

Abstract
In this review, we describe recent empirical and theoretical work on the extent of linkage disequilibrium (LD) in the human genome, comparing the predictions of simple population-genetic models to available data. Several studies report significant LD over distances longer than those predicted by standard models, whereas some data from short, intergenic regions show less LD than would be expected. The apparent discrepancies between theory and data present a challenge—both to modelers and to human geneticists—to identify which important features are missing from our understanding of the biological processes that give rise to LD. Salient features may include demographic complications such as recent admixture, as well as genetic factors such as local variation in recombination rates, gene conversion, and the potential segregation of inversions. We also outline some implications that the emerging patterns of LD have for association-mapping strategies. In particular, we discuss what marker densities might be necessary for genomewide association scans.
Introduction
The extent and distribution of linkage disequilibrium (LD) in humans is a topic of great current interest. LD plays a fundamental role in gene mapping, both as a tool for fine mapping of complex disease genes (e.g., see Horikawa et al. 2000) and in proposed genomewide association studies (Risch and Merikangas 1996). LD is also of interest for what it can reveal about human history and human origins (e.g., see Tishkoff et al. 1996), because the distribution of LD is determined, in part, by population history. Finally, studies of LD may enable us to learn more about the biology of recombination in humans. It is difficult to use pedigrees to estimate rates of homologous gene conversion, or variation in recombination rates over very short distances, because the events of interest occur at very low rates. However, studies of LD may offer insight (e.g., see Chakravarti et al. 1984; Awadalla et al. 1999; Przeworski and Wall 2001).
In the present review article, we describe some predictions about the extent of LD, using simple models of population genetics. After summarizing the main empirical findings in humans, we turn to applications in association mapping and discuss the implications of the data collected thus far. We have not aimed to cover the literature exhaustively but, instead, have highlighted some issues that we think are of particular interest.
Models and Measures of LD
LD refers to the nonindependence of alleles at different sites. For example, suppose that allele A at locus 1 and allele B at locus 2 are at frequencies πA and πB, respectively, in the population. If the two loci are independent, then would we expect to see the AB haplotype at frequency πAπB. If the population frequency of the AB haplotype is either higher or lower than this—implying that particular alleles tend to be observed together—then the two loci are said to be in LD.
A wide variety of statistics have been proposed to measure the amount of LD, and these have different strengths, depending on the context. The measurement of LD is a large and complex topic and will not be reviewed in detail here; but see the work of Devlin and Risch (1995); Jorde (2000) and Hudson (2001). Most of the measures of LD that are in wide use quantify the degree of association between pairs of markers. In part, they differ according to the way in which they depend on the marginal allele frequencies. In the present article, we use one popular measure of LD between pairs of biallelic markers, commonly denoted by r2 (elsewhere, r2 is also denoted by Δ2). We also discuss a multilocus approach, based on an underlying population genetic model, that we feel has some advantages as a summary of the overall amount of LD in a region.
Consider two biallelic loci on the same chromosome, with alleles A and a at the first locus and with alleles B and b at the second locus, where the labeling is arbitrary. The allele frequencies will be written as πA, πa, πB, and πb, and the four haplotype frequencies will be written as πAB, πAb, πaB, and πab. Then,
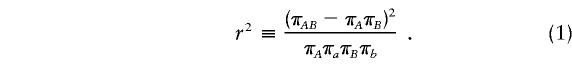 |
In some of the figures, we plot 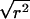 , because this can make it easier to see the data points. For brevity, we will refer to
, because this can make it easier to see the data points. For brevity, we will refer to 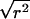 as “r.”
as “r.”
In practice, one typically has a sample of m chromosomes from the population. Then, estimates of r2 (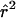 ) are usually obtained by plugging the sample frequencies
) are usually obtained by plugging the sample frequencies 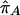 ,
, 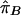 ,
, 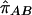 , etc., into equation (1).
, etc., into equation (1).
Besides estimating the amount of disequilibrium between pairs of markers, it is also natural to test the null hypothesis of independence between marker pairs (i.e., linkage equilibrium). This can be done by a χ2 test, and it turns out that, for biallelic markers, 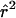 is the standard χ2-test statistic divided by the number of chromosomes in the sample (Weir 1996, p. 113). As shown later in the present article, r2 also arises naturally in the context of association mapping.
is the standard χ2-test statistic divided by the number of chromosomes in the sample (Weir 1996, p. 113). As shown later in the present article, r2 also arises naturally in the context of association mapping.
The actual value of the disequilibrium coefficient r2 (or any other measure of LD) between two loci is drawn from a probability distribution that results from the evolutionary process. This process can be described in terms of a population genetics tool called the “coalescent” (for reviews, see, e.g., Hudson 1993; Nordborg 2001). When we draw a sample of chromosomes from a population, all the chromosomes are related by some unknown ancestral genealogy, known as a “coalescent tree.” Genetic markers that are very close together on a chromosome have either the same or similar genealogies, and this induces dependence between the alleles at different markers. Markers that are farther apart may have different ancestral genealogies, because of recombination. For this reason, the strength of LD between pairs of markers decreases as a function of the genetic distance between markers.
The expected value of r2 is a function of the parameter ρ≡4Nec, where c is the recombination rate between the two markers and where Ne is the effective population size. For large ρ, E(r2)≈1/ρ (reviewed by Hudson 2001). Below, we will show simulations of the distribution of r under various models.
Despite their convenience, the use of r2 and other standard summaries of LD has some shortcomings. Each pair of loci produces an estimate, and it is not clear how to combine these in a sensible way, in part because they are not independent. More seriously, it is not straightforward to compare different regions. For instance, we might want to know whether a difference in the values of r2 in two different regions is biologically meaningful. (There are two kinds of statistical significance in this context: first, we might want to know whether the amount of LD, appropriately defined, in one region is significantly more than that in another [e.g., for repeated sampling of loci from the same region]; second, we might want to know whether there is evidence that the underlying biological parameters—in particular, the recombination rate—differ either between regions or from predicted values. We use the latter sense in this article.) As we will argue below, for these and other reasons, it seems useful to supplement the standard pairwise summaries of LD by a model-based LD measure from population genetics.
As mentioned above, E(r2) is a function of ρ=4Nec. It turns out to be a general result that ρ (also called “C” in the literature) is a key determinant of the extent of LD (Long and Langley 1999), with the strength of LD decreasing as ρ increases. Essentially, ρ is a scaled recombination rate, where the scaling constant 4Ne arises naturally from consideration of the genealogical process (e.g., see Nordborg 2001; also see the Nordborg web site). For a given demographic model of population history, the extent and distribution of LD depends primarily on ρ, and we can readily simulate the distribution of any summary of LD (we also need to specify a mutation model). (Note a matter of notation: when we consider ρ=4Nec for a region containing a series of markers, c is normally taken to refer to the total recombination rate across the entire region.)
The central role of ρ as a determinant of LD suggests that it is of interest to estimate this parameter from data. To do this, we need to assume an explicit model describing the population history and the processes of mutation and recombination. Then, for a given set of haplotype data, we can (in principle) compute the likelihood of the data as a function of ρ, and the mutation rate (Stephens 2001). Unfortunately, computing this likelihood is both technically challenging and computationally intensive, even for relatively small data sets, and methods for doing so are in their infancy. Existing approaches estimate the full likelihood by using either importance sampling (Griffiths and Marjoram 1996; also see the Paul Fearnhead web site) or Markov chain Monte Carlo (Kuhner et al. 2000; Nielsen 2000). To lessen the computational load, other methods simplify the structure of the data to approximate the shape of the likelihood function (Wall 2000; also see the Hudson Lab Home Page); confidence intervals can be obtained by simulation.
Despite the computational difficulties, it is now becoming possible to estimate ρ in cases of interest, and we report some results here. There are several potential advantages to using the model-based estimate 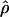 as a summary statistic: (1) We can obtain a single number that summarizes the amount of LD in a region. (2) We can compare the amount of LD observed in studies with different marker spacing or from microsatellite and single-nucleotide polymorphism (SNP) studies. (3) The quantity
as a summary statistic: (1) We can obtain a single number that summarizes the amount of LD in a region. (2) We can compare the amount of LD observed in studies with different marker spacing or from microsatellite and single-nucleotide polymorphism (SNP) studies. (3) The quantity 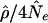 is an estimate of the recombination rate per generation, c. The latter is of particular interest for studying variation in recombination rate at small scales; it also allows us to test the models used to predict patterns of LD.
is an estimate of the recombination rate per generation, c. The latter is of particular interest for studying variation in recombination rate at small scales; it also allows us to test the models used to predict patterns of LD.
Model Predictions
Population history can have a large effect on the distribution of LD. To illustrate this, we plot the decay of 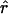 for a sample of 400 chromosomes, under four simple demographic models (see fig. 1). Each plot shows the results for one data set simulated from a neutral coalescent model with recombination (Hudson 1993). Points above the horizontal line are in significant LD at the 5% level. The first column in figure 1 shows realizations of the standard null model of a randomly mating population of constant size Ne=104. (The estimate of Ne is obtained from observed diversity levels, where the mutation rate is assumed to be ≈2×10-8/site/generation [Li and Sadler 1991; Przeworski et al. 2000].) As can be seen by comparison of the five replicates, considerable variability is expected across runs with the same underlying parameters, especially between tightly linked sites.
for a sample of 400 chromosomes, under four simple demographic models (see fig. 1). Each plot shows the results for one data set simulated from a neutral coalescent model with recombination (Hudson 1993). Points above the horizontal line are in significant LD at the 5% level. The first column in figure 1 shows realizations of the standard null model of a randomly mating population of constant size Ne=104. (The estimate of Ne is obtained from observed diversity levels, where the mutation rate is assumed to be ≈2×10-8/site/generation [Li and Sadler 1991; Przeworski et al. 2000].) As can be seen by comparison of the five replicates, considerable variability is expected across runs with the same underlying parameters, especially between tightly linked sites.
Figure 1.

Simulated decay of 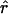 , as a function of genetic distance, for five independent replicates for each of four demographic models. Each plot shows the results for one sample of 400 chromosomes simulated under a particular demographic model of population history. On the X-axis is the genetic distance, in centimorgans, separating pairs of markers; at the genome average recombination rate of ≈1 cM/Mb, this is equivalent to distances in megabases. Each plot shows all pairwise comparisons for 40 biallelic markers, chosen randomly from the available markers whose minor-allele frequencies were ⩾.2. Points above the horizontal lines are in significant LD at the .05 level; for a sample size of 400, this corresponds to a value of r=.098 (see the “Models and Measures of LD” section). The first column shows results for a panmictic population of constant size Ne=104; the second column shows results for the model of population growth considered by Kruglyak (1999) and described in the “Model Predictions” section; and the third and fourth columns show results for a simple model of population structure. In the third column, all individuals are drawn from the same subpopulation; in the last column they are drawn equally from both subpopulations. We used the symmetric two-island migration model (Wright 1951), with Ne=5,000 for each deme, and migration rates of one individual per deme per generation.
, as a function of genetic distance, for five independent replicates for each of four demographic models. Each plot shows the results for one sample of 400 chromosomes simulated under a particular demographic model of population history. On the X-axis is the genetic distance, in centimorgans, separating pairs of markers; at the genome average recombination rate of ≈1 cM/Mb, this is equivalent to distances in megabases. Each plot shows all pairwise comparisons for 40 biallelic markers, chosen randomly from the available markers whose minor-allele frequencies were ⩾.2. Points above the horizontal lines are in significant LD at the .05 level; for a sample size of 400, this corresponds to a value of r=.098 (see the “Models and Measures of LD” section). The first column shows results for a panmictic population of constant size Ne=104; the second column shows results for the model of population growth considered by Kruglyak (1999) and described in the “Model Predictions” section; and the third and fourth columns show results for a simple model of population structure. In the third column, all individuals are drawn from the same subpopulation; in the last column they are drawn equally from both subpopulations. We used the symmetric two-island migration model (Wright 1951), with Ne=5,000 for each deme, and migration rates of one individual per deme per generation.
In the second column of figure 1, we plot the decay of 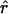 for the model used by Kruglyak (1999). In this scenario, the effective population size increases exponentially, from 104 to 5×109, starting 5,000 generations ago. As pointed out by Kruglyak (1999), very little LD is expected under this model, and, in fact, few significant values of
for the model used by Kruglyak (1999). In this scenario, the effective population size increases exponentially, from 104 to 5×109, starting 5,000 generations ago. As pointed out by Kruglyak (1999), very little LD is expected under this model, and, in fact, few significant values of 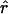 are observed beyond 10 kb. It should be noted, however, that, even in the absence of growth, LD is expected to be smaller in larger populations. In the Kruglyak model, it is assumed that Ne has always been ⩾104, the value for the model of constant population size. If we want to know the effect of population growth per se, rather than that of a large population size, then we need to match the effective population sizes.
are observed beyond 10 kb. It should be noted, however, that, even in the absence of growth, LD is expected to be smaller in larger populations. In the Kruglyak model, it is assumed that Ne has always been ⩾104, the value for the model of constant population size. If we want to know the effect of population growth per se, rather than that of a large population size, then we need to match the effective population sizes.
In figure 2, we plot the expected levels of 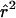 for different growth models with comparable levels of diversity. Under a neutral model, diversity levels are determined by the mutation rate and the effective population size. Thus, matching the diversity levels is one way to match effective population sizes. Here, we fix the current effective population size (taken to be 105) and the time at which population growth started; we then pick the growth rate that yields the same average number of segregating sites (for 100 chromosomes) as does the model of constant population size with Ne=104. Once population sizes are matched in this way, population growth still leads to a reduction in LD, but the effect is smaller than that in the Kruglyak model.
for different growth models with comparable levels of diversity. Under a neutral model, diversity levels are determined by the mutation rate and the effective population size. Thus, matching the diversity levels is one way to match effective population sizes. Here, we fix the current effective population size (taken to be 105) and the time at which population growth started; we then pick the growth rate that yields the same average number of segregating sites (for 100 chromosomes) as does the model of constant population size with Ne=104. Once population sizes are matched in this way, population growth still leads to a reduction in LD, but the effect is smaller than that in the Kruglyak model.
Figure 2.

Decay of expected value of 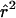 , as a function of genetic distance, for different growth models (sample size of 100 chromosomes). From top to bottom, the four models are as follows: constant population size of Ne=104 (unbroken line); population-growth onset 500 generations ago—that is, 104 years ago, under the assumption that there are 20 years per generation (dotted line); population-growth onset 5,000 generations ago (short-dashed line); and the model used by Kruglyak (1999) (see the “Model Predictions” section) (long-dashed line). In two growth models (dotted line and short-dashed line), the current population was fixed at 105. Except in the Kruglyak model, the parameters were chosen to match the amount of genetic diversity seen in humans (see text). In each of 104 simulations, 10 SNPs with a minor-allele frequency ⩾.2 were chosen.
, as a function of genetic distance, for different growth models (sample size of 100 chromosomes). From top to bottom, the four models are as follows: constant population size of Ne=104 (unbroken line); population-growth onset 500 generations ago—that is, 104 years ago, under the assumption that there are 20 years per generation (dotted line); population-growth onset 5,000 generations ago (short-dashed line); and the model used by Kruglyak (1999) (see the “Model Predictions” section) (long-dashed line). In two growth models (dotted line and short-dashed line), the current population was fixed at 105. Except in the Kruglyak model, the parameters were chosen to match the amount of genetic diversity seen in humans (see text). In each of 104 simulations, 10 SNPs with a minor-allele frequency ⩾.2 were chosen.
It should be noted that, in addition to diversity levels and levels of LD, other aspects of the data (e.g., the frequency spectrum of segregating sites) can be used to gauge the plausibility of particular demographic models. Population growth leads to an excess of low-frequency variants relative to a model of constant population size (Tajima 1989). Under the model used by Kruglyak, >80% of all minor alleles would be found only once in a sample of 400 chromosomes (results not shown). Such a pronounced skew in the frequency spectrum is not seen in actual data. Indeed, the frequency spectrum at synonymous sites (as detected by variant-detection arrays; Cargill et al. 1999) indicates little skew from the predictions of a model of constant population size. Although the size of the human population clearly has increased—at least over recent time—there are not yet enough data to allow us to know which growth model will be appropriate (Wall and Przeworski 2000).
In contrast to population growth, population structure tends to increase levels of LD. It can even lead to significant associations between unlinked markers; however, we still expect the strongest LD between tightly linked markers. An example of this can be seen in the third and fourth columns of figure 1, where we present the results for a model with two subpopulations, corresponding to a level of population differentiation of FST≈.2 (Wright 1951; Hartl and Clark 1997).
In the third column of figure 1, all individuals are sampled from one subpopulation, whereas, in the fourth column of figure 1, they are drawn equally from both. Both situations lead to increased LD, and, particularly in the latter case, there is strong LD across the entire distance plotted.
Empirical Patterns
Long-Distance LD
There are now more than a dozen studies that characterize the extent and range of background LD over large distances (e.g., see Peterson et al. 1995; Laan and Pääbo 1997; Huttley et al. 1999; Dunning et al. 2000; Eaves et al. 2000; Gordon et al. 2000; Taillon-Miller et al. 2000; Wilson and Goldstein 2000; Zavattari et al. 2000; Abecasis et al. 2001; Service et al. 2001). Although a few recent studies consider SNPs (Dunning et al. 2000; Taillon-Miller et al. 2000; Abecasis et al. 2001), most data sets consist of microsatellites. The populations sampled are usually either European or of European descent (e.g., the Amish or Afrikaners). Many are thought to be either relatively isolated or to have experienced rapid growth from a small number of founders (e.g., Finns and Sardinians). Sample sizes vary from 50 to several hundred haplotypes.
Several of these studies find that LD extends over very long distances. For example, Peterson et al. (1995), Laan and Pääbo (1997), Huttley et al. (1999), Gordon et al. (2000), Wilson and Goldstein (2000), and Service et al. (2001) all observe significant associations between pairs of microsatellites separated by ⩾1 cM. Significant LD extends out to 2 cM in Ashkenazim and Bantus, in an X-chromosome survey conducted by Wilson and Goldstein (2000). In a sample of Afrikaners, Gordon et al. (2000) find two pairs of markers in significant LD (after a Bonferroni correction for multiple tests), despite the markers being separated by >3 cM.
Huttley et al. (1999) describe results from a genomewide search for LD in the microsatellite data of the Centre d'Étude du Polymorphisme Humain (CEPH). Those authors used the LD level observed in the major histocompatibility (MHC) region as a benchmark for “high” levels of LD. (The MHC region shows extreme levels of LD, which are thought to reflect the action of natural selection.) By this criterion, eight regions, each spanning several centimorgans, show “excess LD” in a sample of CEPH individuals.
One of the SNP studies (Taillon-Miller et al. 2000) also finds long-range LD in regions Xq25 and Xq28, with highly significant associations between several pairs of markers separated by >500 kb. However, SNP data from four autosomal regions (from Dunning et al. [2000] and Abecasis et al. [2001]) seem to show a different pattern, with a more rapid decay of LD with distance (fig. 3).
Figure 3.

Plots of 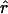 , as a function of physical distance (in kb), for SNP data from five regions (Dunning et al. 2000; Taillon-Miller et al. 2000; Abecasis et al. 2001). On each plot, points above the unbroken line are in significant LD at the .05 level, and points above the dotted line correspond to what Kruglyak (1999) has called “useful LD”; these lines are set at r=.316, the equivalent of r2=.1.
, as a function of physical distance (in kb), for SNP data from five regions (Dunning et al. 2000; Taillon-Miller et al. 2000; Abecasis et al. 2001). On each plot, points above the unbroken line are in significant LD at the .05 level, and points above the dotted line correspond to what Kruglyak (1999) has called “useful LD”; these lines are set at r=.316, the equivalent of r2=.1.
A visual inspection of figure 1 suggests that, under the standard models, the finding that LD extends over multiple centimorgans is surprising, at least in the absence of substantial population structure. Ideally, one would like to compare the patterns of LD found by different studies more formally and, also, to test the predictions of alternative models. However, this is not straightforward. First, the power to detect LD may be higher for markers with many alleles (e.g., microsatellites) than for biallelic markers (Slatkin 1994). Second, the results from different studies are reported in terms of a variety of measures of LD. Most common among these are D′ (Lewontin 1964), r2, and P values from pairwise significance tests of LD. As illustrated in figure 4, D′ and r2 behave very differently, and high values of D′ may not be inconsistent with low values of r2. In particular, there seems to be much more random variation in values of D′ at a given recombination distance.
Figure 4.

Simulated decay of 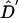 (+) and
(+) and 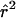 (♦), as a function of genetic distance. These data correspond to one realization from figure 1 (first column, second row), simulated under a model of constant population size (i.e., Ne=104) and random mating.
(♦), as a function of genetic distance. These data correspond to one realization from figure 1 (first column, second row), simulated under a model of constant population size (i.e., Ne=104) and random mating.
The studies also differ in the way in which haplotypes are obtained. In studies of X-linked regions, the haplotypes can be determined in males. For autosomal data, the haplotypes are sometimes reconstructed on the basis of pedigrees. Otherwise, they are usually estimated by statistical methods, with unknown effects on the accuracy of inferences about LD (especially if the populations show pronounced departures from Hardy-Weinberg equilibrium, as found by Dunning et al. [2000]). Significance tests of LD can also be performed on the genotype data directly, at the cost of some loss in power.
Another complication in comparing the data from different studies is that some report only physical distances (as plotted in fig. 4). These are not necessarily comparable, since it is known that recombination rates vary widely across the genome (Payseur and Nachman 2000; Yu et al. 2001). Moreover, in comparisons of LD on the X chromosome and the autosomes, it should be noted that higher levels of LD are expected on the X chromosome. In fact, we expect ρ to be halved, since (1) Ne is 3/4 of the value for autosomes (when a sex ratio of 1:1 and no sexual selection are assumed) and (2) recombination between X chromosomes occurs only in females.
As discussed above, one approach to quantifying the extent of LD in a region is to estimate ρ=4Nec; this estimate, 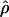 , can be thought of as providing a summary of the amount of LD. We can usually obtain external estimates of Ne (on the basis of diversity data) and of c (on the basis of genetic maps), and this allows us to predict what ρ “should” be. To conclude that there is more LD than what would be predicted under the model, we need to show that
, can be thought of as providing a summary of the amount of LD. We can usually obtain external estimates of Ne (on the basis of diversity data) and of c (on the basis of genetic maps), and this allows us to predict what ρ “should” be. To conclude that there is more LD than what would be predicted under the model, we need to show that 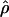 is less than the predicted value of ρ.
is less than the predicted value of ρ.
Estimates of ρ depend on the demographic model. The choice of model influences both the average rate of decay of LD (fig. 2) and the amount of chance variation in the distribution of LD across independent realizations of the evolutionary process (e.g., compare the second and fourth columns of fig. 1). As noted above, there is considerable uncertainty in the choice of an appropriate model for human populations. Here, we report estimates of ρ for a model of constant population size. Since we suspect that there may be an excess of LD, this choice is conservative: more LD is expected with constant population size than is expected in the presence of population growth (fig. 2; Slatkin 1994).
Estimates of ρ are available for three SNP regions (see the first, second, and fifth plots in fig. 3). For the data reported by Taillon-Miller et al. (2000), the maximum-likelihood estimate of ρ is five times lower than would be expected if c=1 cM/Mb and Ne=7,500 (for the X chromosome), lending support to the qualitative view that there is “excess” LD in this region (see the Hudson Lab Home Page). Alternative physical maps yield divergent estimates of the recombination rate for this region (see the data reported by Payseur and Nachman 2000; also see the Human Recombination Rates web site); the rate may be as high as 3 cM/Mb, which would make the discrepancy 15-fold.
In contrast, for the two regions with SNP data reported by Abecasis et al. (2001), the plot of decay of 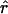 (see fig. 3) appears more like the plots for constant Ne (see the first column of fig. 1). Estimates of ρ were obtained by a pair of similar methods (see the Hudson Lab Home Page and the Gil McVean web site). Because of computational limitations, the analysis was performed on subsets of 50 of the available haplotypes. For Ne=104, the maximum likelihood estimate of c is 1.8 cM/Mb for the region on chromosome 2, and 2.5 cM/Mb for the region on chromosome 14; estimates of c, from comparison of a genetic map and a physical map, are ≈1 and ≈2.5 cM/Mb, respectively (as estimated by Payseur and Nachman [2000]). There are considerable uncertainties in all of these estimates; nonetheless, a model of constant population size roughly predicts the decay of pairwise LD found in the data of Abecasis et al. (2001). We do not have estimates of ρ for the data reported by Dunning et al. (2000), but the plots of decay of
(see fig. 3) appears more like the plots for constant Ne (see the first column of fig. 1). Estimates of ρ were obtained by a pair of similar methods (see the Hudson Lab Home Page and the Gil McVean web site). Because of computational limitations, the analysis was performed on subsets of 50 of the available haplotypes. For Ne=104, the maximum likelihood estimate of c is 1.8 cM/Mb for the region on chromosome 2, and 2.5 cM/Mb for the region on chromosome 14; estimates of c, from comparison of a genetic map and a physical map, are ≈1 and ≈2.5 cM/Mb, respectively (as estimated by Payseur and Nachman [2000]). There are considerable uncertainties in all of these estimates; nonetheless, a model of constant population size roughly predicts the decay of pairwise LD found in the data of Abecasis et al. (2001). We do not have estimates of ρ for the data reported by Dunning et al. (2000), but the plots of decay of 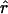 (see fig. 3) also appear to be consistent with either a model of constant population size or a model with some population growth.
(see fig. 3) also appear to be consistent with either a model of constant population size or a model with some population growth.
The parameter ρ has also been estimated for data from a microsatellite study (Laan and Pääbo 1997) that, in the Saami, found strong pairwise LD extending over several centimorgans. The maximum-likelihood estimate of ρ, obtained under a stepwise mutation model, is ∼100 times smaller than that expected on the basis of the genetic distance between markers if Ne=104 (see the Paul Fearnhead web site); use of a geometric mutation model yields similar results (see Pritchard et al. 1999). (Again, because of computational limitations, estimates of ρ were obtained from a series of subsets of the markers.) One explanation for the high LD is simply that the Saami have a tiny effective population size (i.e., Ne≈102). However, the levels of genetic diversity at the same microsatellite loci are similar in the Saami and in neighboring populations that have much lower LD, arguing against the hypothesis of very small Ne. An alternative hypothesis is that recent admixture or gene flow into the Saami is responsible for these observations.
There seems to be mounting evidence that, for some regions, there is more LD than would be expected under simple demographic models. Not all data sets, however, show such departures from the model predictions. For some of the reports of excess LD, there is independent knowledge of demographic departures from model assumptions—for example, of recent admixture between diverged populations of the Lemba (Wilson and Goldstein 2000) or of admixture and inbreeding in a Costa Rican population (Service et al. 2001); for other examples, there is no obvious explanation (e.g., see Laan and Pääbo 1997; Taillon-Miller et al. 2000).
Short-Scale LD
A poor fit of certain regions and/or populations is perhaps not surprising, given that the models are simplifications of the history of human populations. In that light, it is interesting to note that, at short physical distances, there does not appear to be an excess of LD. In fact, polymorphism data from short (i.e., <10 kb) scales appear to show less LD than would be expected if Ne=104 and c=1 cM/Mb. For example, for data on Lpl (Clark et al. 1998), β-globin (Harding et al. 1997), and Dmd44 (Nachman and Crowell 2000), estimates of ρ are all an order of magnitude higher than would be expected under either a model of constant population size or a simple model with population growth (Przeworski and Wall 2001). Recent analysis of SNPs in a series of sequence-tagged-site regions reached a similar conclusion (K. Ardlie and L. Kruglyak, personal communication).
In summary, we have several examples in which large regions exhibit more LD than would be expected under either a model of constant population size or a model with rapid population growth. Yet, at the same time, studies of polymorphism at a small scale reveal less LD than would be expected. These observations at different scales are hard to accommodate in a single explanation, since factors that increase long-distance LD will tend to have an even larger effect on closely linked sites (e.g., see the third and fourth columns of fig. 1). In what follows, we will discuss potentially salient biological features not included in the simple models described thus far.
Demographic Departures from Model Assumptions
LD can be inflated by demographic factors, including inbreeding, population structure, and bottlenecks. The potential importance of inbreeding is highlighted by a recent study that examined genotypes of eight reference families from the CEPH (Broman and Weber 1999). That study found homozygous chromosomal segments of >10 cM in several families, a much larger distance than would be expected with random mating (Clark 1999). As pointed out by the study's authors, these long identical tracts could have resulted from mating of related individuals.
The effect of inbreeding among relatives is to lower diversity levels and to increase LD (i.e., decrease ρ). In this context, it is known that very high rates of inbreeding can have a greater impact on LD than on diversity (as seen in highly selfing plants [Nordborg 2000]). However, this effect is likely to be minor in human populations, in which, by comparison, the rates of inbreeding are modest.
The recent admixture of populations with different allele frequencies is a known factor in the generation of LD. At the time of admixture, there can be LD even between unlinked sites (e.g., see Pritchard and Rosenberg 1999). However, this decays very rapidly: with random mating, LD breaks down at a rate of (1-c) per generation (Hartl and Clark 1997). The net result is that in recently admixed populations, there can be substantial LD over centimorgan distances (e.g., see Wilson and Goldstein 2000; Service et al. 2001).
Note that extensive LD is also seen in populations that appear relatively homogeneous. Low rates of gene flow from more divergent populations could introduce haplotypes that increase LD (as shown in the third column of fig. 1). The importance of this effect is unknown.
The extent of LD may also be increased by temporary reductions in population size (“bottlenecks”). Under some models of changing population size, Ne is given by the harmonic mean of the population size over time (Hartl and Clark 1997). Since the harmonic mean is very sensitive to the smallest terms, severe or long-term bottlenecks can lead to sharp reductions in Ne. Several authors have appealed to a population bottleneck in the history of non-African populations as an explanation for what appear to be higher LD levels outside Africa (e.g., see Tishkoff et al. 1996; Kidd et al. 1998). However, there is still considerable uncertainty about the appropriate model for the evolution of modern humans. In particular, it remains to be tested whether a model with a bottleneck accompanying an emigration from Africa ∼100,000 years ago is consistent with the data on LD, as well as with levels of genetic diversity and with the frequency spectrum.
When recent demographic effects (e.g., the founding events in the Finns or Sardinians) are being considered, it is worth keeping in mind that most surveys of SNPs focus on markers where both alleles are common. These high-frequency variants tend to be older than random polymorphisms and therefore reflect events that occurred farther in the past. Thus, unless they are very severe, recent demographic events may have little impact on the extent and distribution of LD among high-frequency SNPs (see Kruglyak 1999). This might explain why levels of LD appear roughly similar across European populations that probably share most of their evolutionary history (Dunning et al. 2000; Eaves et al. 2000; Taillon-Miller et al. 2000).
Natural Selection
Significant allelic associations over large genetic distances may also result from the action of natural selection (as noted by Peterson et al. 1995; Huttley et al. 1999; Abecasis et al. 2001)—for example, from balancing selection acting in the MHC region. This said, studies of polymorphism in Drosophila and humans suggest that long-lived balancing selection is rare (Hey 1999; Przeworski et al. 2000). A second mode of selection known to generate LD is epistasis (nonadditive interactions between sites [Hartl and Clark 1997; Kelly and Wade 2000]). However, epistatic selection would have to be very strong to maintain allelic associations at the scale of megabases, in the face of substantial recombination. The same is true for “selective sweeps,” in which a rare, favorable mutation is quickly swept to fixation. It is unknown to what extent this mode of selection increases pairwise LD between high-frequency alleles. However, selective sweeps affect sites over a genetic distance on the order of the selection coefficient (Kaplan et al. 1989), so, for a single sweep to affect ⩾1 Mb, the selective advantage of the variant would have to be large (i.e., at least ≈.01).
The Relationship between Physical Distance and Genetic Distance
Estimates of the recombination rate at a centimorgan scale can be obtained by comparison of the genetic and physical maps. Although these are not yet very precise, they indicate that recombination rates vary by an order of magnitude across the human genome (Payseur and Nachman 2000; Yu et al. 2001). Regions of several megabases have rates <0.3 cM/Mb, whereas others have rates >3 cM/Mb. As would be expected, these “jungles and deserts” of recombination correspond to blocks of low and high linkage disequilibrium, respectively (Yu et al. 2001). Thus, comparison of LD levels in different regions may not be meaningful unless the local recombination rates are taken into account.
Recombination rates may also vary at a scale that is not detected by markers separated by megabases (see Yu et al. 2001). Dramatic changes in recombination rate have been reported over distances as short as a few kilobases (e.g., see Chakravarti et al. 1984). The molecular basis of hotspots for recombination remains unknown, but contributing factors might include high GC content (Eisenbarth et al. 2000; Yu et al. 2001) or whether the region is transcribed (Nicolas 1998). Thus, genetic maps may provide limited information about rates at short scales; more-refined estimates can be obtained by means of single-sperm typing (e.g., see Lien et al. 2000).
One known aspect of recombination not taken into account by most models is homologous gene conversion (here, gene conversion and crossing-over are thought of as alternative outcomes of a common recombination mechanism). For markers a megabase or so apart, the contribution of gene conversion to the overall level of genetic exchange is negligible (Andolfatto and Nordborg 1998). As a result, genetic map–based estimates of the recombination rate are essentially estimates of the crossing-over rates alone. The latter should accurately predict the extent of pairwise LD between polymorphisms far apart (given an adequate demographic model). For closely linked markers, however, LD may also be broken up by gene conversion. Indeed, in Drosophila and yeast, it appears that the rate of initiation of recombination events resolved as gene conversions and as crossovers is similar. Thus, pedigree-based estimates may substantially underestimate the total rate of recombination at small scales.
Currently, very little is known about gene conversion in humans. However, as noted above, there appears to be less LD at small scales than would be expected from estimates of crossing-over rates and observed levels of diversity. An intriguing explanation for this pattern is that gene conversion is quite frequent in humans. In support of this, Przeworski and Wall (2001) show that the data that they have analyzed are more likely under a model in which two-thirds of recombination events are gene-conversion events than under a model of crossing-over alone. An analysis of 10 anonymous intergenic regions also finds evidence for extensive gene conversion (A. Di Rienzo, L. Frisse, and R. R. Hudson, personal communication).
The relationship between LD and distance might also be shaped by the segregation of inversions. Inversion polymorphisms are thought to suppress recombination in heterozygotes throughout much of the length of the inverted segment (Roberts 1976; Martin 1999), although the precise details are unknown. Thus, the presence of inversion polymorphisms in a given region will reduce the rate of recombination. For instance, if 50% of the individuals used to construct a genetic map are heterozygous for an inversion, and if there is no recombination in heterozygotes, within the inverted region, then the average recombination rate in the region will be halved.
Moreover, inversion polymorphism can potentially have a second, much stronger effect on the extent of LD. Because recombination between the standard and inverted types is rare or absent, strong LD can develop between the two kinds of chromosomes. In the extreme case where there is complete suppression of recombination, a mutation within the inverted region arises on one type of chromosome and cannot move to the other via recombination. As mutations accumulate on both genetic backgrounds, the two arrangements diverge, potentially leading to a buildup of substantial LD. The extent of the effect will depend on the history and frequency of the inversion—including what kind of natural selection, if any, is acting on the inversion (see Andolfatto et al. 2001).
Little is known about the number, size, and frequency of inversions in the human genome. In particular, inversions shorter than a few megabases are currently difficult to detect by cytological methods but, if they were to reach intermediate frequencies, could have a substantial impact on the extent of LD. There are now a number of findings of common inversion polymorphisms: several studies of disease-associated inversions have reported inversion-heterozygote frequencies of 21%–33% in controls (Small et al. 1997; Saunier et al. 2000; Giglio et al. 2001). The length of the inverted segments varies from ∼50 kb (Small et al. 1997) to 3 Mb (J. Weber, personal communication), long enough for these rearrangements to potentially contribute to islands of extensive LD.
It appears that many of these chromosome rearrangements are mediated by nonhomologous meiotic exchange between inverted repeats (Small et al. 1997; Saunier et al. 2000; Giglio et al. 2001; Tilford et al. 2001). Since much of the human genome consists of repetitive DNA, it is possible that chromosomal rearrangements resulting in inversion polymorphisms are fairly common.
Implications for LD Mapping
The recent interest in LD in humans is due in large part to the prospect of large-scale association studies to locate complex disease genes. Risch and Merikangas (1996) showed that, under ideal circumstances, the power to detect disease mutations of small effect is much greater with association mapping than it is with linkage analysis. On the basis of this result, they argued that the future of complex-disease genetics lies in the use of genomewide screens of association.
However, the practical aspects of genomewide association mapping are currently daunting. It is clear that the number of markers that will be needed to scan the genome for association is very large. Recent progress has expanded the set of available SNPs across the genome (International SNP Map Working Group 2001), but the costs of genotyping a large sample of cases and controls at sufficient marker density would still be extremely high (although dropping). Clearly, we need good estimates of the marker density that will be required to achieve acceptable power in these studies.
The required density will depend on which statistical tests are used to detect association. Currently, it is most common to test for association at each marker in turn (or, sometimes, by combining pairs of nearby markers). In what follows, we consider the case in which only one marker is used, while noting that there seems to be a great need for the development of multilocus tests of association that make use of haplotype information, since these might prove to be much more efficient (see the Nordborg web site).
Density of Markers
Suppose that we test for association at a marker locus that is near a disease-susceptibility mutation. It can be shown that, in order to achieve roughly the same power at the marker locus as we would have if we could test the disease mutation itself, we need to increase the sample size by a factor of 1/r2, where r2 is the coefficient of LD between the marker and the disease mutation (see the sidebar; also see Kruglyak 1999). Hence, for small values of r2, there is little power to detect association at the marker locus.
r2 and the Power of Association Studies.
Several recent articles have referred to the connection between various measures of LD and the power of association studies (Kruglyak 1999; Dunning et al. 2000; Abecasis et al. 2001). Here we clarify this relationship.
Suppose that we genotype a case-control sample of N1 individuals at locus 1, a (true) disease-susceptibility locus, and that we genotype N2 individuals at locus 2, a nearby marker locus. We want to compare the power of association tests at these two loci.
Assume that both loci are biallelic, with alleles A and a at locus 1 and with alleles B and b at locus 2. Let πDA and πCA be the frequencies of allele A in individuals with the disease and in controls, respectively, and let πDB and πCB be the analogous frequencies at locus 2. Let qAB (respectively, qaB) be the probability that a chromosome with allele A (respectively, allele a) at locus 1 has the B allele at locus 2. Then,
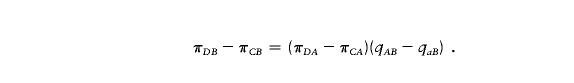 |
The standard χ2 test statistic of association at locus 1 (call this “X21”) can be written as
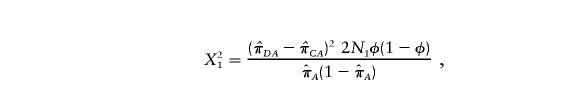 |
where 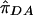 ,
, 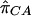 , and
, and 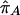 are the sample frequencies of A in affected individuals, in controls, and in the overall sample, respectively, and where φ and 1-φ are the fractions of the sample that are cases and controls, respectively. The test statistic for association at locus 2 (X22) is similar—but with B in place of A and with N2 in place of N1. The distributions of X21 and X22 are approximately the squares of normal random variables, with means
are the sample frequencies of A in affected individuals, in controls, and in the overall sample, respectively, and where φ and 1-φ are the fractions of the sample that are cases and controls, respectively. The test statistic for association at locus 2 (X22) is similar—but with B in place of A and with N2 in place of N1. The distributions of X21 and X22 are approximately the squares of normal random variables, with means
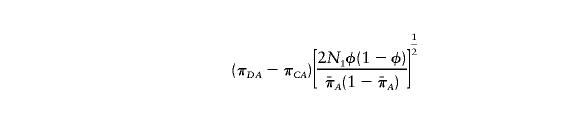 |
and
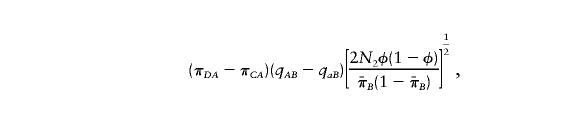 |
where
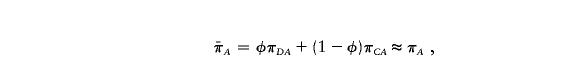 |
and similarly for  , and where the variances are ≈1 if the difference, in frequency of A, between cases and controls is small. Then, since
, and where the variances are ≈1 if the difference, in frequency of A, between cases and controls is small. Then, since
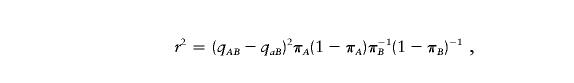 |
it follows that the distributions of X21 and X22 are approximately the same if N2=N1/r2. In other words, to achieve (approximately) the same power at the marker locus as is achieved at the susceptibility locus, the sample size must be increased by a factor of 1/r2.
In a highly influential paper, Kruglyak ran simulations of the coalescent with recombination to predict the rate of decay of LD; he predicted that “a useful level of LD is unlikely to extend beyond an average distance of roughly 3 kb in the general population” (Kruglyak 1999, p. 139). None of his models predicted “useful” LD over >30 kb. His criterion for useful LD was that the sample size necessary to detect association at the marker should not be increased more than 10-fold (in our terms, this corresponds to r2⩾.1). (The formal criterion used by Kruglyak (1999) is slightly different from ours: d2⩾.1.) The predictions of more-realistic models of growth are less drastic (see fig. 2).
As data on LD among SNPs become available, we can start to get an empirical sense of the rate of decay of r2. Figure 3 shows plots of 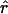 for SNPs in five regions. If we make the assumption that the distribution of r2 between two random SNPs is the same as that between a SNP and a disease mutation, then we can use plots such as those in figure 3 to study the decay of r2 directly (see Dunning et al. 2000; Taillon-Miller et al. 2000; Abecasis et al. 2001). In practice, it seems likely that disease mutations at polymorphic frequencies will usually be deleterious, which would have the helpful effect of increasing the average LD with nearby sites (Pritchard 2001 [in this issue]).
for SNPs in five regions. If we make the assumption that the distribution of r2 between two random SNPs is the same as that between a SNP and a disease mutation, then we can use plots such as those in figure 3 to study the decay of r2 directly (see Dunning et al. 2000; Taillon-Miller et al. 2000; Abecasis et al. 2001). In practice, it seems likely that disease mutations at polymorphic frequencies will usually be deleterious, which would have the helpful effect of increasing the average LD with nearby sites (Pritchard 2001 [in this issue]).
In figure 3, points above the dotted lines meet Kruglyak’s criterion of useful LD. Even at very short distances, many pairs of markers are in low LD (except, possibly, in the data reported by Taillon-Miller et al. (2000)). Thus, even if a disease mutation is very close to the nearest marker, there may be a substantial probability of failing to detect association at that marker.
In a genome-scan for association, there would be a series of markers spaced along the chromosome. For a given location (and frequency) of the disease mutation, there is some probability that each of the nearby markers is in useful LD with the disease mutation; this probability drops as a function of the distance between marker and mutation. In effect, the goal is to choose the marker density in such a way that there is a high probability that the mutation will be in strong LD with at least one of the markers. This density might differ substantially from what would be suggested by consideration of average r2 values.
At this time, it seems premature to provide a formal estimate of the required marker density. However, suppose that the first four plots in figure 3 are typical of LD across the genome. Then the genome scans will need to have numerous (perhaps 5–10 or more) markers within, say, 50 kb of each disease mutation, to ensure a high probability that at least one marker is in strong LD with the mutation.
In the discussion so far, we have implicitly assumed that the value of r2 between the disease mutation and each nearby marker is independent. This is not true: each recombination event in the ancestral genealogy can affect multiple markers. This correlation effect will further increase the required density of markers, although the size of this effect is unknown.
Ultimately, the marker map will also need to reflect variation in the extent of LD across regions, placing more markers in some regions than in others. As discussed above, some of the reasons for variation may be predictable—for example, variation in recombination rate and the predicted difference between the X chromosome and the autosomes. Some may be due to factors that we do not yet understand, whereas some variation will result from the inherent randomness of the coalescent process.
In closing, we should mention one situation in which LD is expected to extend over quite long distances—namely, in admixed populations such as African Americans (Pfaff et al. 2001). It has been proposed that, for diseases that differ in frequency between the parental groups, the resulting “admixture disequilibrium” could enable LD mapping using relatively small numbers of markers, spaced centimorgans apart (Chakraborty and Weiss 1988; McKeigue 1997).
Fine-Scale Mapping
So far, we have discussed the issue of testing for association, but LD also plays a central role in the problem of gene localization. These two goals are rather different: in the first, the aim is either to identify chromosomal regions that contain disease-susceptibility loci (as in the genome-scan model) or to confirm regions with weak evidence from pedigree studies; in fine-scale mapping, one already has strong evidence that a region of interest contains a disease locus, and the goal is to use a series of markers to estimate its location (e.g., see McPeek and Strahs 1999; Horikawa et al. 2000; Morris et al. 2000).
The extent and distribution of LD affect these two kinds of goals in different ways. When one is testing for association, it is helpful if LD extends over long distances around the disease mutation, because then not so many markers are needed to scan for association; but at the later stage, when the goal is to infer location, long-ranging LD is potentially problematic. It means that strong associations might be observed far from the causative site(s), and these could lead to effort being spent on the wrong location.
The possibility of rampant gene conversion is worrying for the use of LD in fine-scale mapping. The presence of gene conversion increases the recombination rate at very-short scales (Andolfatto and Nordborg 1998), which is helpful. However, it may also make the relationship between LD and distance less predictable, since gene conversion affects only short regions, leaving flanking haplotypes unchanged. Currently, little is known about the extent to which the spatial distribution of LD matches model predictions, such as those used in fine-mapping (e.g., see McPeek and Strahs 1999). For now at least, studies at the fine-mapping stage should take great care to map the entire extent of the region of association, in case the region contains local peaks of association far from the causative site(s).
Future Directions
We have aimed to place the new empirical data on LD in the context of various population genetics models. One point that emerges clearly is that, to understand LD in humans, we need to have a much better understanding of human demography. This includes the history of changes in population size, as well as population structure and other forms of nonrandom mating. We also need to have more data on recombination rates across the genome. It is known that recombination rates vary greatly, and accurate data are needed to allow comparisons of LD from different regions. When the time comes to construct marker sets for association mapping, we will need accurate and fine-scale recombination maps, so that the density of markers in each region reflects local rates. As discussed above, little is known about either the rate of gene conversion or the frequency of submicroscopic inversion polymorphisms, but both have the potential to play an important role in determining the distribution of LD.
At this time, our conclusions about LD between SNPs are somewhat limited by the amount of data available. Ideally, one would want a large number of SNPs from many regions, to enable studies of differences among regions. The markers should be placed so that there are data on LD both over short distances (i.e., <5 kb) and over long distances (i.e., ⩾1 Mb). In terms of experimental design, there is probably little to be gained from the genotyping of huge numbers of individuals at each SNP; instead it makes more sense to genotype many SNPs.
Our review has concentrated primarily on the extent of LD, as measured by pairwise measures (mainly r2). This emphasis reflects that of the majority of the literature. However, we feel strongly that, for many problems, it will be important to think about the spatial structure of LD in more detail. For example, how much local variation in LD is there, as one moves through a small region? In LD mapping, how often will there be isolated peaks of association far from the causative site? What is the probability that a disease mutation sits in a local trough of low LD, making it virtually undetectable for any plausible marker density? In other contexts, the use of complete haplotype data may also offer major advantages over pairwise measures—for instance, in the estimation of ρ; in making more efficient use of marker data for detecting associations, particularly if there is allelic heterogeneity (Pritchard 2001 [in this issue]); and in fine-structure mapping (e.g., see McPeek and Strahs 1999).
Acknowledgments
P. Andolfatto, B. Charlesworth, D. Charlesworth, P. Donnelly, P. Fearnhead, R. Hudson, D. Ledbetter, G. McVean, M. Stephens, J. Wall, and J. Weber are thanked for helpful comments or discussions. P. Fearnhead, G. McVean, and J. Wall kindly provided us with estimates of ρ; G. Abecasis, A. Dunning, and P. Taillon-Miller kindly sent us their data. We thank P. Andolfatto, K. Beauregard, P. Fearnhead, and Y. Gilad for comments on the manuscript. J.K.P. is supported by a Hitchings-Elion Fellowship from Burroughs-Wellcome Fund; M.P. is supported by a National Science Foundation postdoctoral grant in bioinformatics.
Note added in proof.—
A recent article by Reich et al. (2001) reports extensive LD among SNPs at 19 loci. They report more LD than would be expected under a model of stepwise population growth (and, also, more than would be expected with a constant population size of 10,000 [D. Reich, personal communication]). As a possible explanation, Reich et al. suggest that there may have been a recent bottleneck in non-African populations.
Electronic-Database Information
URLs for data in this article are as follows:
- Gil McVean web site, http://www.stats.ox.ac.uk/~mcvean/ (for estimation of ρ)
- Hudson Lab Home Page, http://home.uchicago.edu/∼rhudson1/ (for simulation of the coalescent with recombination [see the program “mksamples”] and estimation of ρ [see the article “Two locus sampling distributions and their application”)
- Human Recombination Rates web site, http://eebweb.arizona.edu/nachman/publications/data/microsats.html (for estimation of c)
- Nordborg web site, http://www-hto.usc.edu/people/nordborg/papers.html (for the article “Linkage disequilibrium, haplotype evolution and the coalescent”)
- Paul Fearnhead web site, http://www.stats.ox.ac.uk/∼fhead/index.html (for estimation of ρ [see the article “Estimating recombination rates from population genetic data”])
References
- Abecasis GR, Noguchi E, Heinzmann A, Traherne JA, Bhattacharya S, Leaves NI, Anderson GG, Zhang Y, Lench NJ, Carey A, Cardon LR, Moffatt MF, Cookson WO (2001) Extent and distribution of linkage disequilibrium in three genomic regions. Am J Hum Genet 68:191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto P, Depaulis F, Navarro A (2001) Inversion polymorphisms and nucleotide variability in Drosophila. Genet Res 77:1–8 [DOI] [PubMed] [Google Scholar]
- Andolfatto P, Nordborg M (1998) The effect of gene conversion on intralocus associations. Genetics 148:1397–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awadalla P, Eyre-Walker A, Smith JM (1999) Linkage disequilibrium and recombination in hominid mitochondrial DNA. Science 286:2524–252 [DOI] [PubMed] [Google Scholar]
- Broman KW, Weber JL (1999) Long homozygous chromosomal segments in reference families from the Centre d’Etude du Polymorphisme Humain. Am J Hum Genet 65:1493–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane CR, Lim EP, Kalyanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley GQ, Lander ES (1999) Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet 22:231–238 [DOI] [PubMed] [Google Scholar]
- Chakraborty R, Weiss KM (1988) Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci. Proc Natl Acad Sci USA 85:9119–9123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti A, Buetow KH, Antonararkis SE, Waber PG, Boehm CD, Kazazian HH (1984) Nonuniform recombination within the human β-globin gene cluster. Am J Hum Genet 36:1239–1258 [PMC free article] [PubMed] [Google Scholar]
- Clark AG (1999) The size distribution of homozygous segments in the human genome. Am J Hum Genet 65:1489–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Weiss KM, Nickerson DA, Taylor SL, Buchanan A, Stengard J, Salomaa V, Vartiainen E, Perloa M, Boerwinkle E, Sing CF (1998) Haplotype structure and population genetic inferences from nucleotide-sequence variation in human lipoprotein lipase. Am J Hum Genet 63:595–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Risch N (1995) A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics 29:311–322 [DOI] [PubMed] [Google Scholar]
- Dunning AM, Durocher F, Healey CS, Teare MD, McBride SE, Carlomagno F, Xu CF, Dawson E, Rhodes S, Ueda S, Lai E, Luben RN, Rensburg EJV, Mannermaa A, Kataja V, Rennart G, Dunham I, Purvis I, Easton D, Ponder BA (2000) The extent of linkage disequilibrium in four populations with distinct demographic histories. Am J Hum Genet 67:1544–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaves IA, Merriman TR, Barber RA, Nutland S, Tuomilehto-Wolf E, Tuomilehto J, Cucca F, Todd JA (2000) The genetically isolated populations of Finland and Sardinia may not be a panacea for linkage disequilibrium mapping of common disease genes. Nat Genet 25:320–323 [DOI] [PubMed] [Google Scholar]
- Eisenbarth I, Vogel G, Krone W, Vogel W, Assum G (2000) An isochore transition in the NF1 gene region coincides with a switch in the extent of linkage disequilibrium. Am J Hum Genet 67:873–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglio S, Broman KW, Matsumoto N, Calvari V, Gimelli G, Neumann T, Ohashi H, Voullaire L, Larizza D, Giorda R, Weber JL, Ledbetter DH, Zuffardi O (2001) Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet 68:874–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Simonic I, Ott J (2000) Significant evidence for linage disequilibrium over a 5-cM region among Afrikaners. Genomics 66:87–92 [DOI] [PubMed] [Google Scholar]
- Griffiths RC, Marjoram P (1996) Ancestral inference from samples of DNA sequences with recombination. J Comput Biol 3:479–502 [DOI] [PubMed] [Google Scholar]
- Harding RM, Fullerton SM, Griffiths RC, Bond J, Cox MJ, Schneider JA, Moulin DS, Clegg JB (1997) Archaic African and Asian lineages in the genetic ancestry of modern humans. Am J Hum Genet 60:772–789 [PMC free article] [PubMed] [Google Scholar]
- Hartl DL, Clark AG (1997) Principles of population genetics. Sinauer Associates, Sunderland, MA [Google Scholar]
- Hey J (1999) The neutralist, the fly, and the selectionist. Trends Ecol Evol 14:35–38 [DOI] [PubMed] [Google Scholar]
- Horikawa Y, Oda N, Cox N, Li X, Orho-Melander M, Hara M, Hinokio Y, et al (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26:163–175 [DOI] [PubMed] [Google Scholar]
- Hudson RR (1993) The how and why of generating gene genealogies. In: Takahata N, Clark AG (eds) Mechanisms of molecular evolution. Japan Scientific Societies, Tokyo, pp 23–36 [Google Scholar]
- ——— (2001) Linkage disequilibrium and recombination. In: Balding D, Bishop M, Cannings C (eds) Handbook of statistical genetics. Wiley & Sons, New York, pp 309–324 [Google Scholar]
- Huttley G, Smith M, Carrington M, O’Brien S (1999) A scan for linkage disequilibrium across the human genome. Genetics 152:1711–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International SNP Map Working Group (2001) A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409:928–933 [DOI] [PubMed] [Google Scholar]
- Jorde LB (2000) Linkage disequilibrium and the search for complex disease genes. Genome Res 10:1435–1444 [DOI] [PubMed] [Google Scholar]
- Kaplan NL, Hudson RR, Langley CH (1989) The “hitchhiking effect” revisited. Genetics 123:887–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JK, Wade MJ (2000) Molecular evolution near a two-locus balanced polymorphism. J Theor Biol 204:83–101 [DOI] [PubMed] [Google Scholar]
- Kidd KK, Morar B, Castiglione CM, Zhao H, Pakstis AJ, Speed WC, Bonne-Tamir B, Lu RB, Goldman D, Lee C, Nam YS, Grandy DK, Jenkins T, Kidd JR (1998) A global survey of haplotype frequencies and linkage disequilibrium at the DRD2 locus. Hum Genet 103:211–227 [DOI] [PubMed] [Google Scholar]
- Kruglyak L (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 22:139–144 [DOI] [PubMed] [Google Scholar]
- Kuhner MK, Yamato J, Felsenstein J (2000) Maximum likelihood estimation of recombination rates from population data. Genetics 156:1393–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laan M, Pääbo S (1997) Demographic history and linkage disequilibrium in human populations. Nat Genet 17:435–438 [DOI] [PubMed] [Google Scholar]
- Lewontin RC (1964) The interaction of selection and linkage. I. General considerations: heterotic models. Genetics 49:49–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WH, Sadler L (1991) Low nucleotide diversity in man. Genetics 129:513–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien S, Szyda J, Schechinger B, Rappold G, Arnheim N (2000) Evidence for heterogeneity in recombination in the human pseudoautosomal region: high resolution analysis by sperm typing and radiation-hybrid mapping. Am J Hum Genet 66:557–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AD , Langley CH (1999) The power of association studies to detect the contribution of candidate gene loci to variation in complex traits. Genome Res 9:720–731 [PMC free article] [PubMed] [Google Scholar]
- Martin RH (1999) Sperm chromosome analysis in a man heterozygous for a paracentric inversion of chromosome 14 (q24.1q32.1). Am J Hum Genet 64:1480–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeigue PM (1997) Mapping genes underlying ethnic differences in disease risk by linkage disequilibrium in recently admixed populations. Am J Hum Genet 60:188–196 [PMC free article] [PubMed] [Google Scholar]
- McPeek M, Strahs A (1999) Assessment of linkage disequilibrium by the decay of haplotype sharing, with application to fine-scale genetic mapping. Am J Hum Genet 65:858–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AP, Whittaker JC, Balding DJ (2000) Bayesian fine-scale mapping of disease loci, by hidden Markov models. Am J Hum Genet 67:155–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL (2000) Estimate of the mutation rate per nucleotide in humans. Genetics 156:297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas A (1998) Relationship between transcription and initiation of meiotic recombination: towards chromatin accessibility. Proc Natl Acad Sci USA 6:87–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R (2000) Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics 154:931–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M (2000) Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial self-fertilization. Genetics 154:923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (2001) Coalescent theory. In: Balding D, Bishop M, Cannings C (eds) Handbook of statistical genetics. Wiley & Sons, New York, pp 179–212 [Google Scholar]
- Payseur BA, Nachman MW (2000) Microsatellite variation and recombination rate in the human genome. Genetics 156:1285–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson AC, Rienzo AD, Lehesjoki AE, de la Chapelle A, Slatkin M, Freimer NB (1995) The distribution of linkage disequilibrium over anonymous genome regions. Hum Mol Genet 4:887–894 [DOI] [PubMed] [Google Scholar]
- Pfaff CL, Parra EJ, Bonilla C, Hiester K, McKeigue PM, Kamboh MI, Hutchinson RG, Ferrell RE, Boerwinkle E, Shriver, MD (2001) Population structure in admixed populations: effect of admixture dynamics on the pattern of linkage disequilibrium. Am J Hum Genet 68:198–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK (2001) Are rare variants responsible for susceptibility to common diseases? Am J Hum Genet 69:124–137 (in this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Rosenberg NA (1999) Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet 65:220–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Seielstad MT, Perez-Lezaun A, Feldman MW (1999) Population growth of human Y chromosomes: a study of Y chromosome microsatellites. Mol Biol Evol 16:1791–1798 [DOI] [PubMed] [Google Scholar]
- Przeworski M, Hudson RR, DiRienzo A (2000) Adjusting the focus on human variation. Trends Genet 16:296–302 [DOI] [PubMed] [Google Scholar]
- Przeworski M, Wall JD (2001) Why is there so little intragenic linkage disequilibrium in humans? Genet Res 77:143–151 [DOI] [PubMed] [Google Scholar]
- Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, Lander ES (2001) Linkage disequilibrium in the human genome. Nature 411:199–204 [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 [DOI] [PubMed] [Google Scholar]
- Roberts PA (1976) The genetics of chromosome aberration. In: Ashburner M, Novitski E (eds) The genetics and biology of Drosophila. Academic Press, London, pp 67–184 [Google Scholar]
- Saunier S, Calado J, Benessy F, Silbermann F, Heilig R, Weissenbach J, Antignac C (2000) Characterization of the NPHP1 locus: mutational mechanisms involved in deletions in familial juvenile nephronophthisis. Am J Hum Genet 66:778–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Service SK, Ophoff RA, Freimer NB (2001) The genome-wide distribution of background linkage disequilibrium in a population isolate. Hum Mol Genet 10:545–551 [DOI] [PubMed] [Google Scholar]
- Slatkin M (1994) Linkage disequilibrium in growing and stable populations. Genetics 137:331–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small K, Iber J, Warren ST (1997) Emerin deletion reveals a common X-chromosome inversion mediated by inverted repeats. Nat Genet 16:96–99 [DOI] [PubMed] [Google Scholar]
- Stephens M (2001) Inference under the coalescent. In: Balding D, Bishop M, Cannings C (eds) Handbook of statistical genetics. [Google Scholar]
- Taillon-Miller P, Bauer-Sardina I, Saccone NL, Putzel J, Laitinen T, Cao A, Kere J, Pilia G, Rice JP, Kwok PY (2000) Juxtaposed regions of extensive and minimal linkage disequilibrium in human Xq25 and Xq28. Nat Genet 25:324–328 [DOI] [PubMed] [Google Scholar]
- Tajima F (1989) The effect of change in population size on DNA polymorphism. Genetics 123:597–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilford CA, Kuroda-Kawaguchi T, Skaletsky H, Rozen S, Brown LG (2001) A physical map of the Y chromosome. Nature 409:943–945 [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Dietzsch E, Speed W, Pakstis AJ, Kidd JR, Cheung K, Bonné-Tamir B, Benerecetti ASS, Moral P, Krings M, Pääbo S, Watson E, Risch N, Jenkins T, Kidd KK (1996) Global patterns of linkage disequilibrium at the CD4 locus and modern human origins. Science 271:1380–1387 [DOI] [PubMed] [Google Scholar]
- Wall JD (2000) A comparison of estimators of the population recombination rate. Mol Biol Evol 17:156–163 [DOI] [PubMed] [Google Scholar]
- Wall JD, Przeworski M (2000) When did the human population start increasing? Genetics 155:1865–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS (1996) Genetic data analysis II. Sinauer Associates, Sunderland, MA [Google Scholar]
- Wilson JF, Goldstein DB (2000) Consistent long-range linkage disequilibrium generated by admixture in a Bantu-Semitic hybrid population. Am J Hum Genet 67:926–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S (1951) The genetical structure of populations. Ann Eugenics 15:323–354 [DOI] [PubMed] [Google Scholar]
- Yu A, Zhao C, Fan Y, Jang W, Mungall AJ, Deloukas P, Olsen A, Doggett NA, Ghebranious N, Broman KW, Weber JL (2001) Comparisons of human genetic and sequence-based physical maps. Nature 409:951–953 [DOI] [PubMed] [Google Scholar]
- Zavattari P, Deidda E, Whalen M, Lampis R, Mulargia A, Loddo M, Eaves I, Mastio G, Todd JA, Cucca F (2000) Major factors influencing linkage disequilibrium by analysis of different chromosome regions in distinct populations: demography, chromosome recombination frequency and selection. Hum Mol Genet 9:2947–2957 [DOI] [PubMed] [Google Scholar]