

Abstract
Mutations in the large gene of clotting factor VIII (FVIII) are the most common events leading to severe human bleeding disorder. The high proportion of de novo mutations observed in this gene raises the possibility that a significant proportion of such mutations does not derive from a single germ cell but instead should be attributed to a germline or somatic mosaic originating from a mutation during early embryogenesis. The present study explores this hypothesis by using allele-specific PCR to analyze 61 families that included members who had sporadic severe hemophilia A and known FVIII gene defects. The presence of somatic mosaicisms of varying degrees (0.2%–25%) could be shown in 8 (13%) of the 61 families and has been confirmed by a mutation-enrichment procedure. All mosaics were found in families with point mutations (8 [25%] of 32 families). In the subgroup of 8 families with CpG transitions, the percentage with mosaicism increased to 50% (4 of 8 families). In contrast, no mosaics were observed in 13 families with small deletions/insertions or in 16 families with intron 22 inversions. Our data suggest that mosaicism may represent a fairly common event in hemophilia A. As a consequence, risk assessment in genetic counseling should include consideration of the possibility of somatic mosaicism in families with apparently de novo mutations, especially families with the subtype of point mutations.
Introduction
Hemophilia A (HEMA [MIM 306700]) is the most common severe hereditary bleeding disorder in humans, affecting 1 in 5,000 male newborns. The phenotype is caused by a defective or absent factor VIII protein, which leads to varying degrees of hemorrhage. The factor VIII (FVIII) gene spans 186 kb on chromosome Xq28 and consists of 26 exons. The types of genetic defects vary widely. In severe hemophilia A, 40% of all mutations are caused by the intron 22 inversion (Lakich et al. 1993). Another 30%–35% of severely affected hemophiliacs carry point mutations, either of the nonsense or of the missense type, which are randomly distributed throughout the gene. A minor fraction of mutations comprises small deletions/insertions (10%) and large deletions (5%) (Becker et al. 1996).
Because of the high mutation rate (2.5–4.2 × 10−5) (Strauss 1967; Vogel 1977), ∼50% of families severely affected by hemophilia A include only one member with the disorder, an observation that points to a recent origin (parental or grandparental generation) of the mutation in most affected families. Family studies revealed that mutations in hemophilia A predominantly originated in males; however, some deletions occurred more often in females (Becker et al. 1996). Mutations causing hemophilia A usually appear to have arisen in germ cells, thereby leading to heterozygosity or hemizygosity for the respective mutation in the offspring generation. However, a de novo mutation may also occur during early embryogenesis and thus may represent either germline and/or somatic mosaicism, in which type and degree of mosaicism are determined by the developmental stage and cell lineage.
The significance of mosaic mutations may be underestimated, because they usually remain undetected during routine mutation analysis. Overexposed manual-sequence autoradiographs can detect the mutation only if it is present in >5% of the cells sampled (Ketterling et al. 1999), and the detection limit of Southern blot analysis is 10%–20% (Oldenburg et al. 2000). Special methods, such as allele-specific PCR, are expected to be more sensitive and may detect a mutant allele that is present in as few as 1 of 1,000 samples (Knöll et al. 1996). Somatic and germline mosaics have been documented for only a few X-linked and autosomal dominant single-gene disorders. In neurofibromatosis type 1 (Colman et al. 1996), osteogenesis imperfecta (Lund et al. 1996), and Hunter disease (Froissart et al. 1997), single cases of mosaicism have been described. More-systematic studies to assess the frequency of mosaicism have been reported for retinoblastoma (Sippel et al. 1998), Duchenne muscular dystrophy (Passos-Bueno et al. 1992; van Essen et al. 1992), and hemophilia B (Ketterling et al. 1999), and these studies demonstrate that a significant proportion (10%–20%) of de novo mutations involve proven mosaicism.
In hemophilia A, only seven patients with mosaicism have been reported, and the mutations in this group include three large deletions (Higuchi et al. 1988; Bröcker-Vriends et al. 1990; Levinson et al. 1990), one small deletion (Casey et al. 1999), one inversion of distal intron 22 (Oldenburg et al. 2000), and two point mutations ( Levinson et al. 1990; Schwaab et al. 1993).
In order to elucidate the frequency of mosaicism in hemophilia A, we analyzed 61 families with known type and origin of FVIII gene mutations for the presence of mosaic individuals. A total of eight somatic mosaics were detected, all arising in the patients' mothers and grandmothers of families with point mutations. These findings indicate a significant proportion of mosaicism in hemophilia A, at least in the subgroup of point mutations, that may confound risk estimation during genetic counseling.
Subjects, Material, and Methods
Families
The present study sample comprises 61 families that have at least one member with sporadic severe hemophilia A and known type and origin of the pathogenic mutation. Blood or DNA samples were provided by the following institutions: Department of Experimental Hematology and Transfusion Medicine, Bonn; Department of Human Genetics, Münster, Germany; Department of Human Genetics, Würzburg, Germany; INSERM U143, Hôpital Bicetre, Le Kremlin Bicetre, France; Division of Genomic Medicine, University of Sheffield, Royal Hallamshire Hospital, Sheffield, United Kingdom; and Department of Paediatrics, University Hospital, Malmö, Sweden. All patients gave informed consent, according to the declaration of Helsinki.
Mutation Analysis of the Factor VIII Gene
Mutation analysis was performed using the Southern blot technique for the detection of the intron 22 inversion (Lakich et al. 1993) and using the various mutation-screening methods established in the authors' respective laboratories (Lavergne et al. 1992; Becker et al. 1996; Tavassoli et al. 1998; Williams et al. 1998; Ljung and Sjorin 1999). Mutations localized by the screening methods were characterized by sequencing the corresponding exon.
Allele-Specific PCR and Detection of Fluorescence-Labeled PCR Fragments
The principle of this method is based on the fact that a mismatch at the 3′ end of a primer leads to an insufficient elongation during PCR by the Taq polymerase. A primer that matches a mutation with its 3′ end and concomitantly mismatches to the wild-type allele allows the amplification of the mutated allele but suppresses amplification of the wild-type allele. Allele discrimination can be improved by incorporating an additional mismatching base three bases upstream of the 3′ end of the primers (Hezard et al. 1997). All reverse primers were dye labeled, and the PCR products were detected and sized on a 373A DNA sequencer (ABI).
Allele-specific PCR was performed in a Biometra Trio-Thermoblock. Amplifications were performed using 200 ng of genomic DNA, 20 pmol of allele-specific primer (table 1), 20 pmol of dye-labeled reverse primer (Schwaab et al. 1997), 50 μM of each of the four types of dNTP, and 2.5 U of AmpliTaq-Gold DNA polymerase (PE Applied Biosystems) in a total volume of 50 μl. The PCR program was set to initial denaturation at 94°C for 9 min, followed by 32 cycles of amplification with 45 s of denaturation at 94°C, 45 s of annealing at 55°C, and 90 s of elongation at 72°C. If allele discrimination was insufficient under these conditions, the annealing temperature was raised in steps of 2°C to optimize the performance of the allele-specific PCR; 1 μl of the PCR product was mixed with 1.5 μl of formamide, 0.5 μl LS500 size standard (serac R. Hofmann GmbH), and 0.2 μl of gel-loading buffer. DNA fragments were separated on a 373A DNA sequencer (ABI) using a 6% denaturing polyacrylamide gel. Data were analyzed by GENESCAN 672 software (ABI).
Table 1.
Primers for Allele-Specific PCR
| Family | Allele-Specific Primer Sequence (5′–3′) |
| LU3 | GCCAAGGCCACCCTGGATTGA |
| LÜP2 | CCTACCATCCAGGCTGAAGA |
| LA30 | CCATCCAGGCTGAGGTTTGTC |
| LA27 | CTATAGGAGCTGAATATGTTT |
| ROP1 | GACCTTGGACAGTTTCTTCC |
| TRP1 | ATTACATTGCTGCTGATGG |
| LU6 | TGAAGCTATTCAGCATGATTG |
| JO7 | CATATAACATCTACCCTCTCA |
| LA37 | ATATGTAATTAACAGATAATATA |
| LA6 | GTATTTGATGAGAACCGATGG |
| LA15 | CCATTGTTTTTGCAGGCACCG |
| JO1 | GTGATTTGTTGATGCTCTAGT |
| LA40 | GTGATTTGTTGATGCTCTAGT |
| EGL1 | GAGAATAGTCCATCAGTGTA |
| JO5 | CTTGCCAGAATCAGCAAGGAGA |
| PE1 | CAGATACCAAAAGAAGAATA |
| PE3 | CAGATACCAAAAGAAGAATA |
| EGL2 | AGACAAGTGACAGTACAAGG |
| PE4 | GAGTGTCAGACTCCCCTAGT |
| JO8 | GGCAAAGGACAGTGGGCTCT |
| JO19 | TTAACCCTCCAATTATTGGTT |
| JO10 | CGATACATCCGTTTGCATCG |
| LA28 | ATAGCATTCGCAGCACTCCTT |
| LA38 | CAGCACTCTTCGCATGGACTC |
| LA5 | CCTGGTCTCCTTCAAAAGGTT |
| LA10 | CCTGGTCTCCTTCAAAAGGTT |
| PE2 | AGCATGTATGTGAAGGATTG |
| LA14 | GGTGAACTCTCTAGACCCTCT |
| SCP1 | TTACTGACTCGCTACCTACT |
| RHP1 | TTACTGACTCGCTACCTACT |
| LA29 | TTACTGACTCGCTACCTACC |
| JO9 | CTCAGTCGTGTACAAAAAGTT |
| LA41 | GTGTACAAAAAGACTCTGTAG |
| LU9 | GGGAAGTTGGAGACACACCGA |
| LA18 | GTTGGAGACACACTGTTGTGG |
| LA39 | CTGTAGATCAAAGAGGAAACAG |
| LU5 | ACAATTCCATCAGACAATCTT |
| LU7 | CACACAATCAAGAAAAAATAT |
| MAB1 | CACACAATCAAGAAAAAATAT |
| LA11 | CACACAATCAAGAAAAAATAT |
| LA16 | TCCATCTATTAGACCTATTTC |
| LOP1 | GTGTTCACTGTACGAAAATAG |
| JO4 | TGCCATTCTGAAAAAAGTGT |
Intron 22–Inversion PCR
The PCR for detection of the intron 22 inversion was performed as described by Liu et al. (1998), with the following slight modifications: PCR-amplification started with 100 ng of DNA, and the amplified fragments were separated on 0.8% agarose for 24 h. PCR fragments were detected by Southern blot analysis of the agarose gel, using standard conditions. A digoxigenin-labeled probe (HA-Int22: 5′-digoxigenin-GAGAAGGCGCCCAGG) was used for hybridization, and DNA fragments were visualized according to the protocol of the manufacturer (Roche).
Mutation Enrichment and DNA Sequencing
To obtain evidence of somatic mosaicism, a modified method of the protocol described by Knöll et al. (1996) was established for the enrichment of the mutated FVIII allele in the DNA samples. In a first step, an artificial restriction site was created with specially designed primers. In each case, the 3′ end of the primers flanking the mutation and parts of the following target sequence create a restriction motif that arises only in the wild-type allele. Subsequent digestion with the appropriate restriction endonuclease (table 2) and reamplification resulted in an enrichment of the mutated allele. Because restriction endonucleases do not cleave 100% of their template, the procedure had to be repeated twice to ensure the enrichment of the uncleaved PCR product that corresponded to the mutant allele.
Table 2.
Primers for Incorporation of Artificial Restriction Sites and Sequencing[Note]
| Family, Restriction Endonuclease, and Primer | Primer Sequence |
| JO1, NruIa: | |
| E14JO1ARS/F | AGTGATTTGTTGATGCTCTCG |
| Ex14-2R | ATGGAGCTGTGGCCTGAAGTG |
| M13-Ex14-2R2 | M13-ACTGTCTATTGCTCCAGG |
| JO7, BsiWIa: | |
| E10JO7ARS/F | CCATATAACATCTACCCGTAC |
| Ex10R | GCTATAAACGAGGGAATATTTAC |
| M13-Ex10R | M13-CGAGGGAATATTTACCTTTTG |
| JO10, MunIb: | |
| E23JO10ARS | GCTGCGAATGCTATAATCAATT |
| Ex23F | ACTCTGTATTCACTTTCCATG |
| M13-Ex23F | M13-GCTAATCTCTCCATACAG |
| RHP1, BstBIa: | |
| M13-Ex26F | M13-TTGTCCTGTCAGACAACC |
| Ex26R | TTAGCACAAAGGTAGAAGGC |
| LA3, AvaIIa: | |
| E16LA3ARS | GTTTTTTCTAGGTTCTGGTC |
| Ex16F | TTGTCGTTATTGTTCTACAGG |
| M13-Ex16F | M13-CAGGTAACTTTCAGAAATCAG |
| LA14, AgeIa: | |
| E26LA14ARS | TCGAAGGTAGCGAGTCAGTACC |
| Ex26F | AGCGTCTGTGCTTTGCAGTG |
| M13-Ex26F | M13-TTGTCCTGTCAGACAACC |
| LA28, BstBIa: | |
| E23LA28ARS | ATCACAGCCCATCAACTCCATTC |
| Ex23F | ACTCTGTATTCACTTTCCATG |
| M13-Ex23F | M13-GCTAATCTCTCCATACAG |
| LA15, MunIb | |
| E13LA15ARS | CAAACTATCAAAAACATAGCAAT |
| Ex13FB | GGAAGATATAATATCTCTTCC |
| M13-Ex13FB | M13-GGGAATAAGATAATGGGC |
Note.— The M13-sequence is TGTAAAACGACGGCCAGT.
Supplied by New England Biolabs.
Supplied by Gibco BRL Life Technologies.
PCR was performed on a Biometra Trio-Thermoblock with 1 μl of the initially amplified exon PCR, 100 μM of each of the four types of dNTP, 20 pmol of each primer (table 2), and 1.5 U of AmpliTaq-Gold DNA polymerase (PE Applied Biosystems) in a total volume of 50 μl. PCR parameters were set to initial denaturation at 94°C for 9 min, 32 cycles of amplification with 30 s of denaturation at 94°C, 30 s of annealing at 55°C, and 45 s of elongation at 72°C. Subsequently, 10 μl of the resultant PCR product was digested with 10 U of the appropriate restriction enzyme (table 2), according to the manufacturer's protocol. PCR amplification with primers creating the respective restriction site and subsequent digestion was repeated three times. After the final restriction reaction, 1 μl of the assay solution was amplified with 5 pmol of seminested M13-primers, 50 pmol of the appropriate primers (table 2), 50 μM of each dNTP, and 1.5 U of AmpliTaq-Gold DNA polymerase (PE Applied Biosystems) in a total volume of 50 μl. PCR reaction was carried out on a Biometra Trio-Thermoblock and included initial denaturation at 94°C for 9 min, 32 cycles of amplification with 45 s of denaturation at 94°C, 45 s of annealing at 50°C, and 90 s of elongation at 72°C. Before sequencing, the product was cleaned by using the Qiagen PCR Purification Kit, according to the manufacturer's protocol. Sequencing was performed on an MJ Research PTC-200 thermocycler with the Dye Primer Cycle Sequencing Ready Reaction Kit (PE Applied Biosystems), according to the manufacturer's protocol. Sequence reactions were separated on a 373A DNA sequencer (ABI), using a 7% denaturing polyacrylamide gel. Data were analyzed by the 373A data analysis program (ABI).
Results
Families
Screening for somatic mosaicism was performed in a total of 61 families that included members who had sporadic severe hemophilia A; 32 families had point mutations, 13 families had small deletions or insertions, and 16 families had the intron 22 inversions (table 3).
Table 3.
Families, Mutations, and Origin of Mutation[Note]
| Mutation Type and Patient Number | Family | Exon | Mutation | NucleotidePosition | Type of Mutationa | MutationOrigin |
| Point mutations: | ||||||
| 1 | LU3 | 2 | 266G→A | Gly70Asp | Non-CpG transition | M |
| 2 | LÜP2 | 3 | 296T→A | Val80Asp | Transversion | MGM |
| 3 | LA30 | 3 | 301G→C | Asp82His | Transversion | MGF |
| 4 | LA27 | 4 | 403G→T | Asp116Tyr | Transversion | M |
| 5 | ROP1 | 7 | 980T→C | Leu308Pro | Non-CpG transition | MGM |
| 6 | TRP1 | 8 | 1226A→G | Glu390Gly | Non-CpG transition | M |
| 7 | LU6 | 9 | 1394C→G | Ser446Stop | Transversion | M |
| 8 | JO7 | 10 | 1492G→A | Gly479Arg | CpG transition | M |
| 9 | LA37 | 12 | 1760C→A | Ser568Stop | Transversion | M |
| 10 | LA6 | 12 | 1809C→G | Ser584Arg | Transversion | MGM |
| 11 | LA15 | 13 | 1909A→G | Asn618Asp | Non-CpG transition | MGM |
| 12 | JO1 | 14 | 2440C→T | Arg795Stop | CpG transition | M |
| 13 | LA40 | 14 | 2440C→T | Arg795Stop | CpG transition | M |
| 14 | EGL1 | 14 | 3143G→A | Trp1029Stop | Non-CpG transition | M |
| 15 | JO5 | 14 | 3381G→A | Trp1108Stop | Non-CpG transition | M |
| 16 | PE1 | 14 | 4796G→A | Trp1580Stop | Non-CpG transition | M |
| 17 | PE3 | 14 | 4796G→A | Trp1508Stop | Non-CpG transition | MGM |
| 18b | LA3 | 16 | 5452G→T | Gly1799Stop | Transversion | M |
| 19 | EGL2 | 17 | 5681A→G | Glu1875Gly | Non-CpG transition | M |
| 20 | PE4 | 20 | 6134G→T | Gly2026Val | Transversion | MGF |
| 21 | JO8 | 21 | 6200C→T | Pro2048Leu | Non-CpG transition | M |
| 22 | JO19 | 23 | 6496C→T | Arg2147Stop | CpG transition | MGF |
| 23 | JO10 | 23 | 6515C→G | Pro2153Arg | Transversion | M |
| 24 | LA28 | 23 | 6544C→T | Arg2163Cys | CpG transition | MGM |
| 25 | LA38 | 23 | 6554T→C | Leu2166Ser | Non-CpG transition | M |
| 26 | LA5 | 24 | 6682C→T | Arg2209Stop | CpG transition | M |
| 27 | LA10 | 24 | 6682C→T | Arg2209Stop | CpG transition | M |
| 28 | PE2 | 25 | 6836T→G | Phe2260Cys | Transversion | MGF |
| 29 | LA14 | 26 | 6956C→T | Pro2300Leu | CpG transition | MGM |
| 30 | SCP1 | 26 | 6977G→T | Arg2307Leu | Transversion | MGF |
| 31 | RHP1 | 26 | 6977G→T | Arg2307Leu | Transversion | M |
| 32 | LA29 | 26 | 6977G→C | Arg2307Pro | Transversion | M |
| Small deletions and insertions: | ||||||
| 33 | JO9 | 2 | 202-207 delACTCTG | Thr49 | In frame | M |
| 34 | LA41 | 2 | 209-212 delTTGT | Cys51 | Frameshift | M |
| 35 | LU9 | 9 | 1441 ins-A | Trp462 | Frameshift | MGM |
| 36 | LA18 | 9 | 1443 ins-TG | Trp462 | Frameshift | MGF |
| 37c | LA39 | 11 | 1750 delC | Gln565 | Frameshift | M/F |
| 38b | REP1 | 13 | 1941-1944 delAGTT | Ser628 | Frameshift | M |
| 39 | LU5 | 14 | 2739 ins-T | Trp894 | Frameshift | M |
| 40 | LU7 | 14 | 3637 delA | Lys1194 | Frameshift | M |
| 41 | MAB1 | 14 | 3637 delA | Lys1194 | Frameshift | M |
| 42 | LA11 | 14 | 3637 delA | Lys1194 | Frameshift | M |
| 43 | LA16 | 14 | 4264+4265 delTA | Tyr1403 | Frameshift | M |
| 44 | LOP1 | 18 | 5961 delA | Lys1968 | Frameshift | M |
| 45c | JO4 | 25 | 6876+6877 delCT | Leu273 | Frameshift | F |
| Inversions: | ||||||
| 46 | BII1 | Distal | MGF | |||
| 47 | JO11 | Proximal | MGF | |||
| 48 | JO17 | Distal | MGF | |||
| 49 | JO18 | Proximal | MGGF | |||
| 50 | LA13 | Distal | MGF | |||
| 51 | LA17 | Distal | MGM | |||
| 52 | LA21 | Distal | M | |||
| 53 | LA31 | Proximal | MGF | |||
| 54 | LA32 | Proximal | MGF | |||
| 55 | LU1 | Distal | M | |||
| 56 | LU2 | Distal | M | |||
| 57 | LU4 | Distal | M | |||
| 58 | LU8 | Distal | MGM | |||
| 59 | OSI1 | Distal | M | |||
| 60 | PE5 | Distal | MGF | |||
| 61 | WEI1 | Distal | MGF |
Note.— Data for mosaic individuals are underlined.
M = mother; F = father; MGM = maternal grandmother; MGF = maternal grandfather; MGGF = maternal great grandfather.
Other methods were used for examination.
Female with hemophilia A.
DNA or blood samples were provided by five centers from Germany (24 families), France (23 families), Sweden (9 families), and the United Kingdom (5 families) (see Subjects, Material, and Methods). The families fulfilled the following criteria: (1) presence of apparently sporadic hemophilia, (2) successful characterization of the mutation, (3) availability of blood or DNA samples from the parental and grandparental generation, and (4) determination of the mutation origin by mutation analysis and haplotyping of the patients' mothers and maternal grandparents. The origin of the de novo mutation was assigned to the patient’s mother in 34 families and was assigned to the father in a single family with a female index patient. In another family with a female index patient, the mutation was assigned to one of the patient’s parents (although it was unclear whether it originated in the mother or the father); in 10 families it was assigned to the maternal grandmother, in 14 families to the maternal grandfather, and in 1 family to the maternal great-grandfather (table 4).
Table 4.
Origin of FVIII Mutation Types and Allocation of the Somatic Mosaics[Note]
|
Point Mutations |
|||||||
| Variable | All | CpG Transitions | Non-CpGTransitions | Transversions | SmallDeletions/Insertions | Intron 22 Inversion | Overall |
| Mutation | 32 | 8 | 11 | 13 | 13 | 16 | 61 |
| Mosaic | 8 | 4 | 1 | 3 | … | … | … |
| Mutation origin: | |||||||
| M | 20 (5) | 5 (2) | 8 | 7 (3) | 9 | 5 | 34 |
| F | … | … | … | … | 1 | … | 1 |
| M/F | … | … | … | … | 1 | … | 1 |
| MGM | 7 (3) | 2 (2) | 3 (1) | 2 | 1 | 2 | 10 |
| MGF | 5 | 1 | 4 | 1 | 8 | 14 | |
| MGGF | … | … | … | … | … | 1 | 1 |
| % Mosaics | 25 | 50 | 9 | 23 | … | … | 13 |
Note.— Numbers of mosaic subjects are shown in parentheses.
Detection of Somatic Mosaics
Leukocyte DNA from family members who were identified as the origin of a de novo mutation by routine mutation analysis and haplotyping were investigated by a sensitive allele-specific PCR for the presence of the sequence alteration. The intron 22 inversion was tested by a long-range PCR (Liu et al. 1998). Evidence of somatic mosaicism was found in 8 (13%) of the 61 families with hemophilia A, and the proportion of the mutated allele was 0.2%–25% (figure 1). Each somatic mosaic was confirmed by control experiments that included enrichment and sequence analysis of the mutated alleles.
Figure 1.

Pedigrees of the eight families in which a mosaicism was detected. Blackened symbols indicate the patients with hemophilia; white circles with dots show carriers of the mutation. Those carriers representing mosaic individuals are indicated by the percentage number that corresponds to the proportion of the mutated allele. Values for factor VIII:C and vWF:Ag are shown for those mosaic individuals in whom the information was available. The question mark in family LA28 indicates that it is unknown whether the twins are of monozygotic or dizygotic origin.
All mosaics were detected in families that had point mutations, which suggests a 25% mosaicism rate in families with this type of mutation. In the subgroup of C:G to T:A transitions at a CpG dinucleotide, 50% (4 of 8 families) had evidence of somatic mosaicism. No evidence of mosaicism was obtained in any of the other mutation types, which may result partly from the low numbers of families and partly from differences in the sensitivity of the methods applied. Although the sensitivity of the allele-specific PCR for point mutations was as great as 0.1% in a wild-type background, it decreased to 2%–5% for small deletions/insertions. The sensitivity of the long-range intron 22 inversion PCR was ∼5%. Dilution experiments with the corresponding DNAs of the probands were carried out as positive controls for the PCR performance and for the estimation of the amount of the mutation in the tested individuals. Interestingly, all somatic mosaicisms emerged in women (five patient’s mothers and three patient's grandmothers), although the mutation origin could be assigned to men in 16 (28%) families (table 4).
Detailed descriptions of representative mosaics are given in figures 2, 3, and 4. The index patient of family JO1 had a severe hemophilia A phenotype that was caused by a CpG transition (2440C→T) in exon 14 leading to a stop codon (table 3). The patient's mother was shown, by sequence analysis, to be a noncarrier of the mutation. Allele-specific PCR clearly detected the mutant allele in the patient’s mother at a proportion that corresponded to a 1:200 dilution of the patient’s DNA (fig. 2A). Enrichment and sequence analysis of the mutated allele clearly demonstrated its presence in the patient’s mother (fig. 2B). The maximal sensitivity of the allele-specific PCR for the 2440C→T mutation was shown to be >1:500, corresponding to a sensitivity of <0.2%.
Figure 2.

A, Graph of results of electrophoresis of the allele-specific PCR of the index patient and the mosaic individual of family JO1. The upper three traces show the strength of the mutated allele in the index patient for different dilutions of the allele-specific PCR product. The mutation can be clearly detected in the trace of the 1:500 dilution. The mosaic individual (trace 4) showed a strength of the allele-specific PCR product comparable to a 1:200 dilution (trace 2), thus corresponding to a proportion of 0.5% of the mutated allele. The last two traces represent negative control samples. B, Nucleotide sequence of the reverse strand after mutation enrichment. Results clearly indicate the presence of the mutated allele.
Figure 3.

A, Graph of results of electrophoresis of the allele-specific PCR of the index patient and the mosaic individual of family LA14. The upper four traces show the strength of the mutated allele in the index patient for different dilutions of the allele-specific PCR product. The mutation can be clearly detected in the trace of the 1:200 dilution (trace 3). The mosaic individual (trace 5) showed a strength of the allele-specific PCR product comparable to a 1:20 dilution (trace 2), thus corresponding to a proportion of 5% of the mutated allele. The last trace represents a negative control. B, Nucleotide sequence after mutation enrichment. Results clearly indicate the presence of the mutated allele.
Figure 4.

A, Autoradiograph of the DNA sequence of the mosaic individual of family LA3. DNA sequences of both strands are shown (left panel shows forward sequences; right panel shows reverse sequences). Arrows indicate the mosaic mutation in the mother of the patient (left lane) in comparison with a wild-type control (right lane). B, the sequence traces of the mutated allele before mutation enrichment. C, the sequence traces of the mutated allele after mutation enrichment. Although the mutated nucleotide is not visible in B, it could be clearly identified in C, thus demonstrating that (1) standard automated sequencing analysis is not able to reveal the presence of a mosaicism and (2) that it is less sensitive than the classic radioactive sequencing protocol.
In family LA14, the index patient had severe hemophilia A as a result of a CpG transition (6956C→T) in exon 26 leading to the missense mutation Pro2300Leu (table 3. His mother and aunt were carriers of the mutation. The affected X chromosome was inherited by the grandmother, who did not show the mutation when tested by sequence analysis. Allele-specific PCR revealed that the mutated allele was present in the patient’s grandmother at a proportion of 5%, corresponding to a 1:20 dilution of the patient’s DNA (fig. 3A). Enrichment of the mutated allele clearly showed the presence of the mutation in the grandmother’s leukocyte DNA (fig. 3B). The sensitivity of the allele-specific PCR for the 6956C→T mutation was shown to be >1:200 or <0.5%.
When the proportion of mutated alleles is >20% (families RHP1 and LA3, fig. 1), the allele-specific PCR could not distinguish between mosaicism and normal carrier status. However, at this degree of mosaicism, classical sequence analysis indicated the presence of mosaicism, as was the case for families LA3 and RHP1. The sequence autoradiograph of the patient’s mother in family LA3 is shown in figure 4A. On the basis of the densities of the corresponding signals, the proportion of the mutated allele was estimated to be ∼20%. Notably, the mutated allele was suppressed when sequenced on an ABI system (fig. 4B). The only indirect sign for the presence of mosaicism was a lower peak of the corresponding nucleotide. Mutation enrichment clearly showed the presence of the mutated allele (fig. 4C).
In the present study, allele-specific PCR proved to be a sensitive method for the detection of mosaics. The proportion of mosaic cells was estimated by a semiquantitative approach of diluting the DNA of the corresponding index patients. The degree of dilution that showed a strength of the allele-specific PCR product similar to that of the mosaic individual was taken as a measure of the proportion of mosaic cells. The results of the semiquantitative estimate by allele-specific PCR were supported by manual sequencing autoradiographs from six of the eight families, with varying degrees of mosaicism (0.2%–25%) (fig. 5). The sensitivity of two- to threefold overexposed autoradiographs corresponds to a detection limit of 5% for the proportion of mosaic cells, when compared with the allele-specific PCR. Higher proportions of mosaic cells showed an increased density of the signal of the mutated allele, whereas no signal of the mutated allele could be shown in the autoradiographs for the mosaicisms of 2% and lower degrees. Thus, the data obtained by overexposed manual sequencing autoradiographs correspond to the results from the allele-specific PCR. However, the values given by the allele-specific PCR should not be regarded as exact numbers but, because of the limitations and semiquantitative approach of allele-specific PCR, as an estimate of the order of mosaic cells.
Figure 5.
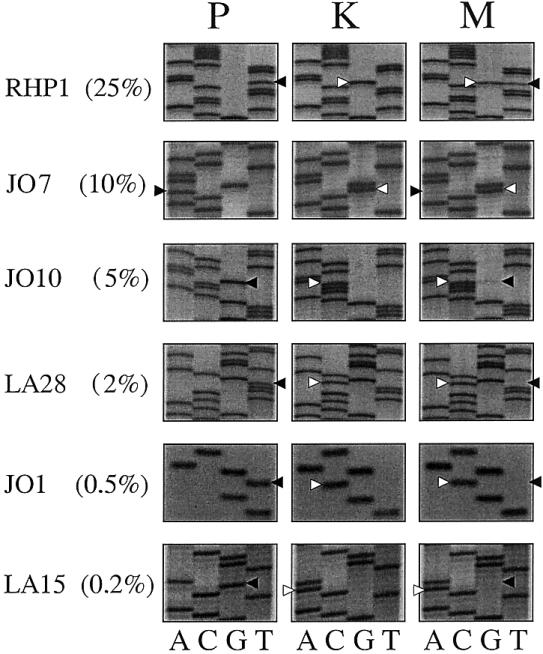
Overexposed manual sequencing autoradiographs for six families, with the percentage of the mutated alleles (obtained by allele-specific PCR ) given in parentheses. From each family, the autoradiograph of the patient (P), a healthy control individual (K), and the mosaic individual (M) are shown. Black arrows indicate the mutated alleles, and white arrows indicate and the healthy alleles. Autoradiographs were exposed for 3–4 d. Although a strong signal for the mutated allele is found in family RHP1, only faint signals are shown in families JO7 and JO10. No signals could be seen in families LA28, JO1, or LA15, which indicated a lower detection limit of 5% of the mutated allele when manual sequencing was performed.
Discussion
Frequency of Mosaicism in Hemophilia A
In the present study a systematic search for mosaicism was performed in 61 families with sporadic hemophilia A. The search revealed 8 (13%) mutations that originated as somatic mosaicism. This high number was not anticipated, because, in spite of extensive mutation analysis (Antonarakis et al. 1995; Kemball-Cook et al. 1998), only seven mosaics have been reported (Higuchi et al. 1988; Bröker-Vriends et al. 1990; Levinson et al 1990; Schwaab et al. 1993; Casey et al. 1999; Oldenburg et al. 2000). Studies that systematically addressed the question of mosaicism in other hereditary diseases reported proportions of 10%–20% mosaicism in retinoblastoma (Sippel et al. 1998), Duchenne muscular dystrophy (Passos-Bueno et al. 1992; van Essen et al. 1992), tuberosis sclerosis complex (Verhoef et al. 1999), and hemophilia B (Ketterling et al. 1999). Thus, somatic mosaicism may represent a fairly common event in human hereditary diseases.
Influence of the Mutation Type
In our study, all mosaics were detected in families with point mutations. This finding could be the result of varying sensitivities of the methods used to detect mosaicism but may also indicate that mosaicism arises preferentially with certain types of mutations. Point mutations or small deletions or insertions are thought to originate during mitotic cell divisions and therefore may lead to somatic mosaicism during early embryogenesis. Somatic mosaicism in these two mutation types, as well as in large deletions, had been anecdotally reported in families with hemophilia A (Higuchi et al. 1988; Bröker-Vriends et al. 1990; Levinson et al. 1990; Schwaab et al. 1993; Casey et al. 1999). Except for the mosaic of a small deletion detected recently by sequencing (Casey et al. 1999), all mosaics reported elsewhere were found by Southern blot analysis, the main tool for mutation detection in the late 1980s. Three of these mosaics were large deletions, which may reflect the fact that Southern blot analysis was mainly applied to the study of this mutation type and of point mutations that affect TaqI restriction sites. In the present study, no families with large deletions were included, probably because Southern blot analysis has became less popular for the detection of large deletions in females at risk of carrying the mutation.
The intron 22 inversion, which is caused by intrachromosomal recombination among the homologous regions of intron 22 (Lakich et al. 1993), is thought to be almost exclusively of meiotic origin, arising predominantly in male germ cells (Rossiter et al. 1994; Becker et al. 1996). This pathogenic mechanism would argue against a somatic origin of an intron 22 inversion during early embryogenesis; however, one instance of mosaicism with this mutation type has recently been observed (Oldenburg et al. 2000).
In other disorders that have been systematically analyzed, somatic mosaicism was frequently found in subjects with large deletions (Passos-Bueno et al. 1992; van Essen et al. 1992) and point mutations (Ketterling et al. 1999). Therefore, a considerable proportion of somatic mosaics might be expected for most mutation types.
In the subgroup of CpG transitions, four of eight families showed evidence of mosaicism. This finding suggests that CpG sites may be especially prone to mosaic mutations. Recently, it was reported that human mature germ cells show a high level of methylation (El-Maarri et al. 1998). Studies of mouse embryos revealed a global demethylation of DNA after the formation of the zygote and during the first cell divisions, which lead to the early blastocyst stage (Buehr 1997; Drost and Lee 1998). One possible mechanism of the global demethylation is the excision of the 5-methylated cytosine nucleotide. The large number of resulting single-strand breaks may overwhelm the capacity of downstream repair enzymes and subsequently lead to the manifestation of mutations at CpG sites during early embryogenesis (Chu and Mayne 1996; Schmutte and Jones 1998). This hypothesis is at variance with a study by Ketterling et al. (1999) of families with hemophilia B, which found no somatic mosaicism for transitions at CpG sites but, instead, found four somatic mosaics for C→T transitions at non-CpG dinucleotides. However, two of these four somatic mosaics occurred at CpNpG trinucleotides. This motif is also known to become methylated in mammalian cells (Clark et al. 1995). Nevertheless, the number of reported somatic mosaics is still too small to allow us to draw any final conclusion about this aspect of its genesis.
Female and Male Origin of Somatic Mosaicism
All somatic mosaicisms in our study were observed in females. This finding may be the result of a selection bias among our families. In only 4 (12.5%) of 32 families with point mutations, the genetic defect originated in males; however, it was derived from females in 28 (87.5%) families. On the basis of studies that showed a 5:1 male:female ratio for de novo point mutations in hemophilia A (Becker et al. 1996), the opposite proportions of mutation origins would be expected. Selection bias probably occurs because blood samples are more likely to be available from patients' mothers than from the patients' grandparents. Although the difference is not statistically significant, it is of note that somatic mosaicism was found in 8 of 28 females but in 0 of 5 males with a de novo point mutation. A less frequent occurrence of mosaicism in males would agree with the hypothesis that the higher mutation rate among males is mainly the result of ongoing cell divisions during male spermatogenesis, which results in a 15–20-fold higher number of cell divisions in the male than in the female germline before fertilization of an oocyte (Vogel and Motulsky 1986). On the basis of this assumption, the proportion of mosaicism originating in males is expected to be the inverse of the male:female ratio of mutation rates, resulting in a proportion of mosaicism that is approximately fivefold lower in males than in females.
Influence of the Method Applied
In the present study we used an allele-specific PCR–based attempt for point mutations and small deletions/insertions. For each mutation the sensitivity of the method was determined by dilution experiments with DNA from the index patient. Comparison of the signal intensity of the carrier of the mosaic with those of various dilutions of the patient’s DNA allowed a semiquantitative assessment of the degree of mosaicism. For point mutations, this approach allowed the detection of 0.1%–0.2% of mutated alleles against a wild-type background, which represents a ratio of 1:500 to 1:1000, which is similar to the sensitivity reported by Knöll et al. (1996) for allele-specific PCR. The presence of the mutated allele was confirmed by mutation-enrichment experiments, as indicated in the Subjects, Material, and Methods section. Although this attempt was highly efficient at very low degrees of mosaicism, it failed to discriminate between mosaicism and carrier status when the proportion of mutated alleles was >20%. When the percentage of mutated alleles is high, the conventional methods (Southern blot analysis and sequencing) are likely to detect the presence of mosaicism (fig. 3). The results obtained by allele-specific PCR were supported by overexposed manual sequencing autoradiographs (fig. 5), which detected the mutant allele at proportions of ⩾5%.
The sensitivity of allele-specific PCR decreased by at least one order of magnitude for small deletions/insertions. Here, the sensitivity was, at best, at 2% of mutated alleles in a wild-type background. It even decreased to 5% when the small deletion/insertion was located within a poly-A stretch (e.g., codons 1192-1194) that is known to represent a mutation hotspot in hemophilia A. Also, the long-range PCR for testing the presence of an intron 22 inversion showed a detection limit of ∼5% of mutant alleles.
When we consider the limited sensitivities of the methods applied for the detection of mosaicism in at least some of the mutation types, together with the fact that mosaicism that affects nearly 50% of the alleles cannot be distinguished from carrier status, it seems likely that some of the somatic mosaics remain undetected and that the true proportion of mosaicism will be even higher than the 13% found in our study.
Ascertainment Bias
As already mentioned, male mutation origin is underrepresented in our cohort of families because of the difficulties of assessing blood samples from the patients' grandparents. Also, the distribution of mutation types is not representative of severe hemophilia A. Becker et al. (1996) found that 37.5% of patients had intron 22 inversions and 32% had point mutations, whereas in our cohort the intron 22 inversions accounted for 26.2% of the index patients, and point mutations accounted for 52.5%. The different mutation profiles may result from the multicenter character of this study. Both types of bias—the underrepresentation of male mutation origin and of the intron 22 inversion—would lead to an overestimation of the proportion of mosaic individuals, because, in both male mutation origin and intron 22 inversion, a higher proportion of germ cell mutations would be expected.
Somatic Mosaicism and Implications for Risk Assessment in Genetic Counseling
In the mouse model, the blastocyst stage consists of the inner cell mass (ICM) and the trophoblast (fig. 6). The ICM develops into the primary ectoderm and the primary endoderm. Only the primary ectoderm (epiblast) that generates the embryonic body retains the capacity to form germ cells (Wylie 1999). An early mutation (before the blastocyst stage) may appear as a complete mutant because, at this stage, very few cells are destined to form the embryonic body, thus masking a somatic origin of the mutation. Mutations occurring at a later stage of embryogenesis will appear as somatic mosaics, with the proportion of mutated alleles depending on the time and location of the mutational event.
Figure 6.

Detailed description of the developmental processes during early embryogenesis in mammals, on a time axis.
A question of special interest in the study of somatic mosaicism is the proportion of germ cells that are affected by the mutation. According to Soriano and Jaenisch (1986), in mice at least three cells, which are allocated before the onset of the somatic cell lineages, are thought to give rise to the germline. In humans, Cohn et al. (1990) estimated—by comparing different proportions of mosaic mutations in sperm and tissue DNA in a family affected with osteogenesis imperfecta—that four progenitor cells make up the germline. The idea that the germline has a limited number of progenitor cells would have a great impact on genetic counseling. Thus, in a somatic mosaic individual, the number of mutant alleles in the germ cells will not decrease below a certain proportion. This lower threshold would be one in eight, when eight monozygotic cells originating from the four putative progenitor cells during the first meiotic cell division are considered. In our study, a considerable recurrence risk is indicated by two of the eight somatic mosaics in whom the mutation had been transmitted to two descendants, in spite of low proportions of the mutated allele (4% and 10%, respectively) in leukocyte DNA (families LA14 and JO7; fig. 1). Family LA28 would represent a third family if the twins were dizygotic; however, the status is not known to the authors). These findings suggest a higher proportion of the defective allele in the germ cells of the mosaic individuals. A high proportion of mutant alleles in germ cells is also suggested by studies of Duchenne muscular dystrophy, which estimate a risk of 7%–11% of transmitting the mutant allele to the first generation of offspring, although the mutation was not detected in the leukocyte DNA of the mosaic individual (Bakker et al. 1989; van Essen et al. 1992).
The occurrence of somatic and germ cell mosaicism represents a matter of concern to the genetic counselor, because it causes uncertainty about the recurrence risk in parents who appear to be noncarriers. Even direct determination of the underlying genetic defect will not detect mosaicism during routine diagnostic procedures. Because past reports of somatic mosaicism in hemophilia A were essentially anecdotal, most genetic counselors may have neglected this problem. Our findings clearly demonstrate the need to consider the potential risk of somatic mosaicism in families with sporadic hemophilia A, especially in those with point mutations.
Acknowledgments
This study was supported by Deutsche Forschungsgemeinschaft grant OL 21/18-2, Swedish grant MFR 13493, and grants from the Stiftung Hämotherapie-Forschung and Baxter Hyland Immuno (to J.O.).
Electronic-Database Information
The accession number and URL for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for HEMA [MIM 306700])
References
- Antonarakis SE, Rossiter JP, Young M, Horst J, de Moerloose P, Sommer SS, Ketterling RP, et al (1995) Factor VIII gene inversions in severe hemophilia A: results of an international consortium study. Blood 86:2206–2212 [PubMed] [Google Scholar]
- Bakker E, Veenema H, Den Dunnen JT, Van Broeckhoven C, Grootscholten PM, Bonten EJ, Van Ommen GJB, Pearson PL (1989) Germinal mosaicism increases the recurrence risk for “new” Duchenne muscular dystrophy mutations. J Med Genet 26:553–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker J, Schwaab R, Möller-Taube A, Schwaab U, Schmidt W, Brackmann HH, Grimm T, Olek K, Oldenburg J (1996) Characterization of the factor VIII defect in 147 patients with sporadic hemophilia A: family studies indicate a mutation type–dependent sex ratio of mutation frequencies. Am J Hum Genet 58:657–670 [PMC free article] [PubMed] [Google Scholar]
- Bröcker-Vriends AHJT, Briet E, Dreesen JCFM, Bakker B, Reitsma P, Pannekoek H, van de Kamp JJP, Pearson PL (1990) Somatic origin of inherited haemophilia A. Hum Genet 85:288–292 [DOI] [PubMed] [Google Scholar]
- Buehr M (1997) The primordial germ cells of mammals: some current perspectives. Exp Cell Res 232:194–207 [DOI] [PubMed] [Google Scholar]
- Casey GJ, Rodgers SE, Hall JR, Rudzki Z, Lloyd JV (1999) Grandpaternal mosaicism in a family with isolated haemophilia A. Br J Haematol 107:560–562 [DOI] [PubMed] [Google Scholar]
- Chu G, Mayne L (1996) Xeroderma pigmentosum, Cockeyne syndrome and trichothi-dystrophy: do these genes explain the disease? Trends Genet 12:187–192 [DOI] [PubMed] [Google Scholar]
- Clark SJ, Harrison J, Frommer M (1995) CpNpG methylation in mammalian cells. Nat Genet 10:20–27 [DOI] [PubMed] [Google Scholar]
- Cohn DH, Starman BJ, Blumberg B, Byers PH (1990) Recurrence of lethal osteogenesis imperfecta due to parental mosaicism for a dominant mutation in a human type I collagen gene (COLIAI). Am J Hum Genet 46:591–601 [PMC free article] [PubMed] [Google Scholar]
- Colman SD, Rasmussen SA, Ho VT, Abernathy CR, Wallace MR (1996) Somatic mosaicism in a patient with neurofibromatosis type 1. Am J Hum Genet 58:484–490 [PMC free article] [PubMed] [Google Scholar]
- Drost JB, Lee WR (1998) The developmental basis for germline mosaicism in mouse and Drosophila melanogaster. Genetica 102/103:421–443 [PubMed] [Google Scholar]
- El-Maarri O, Olek A, Balaban B, Montag M, van der Ven H, Urman B, Olek K, Caglayan SH, Walter J, Oldenburg J (1998) Methylation levels at selected CpG sites in the factor VIII and FGFR3 genes, in mature female and male germ cells: implications for male-driven evolution. Am J Hum Genet 63:1001–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froissart R, Maire I, Bonnet V, Levade T, Bozon D (1997) Germline and somatic mosaicism in a female carrier of Hunter disease. J Med Genet 34:137–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hezard N, Cornillet P, Droulle C, Gillot L, Potron G, Nguyen P (1997) Factor V Leiden: detection in whole blood by ASA PCR using an additional mismatch in antepenultimate position. Thromb Res 88:59–66 [DOI] [PubMed] [Google Scholar]
- Higuchi M, Kochhan L, Olek K (1988) A somatic mosaic detected at the DNA level. Mol Biol Med 5:23–27 [PubMed] [Google Scholar]
- Kemball-Cook G, Tuddenham EGD, Wacey AI (1998) The factor VIII structure and mutation resource site: HAMSReS version 4. Nucleic Acids Res 26:216–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketterling RP, Vielhaber E, Li X, Drost J, Schaid DJ, Kasper CK, Phillips JA 3d, Koerper MA, Kim H, Sexauer C, Gruppo R, Ambriz R, Paredes R, Sommer SS (1999) Germline origins in the human F9 gene: frequent G:C—A:T mosaicism and increased mutations with advanced maternal age. Hum Genet 105:629–640 [DOI] [PubMed] [Google Scholar]
- Knöll A, Ketterling RP, Sommer SS (1996) Absence of somatic mosaicism in 17 families with hemophilia B: an analysis with a sensitivity 10- to 1000-fold greater than that of sequencing gels. Hum Genet 98:539–545 [DOI] [PubMed] [Google Scholar]
- Lakich D, Kazazian HH, Antonarakis SE, Gitschier J (1993) Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet 5:236–241 [DOI] [PubMed] [Google Scholar]
- Lavergne JM, Bahnak BR, Vidaud M, Laurian Y, Meyer D (1992) A direct search for mutations in hemophilia A using restriction enzyme analysis ADN denaturing gel electrophoresis. A study of seven exons of the factor VIII gene of 170 cases. Nouv Rev Fr Hematol 34:85–91 [PubMed] [Google Scholar]
- Levinson B, Lehesjoki AE, de la Chapelle A, Gitschier J (1990) Molecular analysis of hemophilia A mutations in the Finnish population. Am J Hum Genet 46:53–62 [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Nozari G, Sommer SS (1998) Single-tube polymerase chain reaction for rapid diagnosis of the inversion hotspot of mutation in hemophilia A. Blood 92:1458–1459 [PubMed] [Google Scholar]
- Ljung RC, Sjorin E (1999) Origin of mutation in sporadic cases of haemophilia A. Br J Haematol 106:870–874 [DOI] [PubMed] [Google Scholar]
- Lund AM, Schwartz M, Skovby F (1996) Genetic counselling and prenatal diagnosis of osteogenesis imperfecta caused by paternal mosaicism. Prenatal Diagnosis 16:1032–1038 [DOI] [PubMed] [Google Scholar]
- Oldenburg J, Rost S, El-Maarri O, Leuer M, Olek K, Müller CR, Schwaab R (2000) De novo factor VIII intron 22 inversion in a female carrier presents as a somatic mosaicism. Blood 96:2905–2906 [PubMed] [Google Scholar]
- Passos-Bueno MR, Bakker E, Kneppers AL, Takata RI, Rapaport D, den Dunnen JT, Zatz M, van Ommen PJ (1992) Different mosaicism frequencies for proximal and distal Duchenne muscular dystrophy (DMD) mutations indicate difference in etiology and recurrence risk. Am J Hum Genet 51:1150–1155 [PMC free article] [PubMed] [Google Scholar]
- Rossiter JP, Young M, Kimberland ML, Hutter P, Ketterling RP, Gitschier J, Horst J, Morris MA, Schaid DJ, deMoerloose P, Sommer SS, Kazazian HH, Antonarakis SE (1994) Factor VIII gene inversions causing severe hemophilia A originate almost exclusively in male germ cells. Hum Mol Genet 3:1035–1039 [DOI] [PubMed] [Google Scholar]
- Schmutte C, Jones PA (1998) Involvement of DNA methylation in human carcinogenesis. Biol Chem 379:377–388 [DOI] [PubMed] [Google Scholar]
- Schwaab R, Oldenburg J, Lalloz MRF, Schwaab U, Pemberton S, Hanfland P, Brackmann HH, Tuddenham EG, Michaelides K (1997) Factor VIII gene mutations found by a comparative study of SSCP, DGGE and CMC and their analysis on a molecular model of factor VIII protein. Hum Genet 101:323–332 [DOI] [PubMed] [Google Scholar]
- Schwaab R, Oldenburg J, Tuddenham EG, Brackmann HH, Olek K (1993) Mutations in haemophilia A. Br J Haematol 83:450–458 [DOI] [PubMed] [Google Scholar]
- Sippel KC, Fraioli RE, Smith GD, Schalkoff ME, Sutherland J, Gallie BL, Dryja TP (1998) Frequency of somatic and germline mosaicism in retinoblastoma: implications for genetic counseling. Am J Hum Genet 62:610–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P, Jaenisch R (1986) Retroviruses as probes for mammalian development: allocation of cells to the somatic and germ cell lineages. Cell 46:19–29 [DOI] [PubMed] [Google Scholar]
- Strauss HS (1967) The perpetuation of hemophilia by mutation. Pediatrics 39:186–193 [PubMed] [Google Scholar]
- Tavassoli K, Eigel A, Wilke K, Pollmann H, Horst J (1998) Molecular diagnostics of 15 hemophilia A patients: characterization of eight novel mutations in the factor VIII gene, two of which result in exon skipping. Hum Mutat 12:301–303 [DOI] [PubMed] [Google Scholar]
- van Essen AJ, Abbs S, Baiget M, Bakker E, Boileau C, van Broeckhoven C, Bushby K, et al (1992) Parental origin and germline mosaicism of deletions and duplications of the dystrophin gene: a European study. Hum Genet 88:249–257 [DOI] [PubMed] [Google Scholar]
- Verhoef S, Bakker L, Tempelaars AM, Hesseling-Janssen AL, Mazurczak T, Jozwiak S, Fois A, Bartalini G, Zonnenberg BA, van Essen AJ, Lindhout D, Halley DJ, van den Ouweland AM (1999) High rate of mosaicism in tuberosis sclerosis complex. Am J Hum Genet 64:1632–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel F (1977) A probable sex difference in some mutation rates. Am J Hum Genet 29:312–319 [PMC free article] [PubMed] [Google Scholar]
- Vogel F, Motulsky AG (1986) Population genetics. In: Vogel F, Motulsky AG (eds) Human genetics. Springer, Berlin, pp 301–304 [Google Scholar]
- Williams IJ, Abuzenadah A, Winship PR, Preston FE, Dolan G, Wright J, Peake IR, Goodeve AC (1998) Precise carrier diagnosis in families with haemophilia A: use of conformation sensitive gel electrophoresis for mutation screening and polymorphism analysis. Thromb Haemost 79:723–726 [PubMed] [Google Scholar]
- Wylie C (1999) Germ cells. Cell 96:165–174 [DOI] [PubMed] [Google Scholar]