

Abstract
Purpose:
Synaptic communication deficits are central to many neurodevelopmental disorders. However, for rare monogenic conditions, these disorders remain poorly defined, with limited understanding of their molecular etiology. A homozygous frameshift variant in the synaptic cell adhesion molecule ELFN1 was reported in a family with three affected siblings with epileptic encephalopathy, alongside a missense variant of uncertain significance in a cohort study involving a family with intellectual disability. Therefore, we sought to evaluate the role and mechanism of biallelic ELFN1 variants in disease pathogenesis.
Methods:
We describe eight newly identified individuals from five unrelated families, all carrying homozygous ELFN1 variants, including frameshift and in-frame deletions. By integrating data from these cases with clinical details from six previously reported individuals, we delineate the phenotypic spectrum associated with ELFN1 variants.
Results:
Clinical features include varying degrees of developmental delay/intellectual disability, epilepsy, and movement disorders. Molecular investigations reveal that these variants disrupt ELFN1 protein trafficking to the cell surface, resulting in loss of function. Functional modeling in mice and zebrafish demonstrates the role of Elfn1 loss in motor activity abnormalities and seizures.
Conclusion:
Our findings establish ELFN1 deficiency as the cause of a distinct, rare neurodevelopmental disorder, providing a foundation for future investigations into its pathophysiology and therapeutic strategies.
Keywords: ELFN1, synaptic adhesion protein, neurodevelopmental disorder, epilepsy, movement disorder
Graphical Abstract

Introduction:
Synaptic transmission is fundamental to nervous system function. It involves a staggering number of molecular elements that enable formation of physical contacts, neurotransmitter release and its reception.1-3 The process of synaptic transmission is critically tuned by many incompletely understood molecular networks that make synapses plastic and enable scaling of the transmission strength on demand.4,5 Deficits in these processes invariably lead to a range of neuropsychiatric conditions.6,7 However, the genetic etiologies of many nervous system disorders remain poorly understood.
Glutamate is the major excitatory synaptic neurotransmitter. In addition to ion channel receptors, its actions are mediated by the metabotropic G protein coupled receptors (mGlu) that either serve as direct transmitter receptors when localized post-synaptically or autoreceptors that control the extent of glutamate release at the pre-synaptic sites.8 Recent studies have shown that synaptic localization of group III mGlu receptors is mediated by a cell adhesion molecule ELFN1 (extracellular leucine-rich repeat and fibronectin type III domain-containing protein 1, HGNC:33154).9,10 ELFN1 is a trans-membrane protein associated with synaptic maturation with selective expression in several neuronal circuits.10-12 It binds to mGlu receptors directly in a trans-synaptic manner anchoring them at synapses and allosterically modulating mGlu function.9,10,13,14 In mouse hippocampus, deletion of Elfn1 results in loss of synaptic mGlu7 thereby impairing its function as a pre-synaptic autoreceptor.10,14 It results in the increase in the initial glutamate release probability and disrupts synaptic facilitation. In the retina, deletion of Elfn1 leads to the loss of mGlu6 from rod photoreceptor synapses.9 This completely abolishes synaptic communication of rods, impairing dim vision of mice.9 Elfn1 knockout mice also show a range of neuropsychiatric related features including hyperactivity and seizures.10,12
Variants in ELFN1 have been associated with human disease. A homozygous 8 bp frameshift deletion in ELFN1 has been linked to a neurodevelopmental and epileptic encephalopathy with joint laxity in three patients from a consanguineous family.15 Further features included autistic spectrum disorder (ASD), pyramidal signs and facial dysmorphism. In another family, homozygous missense variants of ELFN1 (c.149C>A p.(Pro50His)) were found in three patients with moderate intellectual disability, including one with seizures.16 Four further heterozygous missense variants in ELFN1 have been identified in patients affected by attention-deficit hyperactivity disorder, autism spectrum disorder and epilepsy but their causality have not been established unequivocally.10 Overall, the limitations of these clinical studies and the variable spectrum of associated phenotypes makes attribution of ELFN1 to specific disease uncertain.
In this study, we report eight individuals from five unrelated families with biallelic deletions in ELFN1. We also provide further clinical information regarding a previously described family with biallelic missense variants in ELFN1. We provide detailed clinical phenotyping, neuroimaging and EEG findings. Our functional analysis reveals that all currently and previously reported biallelic ELFN1 variants are either predicted as loss of function, or functionally confirmed via impaired surface trafficking of ELFN1 and/or interaction with group III mGlu receptors. We further model ELFN1 loss of function in mice and zebrafish which demonstrates an epileptogenic and hyperactive. Our results together with published evidence establish ELFN1 deficiency as a distinct neurodevelopmental, autosomal recessive mendelian disorder with a defined clinical spectrum.
Materials and Methods:
Clinical and genetic studies.
We evaluated five new unrelated families of Iranian, Mexican and Palestinian origin. We also evaluated one previously identified Pakistani family, and present data from another Turkish family. This study was approved by the institutional ethics committees from each participating centre, including UCL, Al-Makassed Hospital, Quaid-i-Azam University, Shahid Sadoughi Hospital, KU Leuven, Canton of Geneva and Herbert Wertheim UF Scripps Institute for Biomedical Innovation & Technology. Written informed consent was obtained from each family as per the Declaration of Helsinki. Clinical features were reported from all affected individuals by their local clinicians. Facial features were described by a clinical geneticist based on terminology recommended by Elements of Morphology. Where no term was available for a facial feature seen in a patient, HPO terminology was used instead.17 Brain MR (magnetic resonance) images were reviewed by a pediatric neuroradiologist, and EEGs (electroencephalograms) were reviewed by a pediatric neurologist with a special interest in epilepsy. Patients’ samples either underwent genome or exome sequencing. In-silico variant deleteriousness were predicted using the indel-specific classifier in SIFT (Sorting Intolerant From Tolerant) and CADD (combined annotation dependent depletion).18,19 The frequency of each germline variant was reviewed in relevant population databases including 1KG Structural Variants, DGV, gnomAD, dbSNP, and an in-house database.20-23 The parameters for ELFN1 were reviewed in gnomAD 4.1.0 and RGC Million Exome Variant Browser v1.1.2. These analyses used GRCh38 and the MANE Select transcript NM_001128636.424.
Structural modelling.
Structural modelling was undertaken for the native ELFN1 protein, as well as the predicted ELFN1 protein with c.475_477del p.(Val159del). This was completed using AlphaFold which provides a 3D predicted structure utilising a refined deep learning algorithm.25
cDNA plasmids.
The coding sequence of human ELFN1 (Myc-DDK-tagged) was amplified from a commercial plasmid (CAT#: RC226197, Origene) and inserted into a mammalian expression vector pcDNA3.1 using in-fusion cloning (Takara bio). The c.475_477del p.(Val159del) variant, C terminal Venus tagged ELFN1, Val159del ELFN1 Venus and Val159del-Ecto-ELFN1-Fc Pro50His (P50H)-ELFN1, C terminal Venus tagged P50H-ELFN1, Val591fs (Fs591)-ELFN1, C terminal Venus tagged Fs591-ELFN1, Gln508fs (Fs508)-ELFN1, C terminal Venus tagged Fs508-ELFN1 were synthesized using conventional procedures. Ecto-ELFN1-Fc construct was synthesized using residues 1-399 of human ELFN1 with human codon optimization and a C-terminal Fc fusion and inserted into a mammalian expression vector pEG.26
Cell culture and transfection.
HEK293T/17 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, minimum essential medium non-essential amino acids (Life Technologies), 1 mM sodium pyruvate, penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in a humidified incubator containing 5% CO2. For transfection, cells were seeded in the 3.5cm dishes at a density of 8X105 cells/dish. Four hours later, the cells were transfected with the appropriate expression constructs (2.5 μg for Co-IP and biotinylation experiment; 1 μg for images experiment) with the reagents PLUS (2.5 μL/dish) and Lipofectamine LTX (5 μL/dish). Transfected cells were collected and used for the experiment ~36 hours post transfection.
Cell surface biotinylation and Western blotting
ELFN1 surface proteins were biotinylated using Pierce™ Cell Surface Protein Biotinylation and Isolation Kit (A44390, Thermo Fisher Scientific) according to manufacturer instructions. A total of ~1.2X107 cells were used for each condition. Cells were suspended and washed with 15 mL PBS once. 10 mL of 0.25 mg/mL Sulfo-NHS-SS Biotin was added to biotinylated the cell surface proteins for 10 minutes at room temperature. Cells were then washed with 15 mL ice-cold TBS twice, and subsequently lysed by lysis buffer. After 30 minutes of incubation on ice, lysates were cleared by centrifugation at 15,000 x g for 5 minutes. A small amount of lysates was saved for total protein examination. Lysates were then incubated with NeutrAvidin Agarose for 30 minutes at room temperature. NeutrAvidin Agarose was washed 4 times and then incubated with 200 μL of 10 mM DTT elution buffer. After 30 minutes of incubation, elutes were collected by centrifuge. Elutes and lysates were treated with β-mercaptoethanol-containing sample buffer for 42°C for 10 minutes. SDS-PAGE and Western blotting were performed using 5% milk blocking, then anti-myc antibody (Cell Signaling, 9B11), anti-sodium potassium ATPase (Invitrogen, MA5-32184), and anti-GAPDH (Millipore Sigma, MAB374) in 5% milk at 1:1000. Secondary antibodies were purchased from Kindle Biosciences and used at 1:2000 in 5% milk. Images were obtained using a KwikQuant Imager and KwikQuant ECL Solutions (Kindle Biosciences). For quantification, band intensities were measured by using Image J software.
Confocal fluorescent microscopy.
All microscopy experiments were performed on an Olympus FV3000 Confocal Laser Scanning Microscope. HEK293/17 cells were seeded on PDL-coated glass-bottomed dishes. Live cells were imaged using the 488 laser to excite Venus.
Protein G pulldowns.
Fc tag recombinant proteins were produced in HEK293T/17 cells. After ~12 hours of transfection, their media were replaced with OPTI-MEM and incubated at 30°C to accumulate secreted proteins. After 48 hours, media were collected and incubated with cell lysates from mGluR transfected cells in 1% Triton-X Lysis Buffer rotating for 1 hour at 4°C. Protein G Sepharose Beads (Cytiva) were then added and incubated for 1 hour at 4°C. Samples were washed 3 times with 1% Triton-X Lysis Buffer. Proteins were eluted with SDS sample buffer at 42°C for 10 minutes. SDS-PAGE and Western blotting were performed using 5% milk blocking, then anti-mGlu7 antibody (Thermo Scientific, PIMA531754), anti-mGlu4 (Thermo Scientific, PA550999), anti-flag (Sigma-Aldrich, F1804), or anti-Fc antibody (Invitrogen, MA1-10378) in 5% milk at 1:1000. Secondary antibodies were purchased from Jackson ImmunoResearch and used at 1:2000 in 5% milk. Images were obtained using a KwikQuant Imager and KwikQuant ECL Solutions (Kindle Biosciences).
Zebrafish husbandry.
Zebrafish modelling was used to recapitulate neurodevelopmental traits.27-29 Adult zebrafish (Danio rerio) stocks of AB strain (Zebrafish International Resource Center, Eugene, OR, USA) were maintained at 28.5 °C in UV-sterilized water on a 14h light/10h dark cycle under standard aquaculture conditions. Fertilized embryos were collected via natural spawning and raised in embryo medium (1.5 mM HEPES, pH 7.2, 17.4 mM NaCl, 0.21 mM KCl, 0.12 mM MgSO4, 0.18 mM Ca (NO3)2 and 0.6 μM methylene blue) at 28°C. All zebrafish experiments performed were notified to the Ethics Committee of KU Leuven (notification number M002/2024) for early developmental stages and approved by the Ethics Committee of KU Leuven (approval number P055/2020) and by the Belgian Federal Department of Public Health, Food Safety, and Environment (approval number LA1210261) for developmental stages ≥120 hpf.
Morpholino-based knockdown.
All morpholinos (MOs) were designed and synthesized by GeneTools LLC (Philomath, Oregon, USA).30,31 The MOs were heated at 65°C during 10 min and diluted with DEPC-free water to their working concentration. Injection glass capillaries (WPI, TW100F-4) were pulled with a micropipette puller (Sutter Instruments). MOs were injected in the yolk of 1-2 cell stage embryos using a M3301R Manual Micromanipulator (WPI) and a Femtojet 4i pressure microinjector (Eppendorf). Larvae were selected based on their uniformly spread fluorescence. No commercial antibody was available to verify the MO knockdown and a custom-designed antibody did not show affinity in control experiments. Representative images are available in Supplementary Figure 1.
Maximum tolerated dose analysis.
The maximum tolerated dose (MTD) of MOs was determined at 5 days post fertilization (dpf). Larvae were examined under the microscope for overall morphology, oedema, loss of posture, touch response, swim bladder conditions, and death. The MTD was defined as the highest dose at which the larvae did not exert any sign of toxicity.27
Behavioral analysis (zebrafish).
Locomotor studies were performed as previously published.32 At 5 dpf, MO-injected zebrafish larvae were transferred individually into a 96-well plate containing 100 μl Danieau’s medium per well. The plates were placed in an automated video tracking device, DanioVision™ (Noldus, The Netherlands), in which the behavior of the larvae was monitored for 40 minutes in the dark at 28 °C. The results were calculated for every 5-min period. Ethovision XT16 (Noldus, Netherlands) was used to quantify locomotor behavior as cumulative duration of time spent in highly active state (s/5 minutes). Activity was defined by comparing pixels of the current image (= the entire well) to the pixels of the previous image. If all pixels remained the same, there was 0% activity. Whereas, if all pixels changed, 100% activity was seen. The highly active state was achieved when the threshold of 0.4% activity was exceeded for at least 0.6 seconds. Data are expressed as mean ± SEM (standard error of mean).
Non-invasive local field potential (LFP) recordings.
Non-invasive local field potential (LFP) recordings of 5 dpf larvae, immobilized in 2% low-melting point agarose, were performed as previously described.33-36 LFP recordings were measured via a glass electrode, filled with artificial cerebrospinal fluid (124 mM NaCl, 2 mM KCl, 2 mM MgSO4, 2 mM CaCl2, 1.25 mM KH2PO4, 26 mM NaHCO3 and 10 mM glucose) from the optic tectum of the midbrain for 10 min at room temperature. Clampfit 10.2 software (Molecular Devices Corporation, USA) was used to visually analyse spontaneous epileptiform events, which were defined as an epileptiform event with an amplitude equal to or exceeding three times the baseline and a duration of at least 50 ms. Automated power spectral density (PSD) analysis was performed using Matlab R2020b (The MathWorks, Inc., Natick, MA, USA).37 Electrophysiological data were normalized against the control MO and expressed as mean ± SEM per larva within the 10-90 Hz region.
Statistical analysis
Data are presented as mean ± SEM. For phenotypic evaluation of MO-injected zebrafish larvae at 5 dpf, statistical analysis was performed using non-parametric Kruskal-Wallis test with Dunn’s multiple comparisons test. Outliers were identified via the ROUT test (Q = 1%) and excluded (Graphpad Prism 10, San Diego, CA, USA).
Behavioral analysis (mice).
Locomotor testing was conducted in 40 × 40 × 35-cm chambers (Stoelting Co, Wood Dale, IL), and distance traveled was recorded using Anymaze video-tracking software. Mice (2-3 months old) were handled and habituated to the testing room for 3 days. On testing day, mice were placed in the center of the chambers, and distance traveled was measured for 2 hours and analyzed in 10-minute bins. Mice groups included 12 Elfn1+/+, 15 Elfn1+/−, and 12 Elfn1−/−.
Data availability.
The data from this study are available from the corresponding author upon request. The variants described are deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/)
Results:
Clinical and molecular findings for the patients in this study, alongside those previously reported, are summarized in Supplementary Information 1 (overview patient information table and extended patient information table). This cohort includes eight newly described patients from five unrelated families and six previously reported patients from two families. Notably, we have obtained additional clinical and genetic data from the second reported family (F6). Further detailed descriptions of the clinical and molecular data can be found in Supplementary Information 2 (patient dysmorphology table), and Supplementary Information 3 (extended patient summaries and genomic data). Family pedigrees are illustrated in Figure 1A, clinical photographs in Figure 2A, MR neuroimaging in Figure 2B and summarized clinical findings in Figure 2C.
Figure 1. Pedigrees and variant visualization.

A) Pedigrees demonstrating the seven families and genotypes of tested individuals, indicated using + (pathogenic variant) and – (wild type). B) The ELFN1 transcript (NM_001128636.4) with each exon numbered 1-4 and the coding region depicted in dark blue. The pathogenic deletions identified in this report are represented at their location along the transcript in black text, whilst the previously identified pathogenic deletion is presented in grey text. C) The ELFN1 protein with functional regions depicted by colour and labelled as SP (signal peptide), LRR (leucine rich repeat), LRRCT (leucine rich C-terminal domain), FN3 (fibronectin type III domain), TM (transmembrane domain). The pathogenic deletions identified in this report are represented at their location along the transcript in black text, whilst the previously identified pathogenic deletion is presented in grey text. Regions of putatively translated frameshifts are demonstrated by an extended red line, whilst the downstream new stop codon is shown via a red cross.
Figure 2. Facial photographs and neuroimaging of patients.
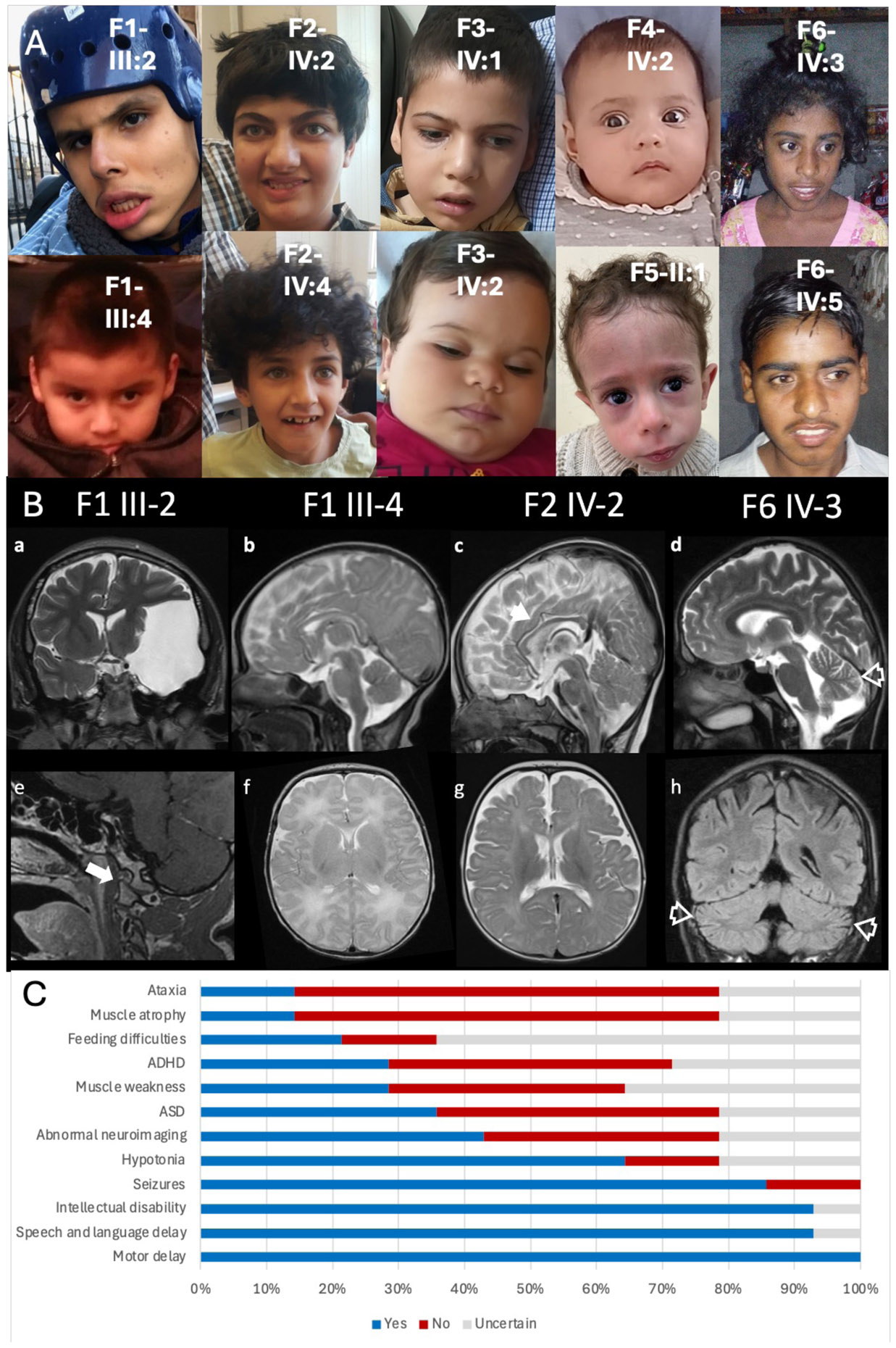
A) Facial photographs of the reported cases. B) Brain MRI. Family 1 III- 2 Coronal T2 weighted image (WI) and Sagittal post gad T1WI (a and e) shows arachnoid cyst in the left middle cranial fossa with mass effect (a) and atlantooccipital subluxation (arrow, e). Family 1 III-4 Sagittal and axial T2WI (b and e) showing normal morphology and signal changes of the brain. Family 2 IV-2 Sagittal and axial T2WI (c and g) showing mild diffuse volume loss with slightly thin corpus callosum (arrowhead, c). Family 6 IV-3 Sagittal T2WI and coronal FLAIR (d and h) showing brain volume loss including involvement of the cerebellar vermis and cerebellar hemispheres (open arrowheads, d and h). C) Common clinical features exhibited by patients, demonstrated by percentage affected.
Clinical features
The patients ranged in age from 10 months to 21 years and included seven males and seven females from seven unrelated families (F1-7). The families had origins in Iran, Mexico, Palestine, and Turkey, with five of the seven families having a known history of consanguinity. No consistent antenatal abnormalities were noted. Most patients had normal growth parameters at birth, except for one patient who exhibited early-onset growth failure and had a concurrent diagnosis of Russell-Silver syndrome. Additionally, three patients experienced feeding difficulties.
All patients exhibited developmental delay, with significant impairments in motor skills and speech/language development. Intellectual disability was reported in all patients, ranging from moderate to severe. One patient was described as mildly affected; however, his/her young age may have precluded the full manifestation of symptoms. Behavioral disturbances were observed in several patients, including ADHD, autism spectrum disorder, stereotypies, biting, and self-mutilation.
Neurological manifestations were common, with nine patients exhibiting hypotonia, four with muscle weakness, two with muscle atrophy, and four displaying ataxia or abnormal gait. Other findings included nystagmus in two patients and slow movement dysmetria in two others.
In terms of facial features, the most common characteristics observed were a thick lower lip vermillion (in eight patients), broad nasal tip (seven patients), narrow forehead (seven patients), broad chin (six patients), and a prominent nasal bridge (six patients). However, expert evaluations did not identify a consistent facial gestalt across the cohort.
Epilepsy: electroclinical features and therapy response.
Epileptic seizures occurred in 12 patients, including all patients with biallelic ELFN1 deletions. Of those who developed seizures, age of onset ranged from 15 days to 8 years. Seizure types included tonic, myoclonic, bilateral tonic-clonic seizures, and infantile spasms. EEGs were reported for 11 of these patients and for one other patient without clinical seizures (Supplementary Information 1). The EEG findings revealed a pattern of abnormal brain activity, characterized by globally slowed and dysregulated background activity, predominantly in the theta and delta/theta ranges. This includes bilateral sharp and slow waves more evident in central and anterior brain areas, diffuse and anterior sharp wave/slow wave complexes, multifocal sharp wave/slow wave complexes, prolonged bursts of high-voltage theta activity, prevalent high-voltage theta/delta activities with brief diffuse bursts of spiky fast rhythm, and high-amplitude irregular theta activity over frontal-temporal regions. Despite these abnormalities, no specific EEG pattern emerged that was unique to these patients, indicating a variety of manifestations without a single defining feature. The response to antiseizure medications was variable and while some patients experience partial seizure control, many continue to have refractory seizures. This suggests that the epileptic component in ELFN1 deficiency may be particularly challenging to manage with conventional medications alone.
Neuroimaging.
MRI studies were performed in six patients; two of them (33%) demonstrated some degree of volume loss and thinning of the corpus callosum (F6 IV-5; F2 IV-2). Patient F6 IV-5 presented also with atrophy of the cerebellar vermis and hemispheres. Furthermore, patient F1 III-2 showed evidence of subluxation of the atlantooccipital join bilaterally along with a large left arachnoid cyst resulting in mass effect and midline shift that had remained unchanged over a 9-month interval. Patient F3-IV:1, showed right maxillary sinusitis and otherwise normal neuroanatomy. Brain MRI for both F5-II:1 and F4-IV:2 was reported as normal.
Genomic investigations.
The variants in the ELFN1 gene are shown in Figure 1B and the resulting protein changes in Figure 1C. All patients were found to have homozygous variants in ELFN1. Of the seven families studied, six were found to have biallelic deletions, of which four variants were predicted to result in a premature termination codon (families F2, F4, F5, F7) and one (F1) resulted in deletion of the 5’ untranslated open reading frame precluding transcription. One family (F5) was found to have a biallelic missense variant. For each variant identified, no matching biallelic variants were identified in both internal and external databases. Additionally, all variants were predicted to be pathogenic utilizing in silico predictions. Notably, patient F5-II:1 was found to have uniparental disomy of chromosome 7 and a diagnosis of Russell-Silver syndrome. The referenced families and the genomic coordinates of the variants identified are as follows: F1 NC_000007.14:g.1648093_1740282del, F2 NC_000007.14:g.1746363del, F3 NC_000007.14:g.1745071_1745073del, F4 NC_000007.14:g.1744724_1744733del, F5 NC_000007.14:g.1746118del, F6 NC_000007.14:g.1744745C>A, F7 NC_000007.14:g.1744638_1744645del.
Truncated ELFN1 variants display major deficits in surface trafficking.
To function as trans-synaptic regulator of mGlu receptors, ELFN1 must localize to the surface of the cell (Fig. 3A). Thus, we focused our analysis of potential deleterious effects associated with variants that cause C-terminal truncations with escape from nonsense-mediated decay by examining their surface trafficking. We have further included a previously reported missense variant that creates a c.149C>A p.(Pro50His) variant in the extracellular domain of ELFN1 as it was not previously functionally examined. All other new and reported frameshift variants of ELFN1 are predicted to completely eliminate the extracellular elements of the protein and its trans-membrane segment which are indispensable for membrane localization of ELFN1 and its function. Western blotting analysis of the myc-tagged constructs showed that all variants were normally expressed at the protein level when transfected in HEK293 cells (Fig. 3B). However, when ELFN1 surface content was probed by surface biotinylation, we found only a small fraction of the protein on the surface of these cells. Quantification of the surface to total ratios, confirmed a severe reduction in the ability of c.1767del p.(Val591CysfsTer93) to reach the surface, while no detectable c.1522del p.(Gln508SerfsTer176) mutant was found to be on the surface at all (Fig. 3C). We further found that the c.149C>A p.(Pro50His) mutant exhibited major defects in surface localization. We confirmed these observations by examining the distribution of ELFN1 using immunofluorescence microscopy (Fig. 3D). While wild-type ELFN1 was robustly detected on the plasma membrane, all examined variants were mostly present at the intracellular sites. These observations indicate that pathogenic ELFN1 variants that cause truncation of its C-terminus as well as the c.149C>A p.(Pro50His) variant are loss of function due to defective localization to the cell surface.
Figure 3. Molecular mechanisms of ELFN1 disruption caused by Pro50His, Val591fs, and Gln508fs variants.

A) Synaptic localization of Elfn1. B) Representative Western Blots of surface and total protein expression level of c.149C>A p.(Pro50His) (P50H), c.1767del p.(Val591CysfsTer93) (Fs591), and c.1522del p.(Gln508SerfsTer176) (Fs508) ELFN1. C) Quantification of the ratio of surface to total protein expression level. D) Representative confocal images showing the WT, Pro50His, Val591fs, and Gln508fs ELFN1 expressed in HEK293 cells.
In frame c.475_477del p.(Val159del) disrupts its surface localization and mGlu binding.
The newly identified variant c.475_477del creates a single amino acid deletion of valine 159 in the extracellular region of ELFN1. To understand the structural implications of c.475_477del p.(Val159del) in ELFN1 we generated structural model of the protein using AlphaFold since no experimental structure of ELFN1 is currently available. Valine 159 maps to the conserved leucine rich repeat (LRR) domain of ELFN1 formed by six parallel β-strands that follow a canonical LRR pattern (LxxLxLxxNxL…). It occupies position 2 of the repeat in β-strand number 5 that forms a ridge with its side chain facing towards the solvent and not predicted to engage into contacts with other residues of ELFN1 (Fig. 4A). c.475_477del p.(Val159del) flattens out the ridge to accommodate the shorter backbone length. This is predicted to cause disturbances to the backbone and side chains in the surrounding sequence and to affect the orientation of side chains in the adjacent β-strands 4 and 5 to a varying degree (Fig. 4B). These perturbations are regional and the model does not predict significant changes in the overall structure of the molecule.
Figure 4. Structural model and functional effects of valine ELFN1 Val159del.
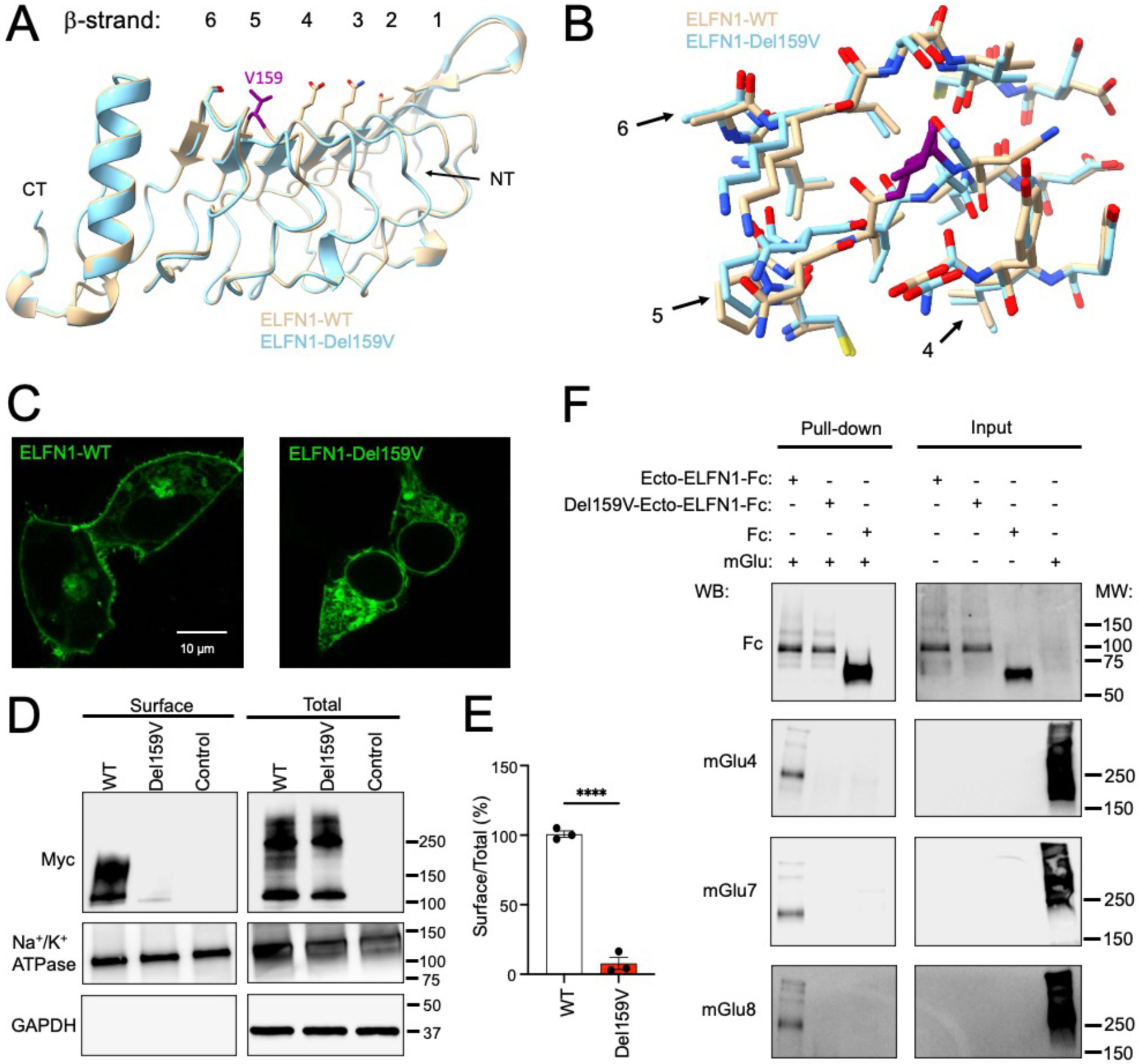
A) Overlay of LRR region of AlphaFold generated structures of WT ELFN1 and ELFN1 c.475_477del p.(Val159del) (Del159V). Position of valine 159 is shown by purple stick model. B) Detailed analysis of backbone and side chain configurations in β-strands of LRR domain in WT ELFN1 and ELFN1 Val159del. C) Analysis of WT and c.475_477del p.(Val159del) ELFN1 Venus localization by confocal microscopy in live HEK293 cells. Representative confocal images are shown. D) Examination of ELFN1 expression and surface localization by biotinylation. Representative Western blots of total and surface content of WT and c.475_477del p.(Val159del) ELFN1 are shown. E) Quantification of the surface biotinylation experiments shown in panel B from three independent experiments. ****p<0.0001, t-test. F) Representative western blots with inputs and pull-downs captured by Ecto-ELFN1-Fc and Val159del-Ecto-ELFN1-Fc by protein G beads.
We next tested whether the c.475_477del p.(Val159del) influences the ability of ELFN1 to traffic to the cell surface. Wild type ELFN1 tagged with Venus was localized predominantly on the plasma membrane in the transfected HEK293 cells when analyzed by live cell confocal microscopy (Fig. 4C). In contrast, we detected a much-reduced signal of Val159del-ELFN1 on the plasma membrane. Instead, most of its Venus fluorescence was found in the intracellular sites. We further corroborated these findings by using surface biotinylation approach that quantitatively determines changes in the surface content of ELFN1 proteins. We found that the abundance of the Val159del-ELFN1 mutant was similar to the wild-type ELFN1 protein as evidenced by the Western blotting of the total cellular lysates (Fig. 4D). However, the levels of Val159del-ELFN1 in the surface exposed fraction were drastically reduced as compared to WT ELFN1 (Fig. 4E). The results indicate that c.475_477del p.(Val159del) severely impairs plasma membrane trafficking of ELFN1.
We further wondered whether a small fraction of mutant ELFN1 that traffics to the cell surface could still interact with group III mGlu receptors. The extracellular ectodomain of ELFN1 that contains the LRR domain was shown to be sufficient for the interaction with mGluRs [1]. Thus, to address this question we used ectodomains of ELFN1 fused with the Fc tag to examine their ability to pull-down full-length group III mGlu receptors (Fig. 4F). We found that WT ectodomain of Elfn1 effectively pulled down mGlu4, mGlu7, and mGlu8. In contrast we detected no specific signal for mGlu receptors present in eluates when Val159del-ELFN1 ectodomain was used as a bait in pull-down assays. These findings indicate that deletion of valine 159 prevents interaction of ELFN1 with group III mGlu in addition to impairment in plasma membrane targeting.
Mouse and zebrafish models confirm that ELFN1 deficiency causes hyperactivity and epileptiform brain activity.
To model ELFN1 deficiency we examined the phenotypes of Elfn1 homozygous knockout mice (Elfn1−/−) comparing them with heterozygous (Elfn1+/−) and wild-type littermates. Using open field test, we found that both Elfn1−/− and Elfn1+/− mice displayed increased locomotor activity that lasted throughout two-hour examination session (Fig. 5A). Indeed, quantitative analysis confirmed hyperactivity (Fig. 5B). These findings confirm previous observations and extend them to demonstrate that haploinsufficiency of Elfn1 may be sufficient to cause hyperactivity in mice.10,12
Figure 5. Studying ELFN1 deficiency in animal models.

A) Time-course of locomotor activity of wildtype, Elfn1+/−, knockout Elfn1−/− and heterozygous Elfn+/− mice in the open field. B) Quantification of the total locomotor activity in the open filed over 2 hours. C) Behavioral and (D,E,F) electrophysiological seizure analysis of MO-injected zebrafish larvae at 5 dpf. C) Behavioral data is compared to control MO (elfn1a+/+/elfn1b+/+) and expressed as cumulative duration in highly active state (mean ± SEM) (n control MO = 73, n elfn1a/elfn1b MO = 29-30). D,E,F) Electrophysiological epileptiform activity is assessed via non-invasive local field potential recordings (n control MO = 18, n elfn1a/elfn1b MO = 15-16). D) Data is visually analysed and expressed as the number of epileptiform events (mean ± SEM) and compared to control MO. E,F) Data is normalized against control MO and expressed as (E) normalized power spectral density (PSD) (mean ±SEM) per larva per 10 Hz frequency band from 1-150 Hz and as (F) normalized PSD (mean ±SEM) per larva within the 10-90 Hz region. Statistical analysis was performed using (C,D,E,F) non-parametric Kruskal-Wallis test with Dunn’s multiple comparisons test. Outliers were removed via the ROUT test (Q = 1%). Significance levels: * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001.
The association between an ELFN1 deficiency and the development of an epileptic phenotype was also investigated in the zebrafish model. A morpholino (MO)-based knockdown was induced to mimic a loss of function of the zebrafish elfn1a or the elfn1b gene, both orthologues of human ELFN1. Both behavioral and electrophysiological analyses were carried out to characterize the phenotype of the MO-injected zebrafish larvae at 5 days post fertilization (dpf). Prior to conducting these analyses, the maximum tolerated dose (MTD) was determined for each MO (3 and 5 ng for elfn1a and elfn1b MO, respectively). Next, behavioral analysis showed a significant increase in cumulative duration in highly active state of 30.9 % for larvae injected with elfn1a MO compared to larvae injected with control MO (p ≤ 0.01), which suggests the presence of seizure-like behavior. Zebrafish larvae injected with elfn1b MO showed a non-significant increase in locomotion of 22.8 % (Fig. 5C).
Furthermore, non-invasive local field potential (LFP) recordings were performed to determine whether the gene knockdowns would induce epileptiform brain activity. Visual analysis showed a significant increase for the mean number of epileptiform events for both larvae injected with elfn1a and elfn1b MOs compared to larvae injected with control MO (Fig. 5D). Representative electrophysiological recordings are demonstrated in Supplementary Figure 2. Similarly, automated power spectral density (PSD) analysis demonstrated significant increases in PSD for larvae with elfn1a and elfn1b knockdown within the 10-90 Hz region (Fig. 5E - 5F). Hence, these observations suggest that knocking down the elfn1a or elfn1b genes induces an epileptic phenotype, characterized by an increase in locomotor behavior and epileptiform brain activity.
Discussion:
In this study we report eight patients from five separate families harboring five unique biallelic deletions in ELFN1. We also provide updated clinical information for a previously described family with biallelic single nucleotide variants in ELFN1. We describe the clinical, radiological, and genetic features with additional functional evidence to provide further understanding of the ELNF1-related disorder phenotype. For all the families presented, we have identified biallelic pathogenic variants in ELFN1 in the affected family members. Most of the pathogenic variants are homozygous deletions, including an in-frame single amino acid deletion, frameshifts with subsequent premature stop codon, and a deletion that encompasses all three exons within the 5’UTR. One previously described family harbored biallelic single nucleotide variants (c.149C>A, p.(Pro50His)).
All identified variants in ELFN1 were predicted to either result in loss of expression or proven loss of function of ELFN1 via functional validation in murine/zebrafish models. Variants with predicted loss of function included the deletion of the 5’UTR and two early frameshift variants. Whereas, this project has confirmed loss of function in the more uncertain variants, inclusive of a missense variant, a single amino acid deletion, and two frameshift variants that are downstream of the main functional motifs. Our functional validation has demonstrated the importance of effective cell surface trafficking of ELFN1 and its localization of mGluRs. This is consistent with its previously identified role as a cell adhesion molecular vital for synaptic maturation and transmission. Intriguingly, despite our findings, ELFN1 is not necessarily predicted to be loss of function intolerant with a pLI of 0.003, LOEUF of 1.84 and Shet of 0.0917. However, there were no homozygous nonsense or frameshift variants identified in gnomAD.
Together with previous reports, our study now provides enough evidence to define key characteristics of the disorder that we propose be named ELFN1-related Deficiency Disorder.
Seizures appear to be the key defining finding which is invariably present in those with biallelic deletions in ELFN1.15 As previously recognized, the type of seizure varied greatly with our patients demonstrating tonic seizures, tonic-clonic seizures, and infantile spasms. However, we observed a wider variability in the onset of seizures ranging from the early neonatal period to 8 years. Notably, the family with biallelic single nucleotide variants, rather than deletions, only exhibited seizures in one of three patients. Interestingly, murine data are also showed a variable age of onset in Elfn1−/− ranging from 11 weeks and 4 months of age.10,12 EEG findings revealed a pattern of abnormal brain activity among the patients studied, characterized by globally slowed and dysregulated background activity, predominantly in the theta and delta/theta ranges. However, no specific EEG pattern emerges that is unique to these patients, indicating a variety of manifestations without a single defining feature. Interestingly, spontaneous epileptiform brain activity was observed in the elfn1a and elfn1b zebrafish morphant models. This further highlights the correlation between an ELFN1 deficiency and the epileptic phenotypes.
Neurodevelopmental delay was found in all patients and was moderate to severe. Only one patient showed mild delay, and this was potentially due to their younger age at assessment. Neuropsychiatric diagnoses such as autism spectrum disorder and ADHD were variable – yet would also be affected by the young age of the patients. Increased locomotion in the Elfn1−/− mice may be reminiscent of an ADHD phenotype, which was also found in previous described murine models.12 Moreover, a MO-based knockdown of the orthologues elfn1a gene in zebrafish larvae showed a significant increase in locomotion, indicative of hyperactivity. Notably, ELFN1 missense monoallelic variants have been identified in three patients with autistic features and ADHD.10 ELFN1 was also a TWAS hit for PTSD and was found to be differentially expressed in these patients.38 ELFN1 has also been identified as a GWAS hit for restless leg syndrome.39 Whilst this phenotype was not explicitly reviewed in these participants, it is notable that there is an association between restless leg syndrome and certain epilepsy subtypes.40
Our observations that haploinsufficiency of Elfn1 in mice results in detectable phenotypes also demonstrates the phenotypic spectrum of ELFN1 deficiency disorder. Common signs included hypotonia, muscle weakness and gait abnormalities. This is compatible with Elfn1−/− mice demonstrating reduced lower limb strength on a hanging test.12 Pyramidal signs such as spasticity and hyperreflexia were only found in two patients. Furthermore, two patients demonstrated ataxia and slow movement dysmetria. Additionally, whilst patients demonstrated multiple facial features, there was no consistent shared facial phenotype. Notably, patient F5-II:1 demonstrated typical features of Russell-Silver syndrome.
In our clinical examination no assessment of dim vision function was performed. Mouse models strongly predict night blindness in patients as Elfn1 loss prevents the synaptic transmission of rod photoreceptor signals 9,41. However, because rods can also transmit light signals through low gain gap junctions, (the secondary rod pathway), ELFN1 deficiency is only expected to affect vision at the dimmest range of the light intensities and be accompanied by electrically negative electroretinograms at those intensities. None of the patients in this report underwent ERG testing and thus this assessment could be considered in future putative cases of ELFN1-related disorders.
The neuroimaging studies did not reveal a consistent pattern of morphological findings. There was a wide variability of phenotypes, with corpus callosum thinning and volume loss being the most common phenotypes in 2 of the 6 patients studied. These findings could be related to the underlying genetic disease and/or superimposed on prolonged episodes of seizures. Notably, one patient presented with evidence of subluxation of the atlanto-occipital joint, which may represent an additional feature of the ELFN1-related disease. This condition is potentially life-threatening, particularly for those patients who tend to have recurrent seizures and should be considered and further evaluated for these patients. Therefore, there does not appear to be a highly penetrant unifying cerebral phenotype, nor do significant structural brain abnormalities appear to be the only reason for their clinical presentation. We also note that we could not confirm the causal role of ELFN1 in joint laxity, reported before. None of the patients we examined had any further notable or consistent joint issues. Neither were these issues apparent in the animal models described here.
In summary, with expanded phenotypic spectrum and several unique variants, we introduce ELFN1 Deficiency Disorder as defined condition featuring distinct neurological and neuropsychiatric manifestations thereby advancing our understanding of the genetic basis for this disease. Given the synaptic dysfunction associated with ELFN1 variants, therapies that address synaptic modulation or neuroplasticity may offer new treatment options.
Supplementary Material
Acknowledgements:
We are grateful for the important support from patients and families, our UK and international collaborators, brainbank and biobanks.
Undiagnosed Diseases Network: Aaron Quinlan, Adeline Vanderver, Adriana Rebelo, Aimee Allworth, Alan H. Beggs, Alden Huang, Alex Paul, Ali Al-Beshri, Alistair Ward, Allyn McConkie-Rosell, Alyssa A. Tran, Andrea Gropman, Andres Vargas, Andrew B. Crouse, Andrew Stergachis, Anita Beck, Anna Hurst, Anna Raper, Anne Hing, Arjun Tarakad, Ashley Andrews, Ashley McMinn, Ashok Balasubramanyam, AudreyStephannie C. Maghiro, Barbara N. Pusey Swerdzewski, Ben Afzali, Ben Solomon, Beth A. Martin, Breanna Mitchell, Brendan C. Lanpher, Brendan H. Lee, Brent L. Fogel, Brett H. Graham, Brian Corner, Bruce Korf, Calum A. MacRae, Camilo Toro, Cara Skraban, Carlos A. Bacino, Carson A. Smith, Cecilia Esteves, Changrui Xiao, Chloe M. Reuter, Christina Lam, Christine M. Eng, Claire Henchcliffe, Colleen E. Wahl, Corrine K. Welt, Cynthia J. Tifft, Dana Kiley, Daniel Doherty, Daniel J. Rader, Daniel Wegner, Danny Miller, Daryl A. Scott, Dave Viskochil, David A. Sweetser, David R. Adams, Dawn Earl, Deborah Barbouth, Deepak A. Rao, Devin Oglesbee, Devon Bonner, Donna Novacic, Dustin Baldridge, Edward Behrens, Edwin K. Silverman, Elaine Seto, Elijah Kravets, Elizabeth A. Burke, Elizabeth Blue, Elizabeth L. Fieg, Elizabeth Rosenthal, Ellen F. Macnamara, Elsa Balton, Emily Glanton, Emily Shelkowitz, Eric Allenspach, Eric Klee, Eric Vilain, Erin Baldwin, Erin Conboy, Erin E. Baldwin, Erin McRoy, Esteban C. Dell’Angelica, Euan A. Ashley, F. Sessions Cole, Filippo Pinto e Vairo, Frances High, Francesco Vetrini, Francis Rossignol, Fuki M. Hisama, Gabor Marth, Gail P. Jarvik, Gary D. Clark, George Carvalho, Gerard T. Berry, Ghayda Mirzaa, Gill Bejerano, Giorgio Sirugo, Gonench Kilich, Guney Bademci, Heidi Wood, Herman Taylor, Holly K. Tabor, Hongzheng Dai, Hsiao-Tuan Chao, Hugo J. Bellen, Ian Glass, Ian R. Lanza, Ingrid A. Holm, Isaac S. Kohane, Ivan Chinn, J. Carl Pallais, Jacinda B. Sampson, James P. Orengo, Jason Hom, Jennefer N. Kohler, Jennifer E. Posey, Jennifer Wambach, Jessica Douglas, Jiayu Fu, Jill A. Rosenfeld, Jimann Shin, Jimmy Bennett, Joan M. Stoler, Joanna M. Gonzalez, John A. Phillips III, John Carey, John J. Mulvihill, Joie Davis, Jonathan A. Bernstein, Jordan Whitlock, Jose Abdenur, Joseph Loscalzo, Joy D. Cogan, Julian A. Martínez-Agosto, Justin Alvey, Kahlen Darr, Kaitlin Callaway, Kathleen A. Leppig, Kathleen Sullivan, Kathy Sisco, Kathyrn Singh, Katrina Dipple, Kayla M. Treat, Kelly Hassey, Kelly Schoch, Kevin S. Smith, Khurram Liaqat, Kim Worley, Kimberly Ezell, Kimberly LeBlanc, Kumarie Latchman, Lance H. Rodan, Laura Pace, Laurel A. Cobban, Lauren C. Briere, Leoyklang Petcharet, LéShon Peart, Lili Mantcheva, Lilianna Solnica-Krezel, Lindsay C. Burrage, Lindsay Mulvihill, Lisa Schimmenti, Lisa T. Emrick, Lorenzo Botto, Lorraine Potocki, Lynette Rives, Lynne A. Wolfe, Manish J. Butte, Margaret Delgado, Maria T. Acosta, Marie Morimoto, Mariko Nakano-Okuno, Mark Wener, Marla Sabaii, Martha Horike-Pyne, Martin G. Martin, Martin Rodriguez, Matt Velinder, Matthew Coggins, Matthew Might, Matthew T. Wheeler, Maura Ruzhnikov, MayChristine V. Malicdan, Meghan C. Halley, Melissa Walker, Michael Bamshad, Michael F. Wangler, Miguel Almalvez, Mohamad Mikati, Monika Weisz Hubshman, Monte Westerfield, Mustafa Tekin, Naghmeh Dorrani, Neil H. Parker, Neil Hanchard, Nicholas Borja, Nicola Longo, Nicole M. Walley, Nina Movsesyan, Nitsuh K. Dargie, Oguz Kanca, Orpa Jean-Marie, Page C. Goddard, Paolo Moretti, Patricia A. Ward, Patricia Dickson, Paul G. Fisher, Pengfei Liu, Peter Byers, Pinar Bayrak-Toydemir, Precilla D'Souza, Queenie Tan, Rachel A. Ungar, Rachel Mahoney, Ramakrishnan Rajagopalan, Raquel L. Alvarez, Rebecca C. Spillmann, Rebecca Ganetzky, Rebecca Overbury, Rebekah Barrick, Richard A. Lewis, Richard L. Maas, Rizwan Hamid, Rong Mao, Ronit Marom, Rosario I. Corona, Russell Butterfield, Sam Sheppeard, Sanaz Attaripour, Seema R. Lalani, Serena Neumann, Shamika Ketkar, Shamil R. Sunyaev, Shilpa N. Kobren, Shinya Yamamoto, Shirley Sutton, Shruti Marwaha, Sirisak Chanprasert, Stanley F. Nelson, Stephan Zuchner, Stephanie Bivona, Stephanie M. Ware, Stephen Pak, Steven Boyden, Suman Jayadev, Surendra Dasari, Susan Korrick, Suzanne Sandmeyer, Tahseen Mozaffar, Tammi Skelton, Tara Wenger, Terra R. Coakley, Thomas Cassini, Thomas J. Nicholas, Timothy Schedl, Tiphanie P. Vogel, Vaidehi Jobanputra, Valerie V. Maduro, Vandana Shashi, Virginia Sybert, Vishnu Cuddapah, Wendy Introne, Wendy Raskind, Willa Thorson, William A. Gahl, William E. Byrd, William J. Craigen, Yan Huang, Yigit Karasozen
Funding Statement:
The work was partially supported by the Swiss National Science Foundation grant (IZSTZ0_216615) to MA and National Institutes of Health grant DA056414 to KAM.
We are also grateful for essential funding from The Wellcome Trust, The MRC, The MSA Trust, The National Institute for Health Research University College London Hospitals Biomedical Research Centre NIHR-BRC), The Michael J Fox Foundation (MJFF), The Fidelity Trust, Rosetrees Trust, The Dolby Family fund, Alzheimer's Research UK (ARUK), MSA Coalition, Parkinson's disease society, Parkinson's Foundation, The Guarantors of Brain, Cerebral Palsy Alliance, FARA, EAN, Victoria Brain bank, the NIH NeuroBioBank, Queen Square BrainBank, The MRC Brainbank Network.
Footnotes
Ethics declaration:
This study was approved by the institutional ethics committees from each participating centre, including UCL, Al-Makassed Hospital, Quaid-i-Azam University, Shahid Sadoughi Hospital, KU Leuven, Canton of Geneva and Herbert Wertheim UF Scripps Institute for Biomedical Innovation & Technology. Written informed consent was obtained from each family as per the Declaration of Helsinki. We have received and archived written consent for participation/publication from every individual whose data is included and attach it to your submission. We have received and archived a valid HIPAA authorization for participation/publication from every individual whose PHI is included and whose photo is included, including consent for photos where applicable.
Conflict of Interest:
The authors declare no conflict of interest.
Data availability:
Data is included in the Supplementary Information. Additional data is available on request to the corresponding authors in accordance with confidentiality policies.
References:
- 1.Südhof TC. The cell biology of synapse formation. J Cell Biol. 2021;220(7). 10.1083/jcb.202103052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurshan PT, Shen K. Synaptogenic pathways. Curr Opin Neurobiol. 2019;57:156–162. 10.1016/j.conb.2019.03.005 [DOI] [PubMed] [Google Scholar]
- 3.Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. 10.1146/annurev.biochem.76.060805.160029 [DOI] [PubMed] [Google Scholar]
- 4.Magee JC, Grienberger C. Synaptic plasticity forms and functions. Annu Rev Neurosci. 2020;43:95–117. 10.1146/annurev-neuro-090919-022842 [DOI] [PubMed] [Google Scholar]
- 5.Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. 2012;4(1):a005736. 10.1101/cshperspect.a005736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Christian KM, Song H, Ming GL. Synaptic dysfunction in complex psychiatric disorders: from genetics to mechanisms. Genome Med. 2018;10(1):9. 10.1186/s13073-018-0518-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Spronsen M, Hoogenraad CC. Synapse pathology in psychiatric and neurologic disease. Curr Neurol Neurosci Rep. 2010;10(3):207–214. 10.1007/s11910-010-0104-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. 10.1146/annurev.pharmtox.011008.145533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao Y, Sarria I, Fehlhaber KE, et al. Mechanism for selective synaptic wiring of rod photoreceptors into the retinal circuitry and its role in vision. Neuron. 2015;87(6):1248–1260. 10.1016/j.neuron.2015.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomioka NH, Yasuda H, Miyamoto H, et al. Elfn1 recruits presynaptic mGluR7 in trans and its loss results in seizures. Nat Commun. 2014;5:4501. 10.1038/ncomms5501 [DOI] [PubMed] [Google Scholar]
- 11.Sylwestrak EL, Ghosh A. Elfn1 regulates target-specific release probability at CA1-interneuron synapses. Science. 2012;338(6106):536–540. 10.1126/science.1222482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dolan J, Mitchell KJ. Mutation of Elfn1 in mice causes seizures and hyperactivity. PLoS One. 2013;8(11):e80491. 10.1371/journal.pone.0080491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunn HA, Patil DN, Cao Y, Orlandi C, Martemyanov KA. Synaptic adhesion protein ELFN1 is a selective allosteric modulator of group III metabotropic glutamate receptors in trans. Proc Natl Acad Sci U S A. 2018;115(19):5022–5027. 10.1073/pnas.1722498115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stachniak TJ, Sylwestrak EL, Scheiffele P, Hall BJ, Ghosh A. Elfn1-induced constitutive activation of mGluR7 determines frequency-dependent recruitment of somatostatin interneurons. J Neurosci. 2019;39(23):4461–4474. 10.1523/jneurosci.2276-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dursun A, Yalnizoglu D, Yilmaz DY, et al. Biallelic mutations in ELFN1 gene associated with developmental and epileptic encephalopathy and joint laxity. Eur J Med Genet. 2021;64(11):104340. 10.1016/j.ejmg.2021.104340 [DOI] [PubMed] [Google Scholar]
- 16.Rasheed M, Khan V, Harripaul R, et al. Exome sequencing identifies novel and known mutations in families with intellectual disability. BMC Med Genomics. 2021;14(1):211. 10.1186/s12920-021-01066-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gargano MA, Matentzoglu N, Coleman B, et al. The Human Phenotype Ontology in 2024: phenotypes around the world. Nucleic Acids Res. 2024;52(D1):D1333–D1346. 10.1093/nar/gkad1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–874. 10.1101/gr.176601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–D894. 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311. 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacDonald JR, Ziman R, Yuen RKC, Feuk L, Scherer SW. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42(D1):D986–D992. 10.1093/nar/gkt958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen S, Francioli LC, Goodrich JK, et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature. 2023;625(7993):92–100. 10.1038/s41586-023-06045-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Auton A, Abecasis GR, Altshuler DM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morales J, Pujar S, Loveland JE, et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature. 2022;604(7905):310–315. 10.1038/s41586-022-04558-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–589. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goehring A, Lee CH, Wang KH, et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat Protoc. 2014;9(11):2574–2585. 10.1038/nprot.2014.173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siekierska A, Stamberger H, Deconinck T, et al. Biallelic VARS variants cause developmental encephalopathy with microcephaly that is recapitulated in vars knockout zebrafish. Nat Commun. 2019;10(1):708. 10.1038/s41467-018-07953-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Meulemeester AS, Heylen L, Siekierska A, Mills JD, Romagnolo A, Van Der Wel NN, et al. Hyperactivation of mTORC1 in a double-hit mutant zebrafish model of tuberous sclerosis complex causes increased seizure susceptibility and neurodevelopmental abnormalities. Front Cell Dev Biol. 2022;10:952832. 10.3389/fcell.2022.952832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Partoens M, De Meulemeester AS, Giong HK, Pham DH, Lee JS, De Witte PA, et al. Modeling neurodevelopmental disorders and epilepsy caused by loss of function of kif2a in zebrafish. eNeuro. 2021;8(5). 10.1523/eneuro.0055-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Summerton J, Weller D. Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997;7(3):187–195. 10.1089/oli.1.1997.7.187 [DOI] [PubMed] [Google Scholar]
- 31.Suls A, Jaehn JA, Kecskés A, Weber Y, Weckhuysen S, Craiu DC, et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet. 2013;93(5):967–975. 10.1016/j.ajhg.2013.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Heylen L, Partoens M, Mills JD, Kaminski RM, Godard P, et al. Connectivity mapping using a novel sv2a loss-of-function zebrafish epilepsy model as a powerful strategy for anti-epileptic drug discovery. Front Mol Neurosci. 2022;15:881933. 10.3389/fnmol.2022.881933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Copmans D, Kildgaard S, Roux E, Partoens M, Steurs G, Wang X, et al. From the North Sea to drug repurposing, the antiseizure activity of halimide and plinabulin. Pharmaceuticals (Basel). 2022;15(2):247. 10.3390/ph15020247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siekierska A, Stamberger H, Deconinck T, Oprescu SN, Partoens M, Zhang Y, et al. Biallelic VARS variants cause developmental encephalopathy with microcephaly that is recapitulated in vars knockout zebrafish. Nat Commun. 2019;10(1):708. 10.1038/s41467-018-07953-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheldeman C, Mills JD, Siekierska A, Serra I, Copmans D, Iyer AM, et al. mTOR-related neuropathology in mutant tsc2 zebrafish: phenotypic, transcriptomic and pharmacological analysis. Neurobiol Dis. 2017;108:225–237. 10.1016/j.nbd.2017.09.004 [DOI] [PubMed] [Google Scholar]
- 36.Partoens M, De Meulemeester AS, Giong HK, Pham DH, Lee JS, De Witte PA, et al. Modeling neurodevelopmental disorders and epilepsy caused by loss of function of kif2a in zebrafish. eNeuro. 2021;8(5):ENEURO.0055-21.2021. 10.1523/eneuro.0055-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunyadi B, Siekierska A, Sourbron J, Copmans D, de Witte PAM. Automated analysis of brain activity for seizure detection in zebrafish models of epilepsy. J Neurosci Methods. 2017;287:13–24. 10.1016/j.jneumeth.2017.05.024 [DOI] [PubMed] [Google Scholar]
- 38.Girgenti MJ, Wang J, Ji D, Cruz DA, Alvarez VE, Benedek D, et al. Transcriptomic organization of the human brain in post-traumatic stress disorder. Nat Neurosci. 2021;24(1):24–33. 10.1038/s41593-020-00748-7 [DOI] [PubMed] [Google Scholar]
- 39.Akçimen F, Chia R, Saez-Atienzar S, Ruffo P, Rasheed M, Ross JP, et al. Genomic analysis identifies risk factors in restless legs syndrome. Ann Neurol,2024;95(5):994–1005. 10.1002/ana.27040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geyer JD, Geyer EE, Fetterman Z, Carney PR. Epilepsy and restless legs syndrome. Epilepsy Behav. 2017;68:41–44. 10.1016/j.yebeh.2016.12.010 [DOI] [PubMed] [Google Scholar]
- 41.Cao Y, Wang Y, Dunn HA, Orlandi C, Shultz N, Kamasawa N, et al. Interplay between cell-adhesion molecules governs synaptic wiring of cone photoreceptors. Proc Natl Acad Sci U S A. 2020;117(38):23914–23924. 10.1073/pnas.2009940117 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data from this study are available from the corresponding author upon request. The variants described are deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/)
Data is included in the Supplementary Information. Additional data is available on request to the corresponding authors in accordance with confidentiality policies.