Sml1 Ab, respectively. (c) Wild-type, dun1Δ, and mec1Δ strains, all containing a RNR1 plasmid, were treated as in a. Proteins were extracted by trichloroacetic acid methods (5), and phosphorylated Sml1 bands (bracket) were visualized by immunoblot with anti-Sml1 Ab. Note that the crossreacting band above Sml1 in a does not appear under these conditions.
Next, we examined a kinase-deficient dun1-D328A strain (6) and found that, similar to a dun1Δ strain, there is no reduction of Sml1 protein levels after the cells were exposed to DNA-damaging agents (Fig. 3b and data not shown). Thus, the kinase activity of Dun1 is necessary for regulating Sml1.
Finally, no phosphorylated Sml1 can be detected in dun1Δ strains after DNA damage even when Sml1 levels are artificially elevated because of the presence of an RNR1 plasmid or a GAL-SML1 construct (5) (Fig. 3c). Thus, like Mec1 and Rad53, Dun1 also controls Sml1 phosphorylation and protein levels during the DNA damage response. It is noteworthy that, unlike the situation in S phase, this regulation completely depends on Dun1. The fact that deletion of SML1 suppresses the DNA damage sensitivity of dun1Δ strains without inducing RNR transcription (3) indicates that the regulation of Sml1 by Dun1 is an important component of the DNA damage response.
Dun1 Interacts with Sml1 in Two-Hybrid and Phosphorylates Sml1 in Vitro.
The observations described above have led us to test the possibility that Sml1 is a substrate of the Mec1/Rad53/Dun1 kinase cascade. Because Dun1 and Rad53 function downstream of Mec1 (13, 21), we first examined whether either kinase interacts with Sml1 by using a two-hybrid assay. All fusion proteins used are fully functional as determined by complementation analysis (see Materials and Methods). As shown in Fig. 4a, the coexpression of GBD-Dun1 and GAD-Sml1 activates both two-hybrid reporters (GAL1-HIS3 and GAL2-ADE2), indicating that Dun1 physically associates with Sml1. No interaction between Rad53 and Sml1 was detected (Fig. 4a).
Figure 4.

Dun1 interacts with and phosphorylates Sml1. (a) Plasmids containing different GBD- and GAD-fusion proteins were cotransformed into a two-hybrid strain (PJ69-4A, 14), and the transformants were selected on medium lacking tryptophan and leucine (-T-L). Interactions between fusion proteins were indicated by growth on histidine-less (-T-L-H) and adenine-less media (-T-L-A) to examine the activation of the two-hybrid reporters. The interaction between GBD-Rnr1 and GAD-Sml1 (3) was used as a positive control. (b) dun1Δ sml1Δ strains that overexpress GST-Dun1 and GST-Dun1-D328A fusion proteins were irradiated with γ-rays (30 krads). Before and after DNA damage, the fusion proteins were purified by using glutathione beads and then incubated with Sml1 protein in kinase reactions. The levels of the fusion proteins and Sml1 were visualized by immunoblot (Left). The brackets indicate the phosphorylated forms of Sml1, and the arrows below show the position of unphosphorylated Sml1 protein. The phosphorylation levels (32P) were visualized after autoradiography (Right).
Next, we tested whether Dun1 can phosphorylate Sml1 in vitro. To this end, Dun1 and the kinase-inactive form, Dun1-D328A, were fused to GST, and both fusion proteins were overexpressed and purified from yeast cells before and after DNA damage. These fusion proteins were next incubated in kinase reactions with purified Sml1 protein. Consistent with previous reports (6), autophosphorylation was observed for the wild-type Dun1 kinase but was barely seen with the kinase-deficient mutant GST-Dun1-D328A, irrespective of the DNA damage state of the cellular extracts (Fig. 4b). Similarly, only GST-Dun1 but not GST-Dun1-D328A phosphorylates Sml1 (Fig. 4b). Phosphorylated Sml1 proteins can be detected on a protein blot as slower migrating bands both visually and after autoradiography (Fig. 4b). Several unknown E. coli proteins that copurify with Sml1 are not phosphorylated by GST-Dun1 (data not shown). In addition, the phosphorylation of Sml1, as well as Dun1 itself, is more prominent by using cell extracts made after DNA damage. These results show that Sml1 is an excellent substrate of the Dun1 protein kinase in vitro.
Discussion
Sufficient and balanced dNTP pools are critical for successful DNA replication and DNA repair, and subsequently contribute greatly to genomic integrity. As the critical enzyme in dNTP synthesis, RNR is regulated by the evolutionarily conserved checkpoint proteins, Mec1 and Rad53, at two levels: transcriptional induction as well as phosphorylation and removal of its inhibitor Sml1. Here, we present evidence that strongly supports the notion that the Dun1 kinase functions after Mec1 and Rad53 to phosphorylate and remove the RNR inhibitor Sml1 both at S phase and after DNA damage.
We show that although dun1Δ itself does not cause any obvious growth defect, it leads to synthetic growth defects and synthetic lethality in combination with hypomorphic mec1 and rad53 alleles, respectively. This result extends the previous observation that dun1Δ is synthetically lethal with rad53-11 (22). In addition, dun1Δ strains exhibit an increased rate of petite formation, and dun1Δ mec1-3 or -8 strains exhibit even greater levels of petite formation. These defects in growth and petite formation are completely suppressed by deletion of SML1, suggesting that Dun1 is involved in dNTP regulation during normal cell growth. This idea is further supported by the fact that Dun1 is required to efficiently phosphorylate and remove Sml1 at S phase. However, the residual Sml1 phosphorylation and the slight decrease of Sml1 levels at S phase in dun1Δ strains suggest that a redundant pathway exists during DNA replication. This functional redundancy may account for the fact that, unlike mec1Δ and rad53Δ, dun1Δ strains are viable. Nevertheless, the function of Dun1 in Sml1 regulation at S phase is important, as a dun1Δ strain has a longer S phase, which is completely suppressed by sml1Δ (Fig. 2).
After DNA damage, the regulation of Sml1 depends completely on the Dun1 kinase. In addition, phosphorylation and the concomitant decrease of Sml1 protein require the kinase activity of Dun1. Finally, Dun1 associates with Sml1 in vivo and directly phosphorylates Sml1 in vitro. These results are consistent with a recent observation that FLAG-tagged Sml1 protein can “pull down” Dun1 protein (23). These genetic and biochemical interactions provide a molecular basis for the Sml1 regulation of RNR activity and reveal a new function for the Dun1 kinase. Based on these results, we refine the model that we proposed (5), focusing only on the regulation of RNR after DNA damage as shown in Fig. 5. DNA lesions activate the Mec1/Rad53/Dun1 kinase cascade via the bifurcated Rad9 and Rad17/Rad24/Mec3 pathways. The most downstream kinase, Dun1, can then phosphorylate Sml1, leading to its disappearance. We do not exclude the possibility, however, that other kinases can also phosphorylate Sml1. In addition, this kinase cascade leads to the transcriptional induction of the RNR genes. The regulation of Sml1 provides a means to rapidly increase RNR activity, which acts to complement the inherently slower transcriptional induction. Such dual control of RNR activity by an evolutionarily conserved checkpoint pathway underscores the importance of RNR in cell survival. It will be of interest to see how conserved this regulatory circuitry is in other organisms.
Figure 5.

A model for the role of Dun1 in regulation of RNR activity after DNA damage. DNA lesions provoke the activation of Mec1/Rad53/Dun1 via the bifurcated Rad9 and Rad17/Rad24/Mec3 pathways (5). Dun1 kinase phosphorylates Sml1 directly leading to its degradation. We suspect that the unbound form of Sml1 is targeted for destruction (6). At the same time, Dun1 activates the transcriptional induction of RNR genes although its direct substrate remains unknown. The transcriptional induction likely causes increased production of RNR proteins (dashed arrow). The dual mode of Dun1 function outlined here ensures a rapid yet lasting increase in RNR activity likely necessary for efficient DNA repair.
Acknowledgments
We thank A. Chabes and L. Thelander (Umeå University) for providing Sml1 protein; S. Elledge (Baylor University) for plasmid pZZ99; H. Zou (Univ. of Texas, Southwestern) for the rad53-17 allele; T. Weinert (Univ. of Arizona) for the mec1-3, mec1-8, and rad53-1 alleles; and S. Chaves, E. Shor, M. Lisby, M. Wagner, B. Georgieva, and T. Lamb for comments on the manuscript. This work was supported by National Institutes of Health Grant GM50237 (to R.R.).
Abbreviations
- RNR
ribonucleotide reductase
- GST
glutathione S-transferase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Reichard P. Annu Rev Biochem. 1988;57:349–374. doi: 10.1146/annurev.bi.57.070188.002025. [DOI] [PubMed] [Google Scholar]
- 2.Elledge S J, Zhou Z, Allen J B. Trends Biochem Sci. 1992;17:119–123. doi: 10.1016/0968-0004(92)90249-9. [DOI] [PubMed] [Google Scholar]
- 3.Zhao X, Muller E G, Rothstein R. Mol Cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- 4.Chabes A, Domkin V, Thelander L. J Biol Chem. 1999;274:36679–36683. doi: 10.1074/jbc.274.51.36679. [DOI] [PubMed] [Google Scholar]
- 5.Zhao X, Chabes A, Domkin V, Thelander L, Rothstein R. EMBO J. 2001;20:3544–3553. doi: 10.1093/emboj/20.13.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Z, Elledge S J. Cell. 1993;75:1119–1127. doi: 10.1016/0092-8674(93)90321-g. [DOI] [PubMed] [Google Scholar]
- 7.Elledge S J. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- 8.Huang M, Elledge S J. Mol Cell Biol. 1997;17:6105–6113. doi: 10.1128/mcb.17.10.6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang M, Zhou Z, Elledge S J. Cell. 1998;94:595–605. doi: 10.1016/s0092-8674(00)81601-3. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, Takei Y, Nakamura Y. Nature (London) 2000;404:42–49. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 11.Caspari T. Curr Biol. 2000;10:R315–R317. doi: 10.1016/s0960-9822(00)00439-5. [DOI] [PubMed] [Google Scholar]
- 12.Craven R J, Petes T D. Mol Cell Biol. 2000;20:2378–2384. doi: 10.1128/mcb.20.7.2378-2384.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanchez Y, Desany B A, Jones W J, Liu Q, Wang B, Elledge S J. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- 14.James P, Halladay J, Craig E A. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas B J, Rothstein R. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- 16.Mitchell D A, Marshall T K, Deschenes R J. Yeast. 1993;9:715–722. doi: 10.1002/yea.320090705. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez Y, Zhou Z, Huang M, Kemp B E, Elledge S J. Methods Enzymol. 1997;283:398–410. doi: 10.1016/s0076-6879(97)83033-9. [DOI] [PubMed] [Google Scholar]
- 18.Adams A, Gottschling D E, Kaiser C A, Stearns T. The Methods in Yeast Genetics, A Cold Spring Harbor Laboratory Course Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1997. [Google Scholar]
- 19.Zhao X, Georgieva B, Chabes A, Domkin V, Ippel J H, Schleucher J, Wijmenga S, Thelander L, Rothstein R. Mol Cell Biol. 2000;20:9076–9083. doi: 10.1128/mcb.20.23.9076-9083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elledge S J, Davis R W. Mol Cell Biol. 1987;7:2783–2793. doi: 10.1128/mcb.7.8.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen J B, Zhou Z, Siede W, Friedberg E C, Elledge S J. Genes Dev. 1994;8:2401–2415. doi: 10.1101/gad.8.20.2401. [DOI] [PubMed] [Google Scholar]
- 22.Gardner R, Putnam C W, Weinert T. EMBO J. 1999;18:3173–3185. doi: 10.1093/emboj/18.11.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho Y, Gruhler A, Heilbut A, Bader G D, Moore L, Adams S-L, Millar A, Taylor P, Bennett K, Boutilier K, et al. Nature (London) 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 24.Desany B A, Alcasabas A A, Bachant J B, Elledge S J. Genes Dev. 1998;12:2956–2970. doi: 10.1101/gad.12.18.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]