

Abstract
Hereditary coproporphyria (HCP) is the least common of the autosomal dominant acute hepatic porphyrias. It results from mutations in the CPO gene that encodes the mitochondrial enzyme, coproporphyrinogen oxidase. A few patients have also been reported who are homoallellic or heteroallelic for CPO mutations and are clinically distinct from those with HCP. In such patients the presence of a specific mutation (K404E) on one or both alleles produces a neonatal hemolytic anemia that is known as “harderoporphyria”; mutations on both alleles elsewhere in the gene give rise to the “homozygous” variant of HCP. The molecular relationship between these disorders and HCP has not been defined. We describe the molecular investigation and clinical features of 17 unrelated British patients with HCP. Ten novel and four previously reported CPO mutations, together with three previously unrecognized single-nucleotide polymorphisms, were identified in 15 of the 17 patients. HCP is more heterogeneous than other acute porphyrias, with all but one mutation being restricted to a single family, with a predominance of missense mutations (10 missense, 2 nonsense, 1 frameshift, and 1 splice site). Of the four known mutations, one (R331W) has previously been reported to cause disease only in homozygotes. Heterologous expression of another mutation (R401W) demonstrated functional properties similar to those of the K404E harderoporphyria mutation. In all patients, clinical presentation was uniform, in spite of the wide range (1%–64%) of residual coproporphyrinogen oxidase activity, as determined by heterologous expression. Our findings add substantially to knowledge of the molecular epidemiology of HCP, show that single copies of CPO mutations that are known or predicted to cause “homozygous” HCP or harderoporphyria can produce typical HCP in adults, and demonstrate that the severity of the phenotype does not correlate with the degree of inactivation by mutation of coproporphyrinogen oxidase.
Introduction
The 14-kb human CPO gene on chromosome 3q11.2 contains seven exons that encode the mitochondrial enzyme, coproporphyrinogen oxidase (CPOX) (EC 1.3.3.3), which catalyses the stepwise oxidative decarboxylation of the heme precursor, coproporphyrinogen III, to protoporphyrinogen IX via a tricarboxylic intermediate known as “harderoporphyrinogen” (Elder et al. 1978; Delfau-Larue et al. 1994). Mutations in the CPO gene cause three clinically distinct disorders: hereditary coproporphyria (HCP [MIM 121300]), its homozygous variant, and harderoporphyria (Nordmann and Deybach 1990; Kappas et al. 1995; Martasek 1998). HCP is the least common of the three autosomal dominant acute porphyrias. As in the others—acute intermittent porphyria (AIP) and variegate porphyria (VP)—clinical penetrance is low, and symptoms are very rare before puberty. Patients almost always present with an acute neurovisceral crisis, often provoked by drugs, alcohol, or endocrine factors (Brodie et al. 1977; Kuhnel et al. 2000). If skin lesions caused by photosensitization by porphyrins are absent, as occurs in ∼70% of patients, HCP is clinically indistinguishable from AIP. If they are present, HCP resembles VP, although in the latter condition ∼70% of patients have only skin lesions (Kirsch et al. 1998; Whatley et al. 1999a). Presentation with skin lesions alone is uncommon among patients with HCP; the few cases reported have usually been associated with hepatobiliary dysfunction (Brodie et al. 1977; Hawk et al. 1978; Martasek 1998). In contrast, the very rare homozygous form of HCP presents in childhood with skin lesions (Grandchamp et al. 1977; Martasek et al. 1994b; Doss et al. 1999). Harderoporphyria is characterized by neonatal hemolytic anemia, sometimes accompanied by skin lesions and accumulation of harderoporphyrin in feces (Nordmann et al. 1983; Lamoril et al. 1995; Lamoril et al. 1998). All affected individuals described to date have been homoallelic or heteroallelic for the same missense mutation in the CPO gene (K404E in exon 6), suggesting that disruption of this region of CPOX impairs the second stage of the conversion of coproporphyrinogen III to protoporphyrinogen IX (Lamoril et al. 1998). Missense mutations of this region of exon 6 have not been described in HCP.
Detection of asymptomatic affected individuals—so they can be advised to avoid factors that provoke acute attacks, and so they can receive prompt treatment if an attack occurs—is an important part of the management of HCP. Asymptomatic affected individuals can be detected at any stage of life by measuring the level of CPOX activity in lymphocytes, which is 50% of the mean level in control subjects (Nordmann and Deybach 1990; Martasek 1998). After the age of 7 years, asymptomatic affected individuals can be identified by demonstration of an increased coproporphyrin isomer III:I ratio in feces (Blake et al. 1992; Kuhnel et al. 2000). Unfortunately, these methods are technically complex, imprecise, or insensitive. Mutational analysis has potential advantages but, because of the rarity of HCP, has been applied to only a few families from five countries; mutations have been identified in 17 families (Delfau-Larue et al. 1994; Fujita et al. 1994; Daimon et al. 1997; Lamoril et al. 1997; Schreiber et al. 1997; Susa et al. 1998; Rosipal et al. 1999; also see Human Gene Mutation Database). The extent of allelic heterogeneity, the distribution and functional expression of mutations within the CPO gene, and genotype-phenotype correlations have yet to be studied in a large, unselected group of patients from a single country. We report an investigation of the CPO gene in 17 apparently unrelated patients from the United Kingdom.
Patients and Methods
Patients
We studied 17 apparently unrelated patients with HCP who presented sequentially for confirmation of diagnosis and/or for family investigation. Thirteen patients had a current or past attack of acute porphyria; in two of these, skin lesions were present during the acute attack. Four patients had never had symptoms of porphyria but had a family history of porphyria. Three of the four came from families in which at least one member was known to have overt HCP (including a large family described by Andrews et al. [1984]). One family had never had symptoms, the proband having been detected during routine urine testing for another disease. In addition, samples for mutational analysis were obtained from 29 asymptomatic relatives from eight families. The diagnosis was established by measurement of fecal porphyrin: total fecal porphyrin concentrations were 360–3,649 nmol/g dry wt (mean 2,184 nmol/g dry wt vs. <200 nmol/g dry wt in normal subjects), with coproporphyrin accounting for >80% of the total porphyrin. Coproporphyrin isomer III:I ratios, measured in nine patients, were 23–87 (mean 36 vs. <1.4 in unaffected subjects). VP was excluded by fluorescence-emission spectroscopy of plasma (Poh-Fitzpatrick 1980). Fecal coproporphyrin isomer ratios were measured in six asymptomatic family members who were found, by use of mutational analysis, to be affected. The mode of presentation was recorded for all symptomatic family members. Unrelatedness was determined by family inquiries; none of the patients were known to be related. Patients resided in various locations throughout the United Kingdom; all patients were white.
Identification of Mutations
DNA was extracted from peripheral blood leukocytes. Exons 1–7 of the CPO gene, along with 50–150 bp of their flanking regions, were amplified by means of the primers and conditions described elsewhere (Rosipal et al. 1999) and were screened for mutations by denaturing gradient gel electrophoresis (DGGE). Regions showing abnormal patterns were sequenced to identify mutations and polymorphisms. When no abnormality was detected by DGGE, the gene was sequenced.
For DNA sequencing, the CPO gene was amplified with primers different from those used for DGGE analysis (Rosipal et al. 1999). DNA fragments were purified with the Qiaquick PCR purification kit (Qiagen) and were directly sequenced using the Thermo Sequenase radiolabeled dye terminator (α-33[P] dideoxyribonucleotide triphosphate) cycle sequencing kit (Amersham Life Science). The presence of mutations was confirmed by sequencing of both DNA strands of amplified DNA.
Construction and Prokaryotic Expression of Normal and Mutated Human CPOX cDNA
Normal human cDNA was expressed using the pGEX-2T expression vector (Pharmacia Biotech) in the recombinant bacteria Escherichia coli DH5α, as described elsewhere (Lamoril et al. 1998). Site-directed mutagenesis was performed using normal cloned CPOX cDNA (pGEX-2T:CPOX) as the template. We used the Transformer site-directed mutagenesis kit (Clontech Laboratories), as described elsewhere (Rosipal et al. 1999). The entire sequence of each mutated plasmid was verified by sequencing.
Other Methods
The recombinant bacteria (E. coli DH5α) were grown, and CPOX activities of control and mutant enzymes were determined in bacterial lysates, as described elsewhere (Lamoril et al. 1995). Specific activity was expressed as the mean of two duplicate experiments. Fecal porphyrins were quantified (Lockwood et al. 1985), and coproporphyrin isomer ratios were determined (Blake et al. 1992) by standard methods; fluorescence-emission spectroscopy of plasma was performed as described elsewhere (Long et al. 1993).
Phylogenetic Analysis
Protein sequences were obtained from SwissProt, and the alignment of the protein sequences was made with Clustal W software (Thompson et al. 1994). Phylogenetic reconstructions were generated by using the neighbor-joining method in the Phylogeny Inference Package (PHYLIP, version 3.5c) (Felsenstein et al. 1989) with a Kimura two-parameter distance matrix program (PROTDIST and NEIGHBOR). Nonsynonymous phylogenetic reconstruction was generated with a Kimura formula (PROTDIST). A consensus phylogenetic tree was elaborated with the program CONSENSE.
Polymorphisms
After informed consent was obtained, the frequencies of known and suspected single-nucleotide polymorphisms (SNPs) in the CPO gene were determined by studying 84 white families who did not have porphyria. The families were drawn from the Centre d’Etude du Polymorphisme Humain (Paris), and frequencies were determined by means of direct sequencing, restriction-enzyme digestion, or DGGE.
Numbering System
Nucleotides are numbered according to the cDNA sequence derived from the CPO genomic sequence (Delfau-Larue et al. 1994), in which the A of the ATG initiation codon is numbered as +1. Comparison of the sequence of the CPO gene (Delfau-Larue et al. 1994) with cDNA sequences (Taketani et al. 1994) revealed that the first in-frame AUG codon lies within the first exon, 300 bp upstream from the putative initiating codon described elsewhere (Martasek et al. 1994a; Taketani et al. 1994). CPO mutations reported in 1994–95 that were numbered from the downstream AUG have been renumbered here: for example, R231W and K304E become R331W and K404E, respectively.
Results
Identification of Mutations
Initially, exons 2–7 and their flanking regions, the 3′ part of exon 1 and one-fifth of the 3′ noncoding sequence, were amplified from genomic DNA from all 17 unrelated patients with HCP. Amplified fragments were screened by DGGE, and any that showed abnormal patterns were sequenced. The promoter, nonanalyzable regions of exon 1, and the noncoding sequence of exon 7 were directly sequenced. This approach identified 10 novel and 4 previously reported mutations in 15 patients (table 1). All mutations except one (1277 G→A, which was previously identified in a French patient [Delfau et al. 1994]) were restricted to a single family; no base change was detected in the two remaining patients, despite DGGE analysis and sequencing of all exons, their flanking regions, and 530 bp upstream from the major transcriptional sites (Delfau-Larue et al. 1994). Major deletions were excluded by showing that each patient was heterozygous for at least one intragenic SNP. This method of mutational analysis therefore detected mutations in 15 of 17 patients, giving a sensitivity of 88% (95% confidence interval 70%–99%). Mutations were present in all exons except exon 3, with no exon having >3 mutations (fig. 1); 10 mutations (71%) were missense, 2 (14%) produced stop codons, 1 changed an invariant nucleotide at the splice donor site of exon 6, and 1 (1133delC) produced a frameshift leading to a stop codon (table 1; fig. 1).
Table 1.
Mutations in the CPO Gene in British Patients with HCP
| Exon and Mutation | Sequence Modification | Reference |
| 1: | ||
| 485A→C | Q162P | Present study |
| 511G→A | D171N | Present study |
| 2: | ||
| 601G→A | E201K | Schreiber et al. 1997 |
| 607G→A | A203T | Present study |
| 4: | ||
| 877G→A | A293T | Present study |
| 916C→T | Q306X | Rosipal et al. 1999 |
| 923G→T | G308V | Present study |
| 5: | ||
| 991C→T | R331W | Martasek et al. 1994b |
| 1133delC | Frameshift (stop codon +42) | Present study |
| 1171C→T | R391W | Present study |
| 6: | ||
| 1201C→T | R401W | Present study |
| 1276C→T | R426X | Present study |
| 1277G→A | Exon 6 skipping | Delfau-Larue et al. 1994 |
| 7: | ||
| 1339C→T | R447C | Present study |
Figure 1.

Localization of all mutations identified in CPO gene. Exons are represented by rectangles and are numbered 1–7. Filled rectangles indicate protein coding, and unfilled rectangles indicate untranslated sequences. The mutations reported in this study of patients in the United Kingdom are depicted above the diagram, and the mutations reported elsewhere are depicted below the diagram (Delfau-Larue et al. 1994; Fujita et al. 1994; Martasek et al. 1994b; Lamoril et al. 1995; Daimon et al. 1997; Lamoril et al. 1997, 1998; Schreiber et al. 1997; Susa et al 1998; Doss et al. 1999; Rosipal et al. 1999). ∇ = frameshift ; ● = missense; □ = nonsense; O = splice mutation; H = harderoporphyrinogen overproduction, found in R401W and K404E mutations.
A total of 29 asymptomatic relatives of 8 patients were screened for the presence of the appropriate mutation. Mutations were present in 15 of these relatives. Among the affected relatives who provided fecal samples, fecal porphyrin concentrations were 112–261 nmol/g dry wt (coproporphyrin isomer III/I ratio 1.5:2.2) in three adults and were 58–101 nmol/g dry wt (isomer ratio 0.7:2.4) in three children aged 7–9 years.
Heterologous Expression of Mutations
To further characterize CPO missense mutations, pGEX-2T: CPO expression vectors were constructed for each mutant allele and expressed in E. coli. Specific activity was expressed in pmol of protoporphyrin/h/mg protein at 37°C, as mean of two duplicate experiments. Residual activity was determined by the formula
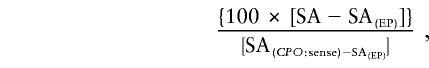 |
where SA(EP) indicates specific activity for empty plasmid. Table 2 shows that the CPOX activities of the mutant proteins, determined by measuring protoporphyrin(ogen) formation, were 1%–63% of wild-type activity, with all mutations except one decreasing activity by ⩾ 60%. Protoporphyrin(ogen) was the main reaction product in all cases. However, one mutation (R401W) also caused substantial accumulation (44%) of the tricarboxylic intermediate harderoporphyrinogen (table 3; fig. 3). Analysis of fecal porphyrins from the patient with this mutation showed no significant accumulation of harderoporphyrin (fig. 3) and a pattern indistinguishable from that of other patients with HCP.
Table 2.
Expression of Mutant CPOX cDNAs in E. coli
|
Activity |
|||
| Constructa | CPOXb | Overallc | Harderoporphyrin (%) |
| CPO/sense | 18,460 | 100 | 11 |
| CPO/antisense | 789 | … | 6 |
| Empty plasmid | 728 | … | 9 |
| CPO-Q162P | 1,786 | 6 | 10 |
| CPO-D171N | 6,595 | 33 | 5 |
| CPO-E201K | 969 | 1 | 15 |
| CPO-A203T | 6,812 | 34 | 7 |
| CPO-A293T | 5,930 | 29 | 8 |
| CPO-G308V | 7,730 | 39 | 6 |
| CPO-R331Wd | 6,394 | 32 | 9 |
| CPO-R391W | 4,710 | 22 | 12 |
| CPO-R401W | 11,970 | 63e | 44e |
| CPO-R447C | 3,240 | 14 | 9 |
Expression vector: pGEX-2T.
Specific activity was expressed in pmol of protoporphyrin/h/mg protein at 37°C, as mean of two duplicate experiments (see text for formula used to calculate residual activity).
Overall activity is shown as percent of wild-type activity.
Previously reported in homozygous HCP (Martasek et al. 1994b).
Table 3.
HCP Clinical Features and Type of Mutation
|
No. (%) of Patients from |
No. (%) of Patients with |
||||
| Clinical Featuresa | Present Study (n = 15) | Western Europeb(n = 13) | Missensea (n = 16) | Splice Sitea (n = 3) | Nonsense/ Frameshifta (n = 9) |
| Acute attack only | 11 (73%) | 10 (77%) | 13 (81%) | 2 (67%) | 6 (67%) |
| Skin lesions only | 0 (0%) | 1 (8%) | 0 (0%) | … | 1 (11%) |
| Both together | 4 (27%) | 2 (15%) | 3 (19%) | 1 (33%) | 2 (22%) |
Figure 3.

A, Coproporphyrin III/I ratio in samples of feces from the patient with the R401W heterozygous mutation. B, High-performance liquid chromatography of porphyrins in vitro from expression studies of this R401W mutation in E. coli. CI = coproporphyrin I; CIII = coproporphyrin III; P = protoporphyrin; H = harderoporphyrin; C = coproporphyrin.
Phylogenetic analysis of amino acid sequences for CPOX showed a large distance between bacteria, yeast, plants, and mammals (fig. 2A). Four mutations (D171N, E201K, R391W, and R401W) alter residues in regions of the enzyme that are conserved throughout evolution (fig. 2B). Other mutations are in regions that are less highly conserved or are conserved only between mammals (fig. 2B). There was no close correlation between degree of conservation and effect on activity.
Figure 2.

A, Phylogenetic tree of amino acid sequences for CPOX. PHYLIP software (Phylogeny Inference Package, version 3.5c) was used to draw the phylogenetic tree and evaluate the protein sequence distance, using the protein sequence parsimony method (Felsenstein 1989). The reference in brackets indicates the SwissProt accession number of the protein sequence. The numbers above the lines indicate the relative distance between species. B, Comparison of amino acid sequences deduced from nucleotide sequences of human (HS), mouse (MM), Saccharomyces cerevisiae (SC), E. coli (EC), Salmonella typhimurium (ST), soybean (GM), Pseudomonas aeruginosa (PS), common tobacco (Nicotiana tabacum [TB]), barley (Hordeum vulgare [HO]), and Synechocystis species (SY). The alignment of the sequences was made with Clustal W software (Thompson et al. 1994). Asterisks indicate identical amino acids. Diamonds indicate similar amino acids. Point mutations are represented above the human sequence. The number indicates the position of the amino acid in the human sequence.
Phenotype-Genotype Relationship
Of the 15 patients in whom mutations were identified, 11 had clinically overt HCP; 3 patients had a first-degree relative with overt disease, and 1 patient was from a family with no clinical manifestations of HCP. Table 3 compares the mode of presentation of the 14 probands who had overt HCP with that of other patients with HCP in whom CPO mutations have been identified. The data confirm that HCP resembles AIP in being characterized mainly by acute neurovisceral attacks (Brodie et al. 1977); 4 of the patients we studied were originally considered to have AIP.
Table 3 shows the relationship between mode of presentation and type of mutation. Truncating mutations (frameshift, nonsense, and splice site) or missense mutations are not associated with a particular mode of presentation (χ2=4.31; P=.09). In two families, different missense mutations gave rise, in different relatives, to more than one type of presentation.
Four patients had mutations that were found, by heterologous expression, to decrease CPOX activity to 33%–39% of the level in control subjects. The mode of presentation in these patients (three had acute attacks only; one had an acute attack with skin lesions) did not differ from the mode of presentation in those with truncating mutations likely to lead to no residual activity (three had acute attacks only; two had acute attacks with skin lesions).
SNPs
Three SNPs, in addition to the five SNPs reported elsewhere (Martasek et al. 1994b; Rosipal et al. 1999), were identified (table 4). All eight SNPs found in the population that we studied gave abnormal patterns on DGGE, thereby facilitating screening of sufficient subjects to establish allele frequencies (table 2). The three novel SNPs were as follows: a C→T transition in exon 5 at nucleotide position 1054, which predicted substitution R351C (frequencies of .97 for allele C and .03 for allele T, with 5.8% observed heterozygosity); an A→C transversion 159 bp downstream from the last base of stop codon (X+159A/C) in exon 7 (frequencies of .68 for allele A and .32 for allele C, with 43.5% observed heterozygosity); and an A→G transition 928 bp downstream of the last base of stop codon (X+928A/G) in exon 7 (frequencies of .69 for allele A and .31 for allele G, with 42.8% observed heterozygosity) (table 4).
Table 4.
Polymorphisms Reported in the Human CPO Gene
| Region and Sequence Modification | Allele Frequency | References |
| Promoter: | ||
| —142 C/Ga | .69 C/.31 G | Rosipal et al. 1999 |
| Exon 4: | ||
| 814 A/C | .80 A/.20 C | Martasek et al. 1994a |
| 880 G/A | .70 G/.30 A | Martasek et al. 1994b |
| Exon 5: | ||
| 990 G/A | .75 G/.25 A | Martasek et al. 1994b |
| 1054 C/T | .97 C/.03 T | Present study |
| Exon 7: | ||
| X+159 A/Cb | .68 A/.32C | Present study |
| X+574 del ATTCTTc | .85 undel/.15 del | Rosipal et al. 1999 |
| X+928 A/Gd | .69A/.31G | Present study |
Numbering according to the first base of transcription initiation site (Delfau-Larue et al. 1994).
At position stop codon +159.
At position stop codon +574.
At position stop codon +928.
Discussion
Using DGGE and direct sequencing, we have identified mutations in the CPO genes of 15 of 17 unrelated British patients with HCP (table 1). Both patients in whom mutations were not identified had an unequivocal diagnosis of HCP. Complete deletions of the CPO gene were excluded by showing that both patients were heterozygous for at least one intragenic SNP. It is probable that the causative mutations either lie outside the regions that we sequenced or are partial deletions or insertions not detected by our PCR-based methods.
Of the 14 mutations in the patients with HCP described here, 10 are novel, bringing the total number of mutations reported in HCP to 27 (table 1; fig. 1) (Delfau-Larue et al. 1994; Fujita et al. 1994; Daimon et al. 1997; Lamoril et al. 1997; Schreiber et al. 1997; Susa et al. 1998; Rosipal et al. 1999), whereas a further 5 mutations have been identified in homozygous patients with HCP (Martasek et al. 1994b; Doss et al. 1999) or harderoporphyria (Lamoril et al. 1995, 1998). Eight of the novel mutations are missense and are located in GC-rich regions containing CpG dinucleotides known to be hot spots for mutation (Cooper and Krawczak 1993). The probable functional importance of the mutated amino acids is indicated by the finding that all are conserved between species (fig. 2B), and four of the eight residues are highly conserved throughout evolution, despite the large distance between bacterial, plant, yeast, and mammalian CPOXs that is revealed by phylogenetic analysis (fig. 2A). Heterologous expression in E. coli of these missense mutations showed that all decreased protoporphyrinogen IX formation by >60% (table 2). The other two novel mutations introduce premature stop codons (table 1) and are likely to abolish CPOX activity, primarily by accelerating mRNA decay (Culbertson 1999).
Previous studies of mutations in patients with typical HCP have not revealed any relationship between this disorder and other clinically distinct conditions caused by CPO mutations. Here we show that a mutation (R331W), previously identified only in homozygous patients with HCP (Martasek et al. 1994b) and having sufficient residual activity (table 2) to support life, can cause overt HCP in a heterozygote. This finding contrasts with data from mutational analysis of homozygous VP, in which there is as yet no evidence that mutations that preserve significant protoporphyrinogen oxidase activity produce symptoms in heterozygotes (Kirsch et al. 1998; Roberts et al. 1998; ), but resembles the relationship between AIP and its homozygous counterpart, in which the same mutations are found in both conditions (Elder 1997).
More unexpectedly, a mutation (R401W) that we identified in a patient with HCP was found, by heterologous expression, to impair decarboxylation of harderoporphyrinogen in a pattern indistinguishable from that shown by a neighboring mutation (K404E) that, in homozygotes or compound heterozygotes, causes harderoporphyria (Lamoril et al. 1998) (table 2; fig. 3B). Of the 16 missense mutations in the CPO gene that have been expressed in vitro, these are the only two that mainly affect the second decarboxylation reaction during the conversion of coproporphyrinogen III to protoporphyrinogen IX (Martasek et al. 1997; Rosipal et al. 1999) (table 2). Both mutations lie in a highly conserved region of CPOX (fig. 2B) that may form part of its single catalytic site (Lamoril et al. 1998). Our data support the hypothesis that the short region around R401–K404 is important for catalysis of the oxidative decarboxylation of harderoporphyrinogen. There is evidence that this intermediate remains adjacent to the catalytic site during the conversion of coproporphyrinogen III to protoporphyrinogen IX (Elder et al. 1978). The R401–K404 region may be important in preventing diffusion of harderoporphyrinogen away from the catalytic site during the sequential reaction.
The patient described here who had the R401W mutation trans to a normal allele had typical HCP without the hemolytic anemia or increased fecal excretion of harderoporphyrin that characterizes harderoporphyria in adults (fig. 3A) (Lamoril et al. 1998). Similarly, those asymptomatic parents of harderoporphyric patients who were heterozygous for the K404E mutation had a fecal porphyrin excretion pattern identical to that found in patients with HCP (Lamoril et al. 1998; C. Martin, personal communication). Our finding that a single copy of a gene carrying a mutation in this region (a mutation that is functionally indistinguishable, by heterologous expression in vitro, from the harderoporphyria mutation) strongly suggests that K404E heterozygotes also may develop acute attacks of porphyria and should therefore be managed in the same way as individuals with clinically latent HCP. Still to be determined is the mechanism by which CPOX from the normal allele produces a pattern of porphyrin excretion in these heterozygotes that is typical of HCP rather than harderoporphyria or is intermediate between these two conditions. Human CPOX is a 76-kD protein that is active as a homodimer in the mitochondrial intermembrane space (Martasek et al. 1997). It is possible that the R401W and K404E mutations impair dimerization or the function of mutant-normal heterodimers so that activity is decreased to the levels ∼50% of normal that are observed in heterozygotes (Lamoril et al. 1998).
Our findings also reveal differences between the pattern of mutations in HCP and in other acute hepatic porphyrias. Only one of the mutations that we identified (1277G→A, which was previously reported in a Czech family [Delfau-Larue et al. 1994]) was present in more than one family. The least common of the acute porphyrias in the United Kingdom thus shows more allelic heterogeneity than either of the other two, AIP (Whatley et al. 1999b) and VP (Whatley et al. 1999a). Mutations were spread throughout the gene (fig. 1), unlike mutations in AIP (in which 60% of mutations are clustered in three exons of the HMBS gene [Puy et al. 1997; Whatley et al. 1999b]), and a higher proportion (72% of the patients reported here [table 1]) are missense than in AIP or VP (Puy et al. 1997; Whatley et al. 1999a, 1999b).
The probands of all the families we studied presented with acute attacks of porphyria, except for one family in which latent HCP was discovered fortuitously by urine testing. Skin lesions, when present (table 3), were a minor feature. This uniformity of presentation adds to previous evidence (Brodie et al. 1977; Kuhnel et al. 2000) that the clinical characteristics of HCP are closer to those of AIP than to those of other mixed porphyria (e.g., VP or porphyria cutanea tarda [PCT]) (fig. 4). Neither the type of mutation (table 3) nor the extent of inactivation of CPOX (table 2), as assessed by heterologous expression in E. coli, influenced the mode of presentation to any major extent. The wide range of residual activity in vitro (table 2) was unexpected, because levels of CPOX activity in cells from patients with latent or overt HCP are ∼50% of normal (Elder et al. 1976; Grandchamp and Nordmann 1977; Grandchamp et al. 1996). This discrepancy may be explained, at least for some mutations, by the presence of factors in human cells that further decrease activity by affecting enzyme processing or stability.
Figure 4.

Clinical features in acute hepatic porphyrias in British patients.
In conclusion, this study highlights differences between the molecular epidemiology of HCP and that of other autosomal dominant acute porphyrias, demonstrates that heterozygotes for mutations previously known to cause only homozygous HCP or harderoporphyria may develop clinically overt HCP, shows that there is no major correlation between genotype and severity of disease in HCP, and adds to the evidence that a few amino acid residues in exon 6 determine the efficiency of decarboxylation of harderoporphyrinogen.
Acknowledgments
This work was supported by grants from INSERM (U409) and Université Paris VII.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- CONSENSE, http://evolution.genetics.washington.edu/phylip.html
- Human Gene Mutation Database,http://www.uwcm.ac.uk/uwcm/mg/hgmd0.html (for mutations in CPO gene)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for HCP [MIM 121300]) [PubMed]
- Phylogeny Inference Package, http://evolution.genetics.washington.edu/phylip.html
References
- Andrews J, Erdjument H, Nicholson DC (1984) Hereditary coproporphyria: incidence in a large English family. J Med Genet 21:341–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake D, McManus J, Cronin V, Ratnaike S (1992) Fecal coproporphyrin isomers in hereditary coproporphyria. Clin Chem 38:96–100 [PubMed] [Google Scholar]
- Brodie MJ, Thompson GG, Moore MR, Beattie AD, Goldberg A (1977) Hereditary coproporphyria: demonstration of the abnormalities in haem biosynthesis in peripheral blood. Q J Med 46:229–241 [PubMed] [Google Scholar]
- Cooper DN, Krawczak M (1993) Human gene mutation. BIOS Scientific, Oxford [Google Scholar]
- Culbertson MR (1999) RNA surveillance: unforeseen consequences for gene expression, inherited genetic disorders and cancer. Trends Genet 15:74–80 [DOI] [PubMed] [Google Scholar]
- Daimon M, Gojyou E, Sugawara M, Yamatani K, Tominaga M, Sasaki H (1997) A novel missense mutation in exon 4 of the human coproporphyrinogen oxidase gene in two patients with hereditary coproporphyria. Hum Genet 99:199–201 [DOI] [PubMed] [Google Scholar]
- Delfau-Larue MH, Martasek P, Grandchamp B (1994) Coproporphyrinogen oxidase: gene organization and description of a mutation leading to exon skipping. Hum Mol Genet 3:1325–1330 [DOI] [PubMed] [Google Scholar]
- Doss MO, Gross U, Lamoril J, Kranl C, Jacob K, Doss M, Da Silva V, Freesemann AG, Deybach JC, Sepp, Nordamnn Y (1999) Compound heterozygous hereditary coproporphyria with fluorescing teeth. Ann Clin Biochem 36:680–682 [DOI] [PubMed] [Google Scholar]
- Elder GH (1997) Hepatic porphyrias in children. J Inherit Metab Dis 20:237–246 [DOI] [PubMed] [Google Scholar]
- Elder GH, Evans JO, Jackson JR, Jackson AH (1978) Factors determining the sequence of oxidative decarboxylation of the 2- and 4-propionate substituents of coproporphyrinogen III by coproporphyrinogen III oxidase in rat liver. Biochem J 169:215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder GH, Evans JO, Thomas N (1976) The primary enzyme defect in hereditary coproporphyria. Lancet 2:1217–1219 [DOI] [PubMed] [Google Scholar]
- Felsenstein J (1989) Phylip: Phylogeny inference package (version 3.2). Cladistic 5:164–166 [Google Scholar]
- Fujita H, Kondo M, Taketani S, Nomura N, Furuyama K, Akagi R, Nagai T, Terajima M, Galbraith RA, Sassa S (1994) Characterization and expression of cDNA encoding coproporphyrinogen oxidase from a patient with hereditary coproporphyria. Hum Mol Genet 10:1807–1810 [DOI] [PubMed] [Google Scholar]
- Grandchamp B, Nordmann Y (1977) Decreased lymphocyte coproporphyrinogen III oxidase activity in hereditary coproporphyria. Biochem Biophys Res Commun 74:1089–1095 [DOI] [PubMed] [Google Scholar]
- Grandchamp B, Phung N, Nordmann Y (1977) Homozygous case of hereditary coproporphyria. Lancet 2:1348 [DOI] [PubMed] [Google Scholar]
- Grandchamp B, Puy H, Lamoril J, Deybach JC, Nordmann Y (1996) Molecular hepatology: molecular pathogenesis of hepatic acute porphyrias. J Gastroenterol Hepatol 11:1046–1052 [DOI] [PubMed] [Google Scholar]
- Hawk JLM, Magnus IA, Parkes A, Elder GH, Doyle M (1978) Deficiency of hepatic copro-porphyrinogen oxidase in hereditary coproporphyria. J R Soc Med 71:775–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappas A, Sassa S, Galbraith RA, Nordmann Y (1995) The porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular bases of inherited disease, 7th ed. McGraw-Hill, New York, pp 2103–2159 [Google Scholar]
- Kirsch RE, Meissner PN, Hift RJ (1998) Variegate porphyria. Semin Liver Dis 18:33–41 [DOI] [PubMed] [Google Scholar]
- Kuhnel A, Gross U, Doss MO (2000) Hereditary coproporphyria in Germany: clinical-biochemical studies in 53 patients. Clin Biochem 33:465–473 [DOI] [PubMed] [Google Scholar]
- Lamoril J, Deybach JC, Puy H, Grandchamp B, Nordmann Y (1997) Two novel mutations in the coproporphyrinogen oxidase gene. Hum Mutat 9:78–80 [DOI] [PubMed] [Google Scholar]
- Lamoril J, Martasek P, Deybach JC, Da Silva V, Grandchamp B, Nordmann Y (1995) A molecular defect in coproporphyrinogen oxidase gene causing harderoporphyria, a variant form of hereditary coproporphyria. Hum Mol Genet 4:275–278 [DOI] [PubMed] [Google Scholar]
- Lamoril J, Puy H, Gouya L, Rosipal R, Da Silva V, Grandchamp B, Foint T, Bader-Meunier B, Dommergues JP, Deybach JC, Nordmann Y (1998) Neonatal hemolytic anemia due to inherited harderoporphyria: clinical characteristics and molecular basis. Blood 91:1453–1457 [PubMed] [Google Scholar]
- Lockwood WH, Poulos V, Rossi E, Curnow DH (1985) Rapid procedure for fecal porphyrin assay. Clin Chem 31:1163–1167 [PubMed] [Google Scholar]
- Long C, Smyth SJ, Woolf J, Murphy GM, Finlay AY, Newcombe RG, Elder GH (1993) The detection of latent variegate porphyria by fluorescence emission spectroscopy of plasma. Br J Dermatol 129:9–13 [DOI] [PubMed] [Google Scholar]
- Martasek P (1998) Hereditary coproporphyria. Semin Liver Dis 18:25–32 [DOI] [PubMed] [Google Scholar]
- Martasek P, Camadro JM, Delfau-Larue MH, Dumas JB, Montagne JJ, De Verneuil H, Labbe P, Grandchamp B (1994a) Molecular cloning sequencing and functional expression of a cDNA encoding human coproporphyrinogen oxidase. Proc Natl Acad Sci USA 91:3024–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martasek P, Camadro JM, Raman CS, Lecomte MC, Le Caer JP, Demeler B, Grandchamp B, Labbe P (1997) Human coproporphyrinogen oxidase: biochemical characterization of recombinant normal and R231W mutated enzymes expressed in E. coli as soluble, catalytically active homodimers. Cell Mol Biol (Noisy-le-grand) 43:47–58 [PubMed] [Google Scholar]
- Martasek P, Nordmann Y, Grandchamp B (1994b) Homozygous hereditary coproporphyria caused by an arginine to tryptophan substitution in coproporphyrinogen oxidase and common intragenic polymorphisms. Hum Mol Genet 3:477–480 [DOI] [PubMed] [Google Scholar]
- Nordmann Y, Deybach JC (1990) Human hereditary porphyrias. In: Dailey HA, ed. Biosynthesis of heme and chlorophylls. McGraw-Hill, New York pp 491–542 [Google Scholar]
- Nordmann Y, Grandchamp B, de Verneuil H, Phung L, Cartigny B, Fontaine G (1983) Harderoporphyria: a variant hereditary coproporphyria. J Clin Invest 72:1139–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh-Fitzpatrick MB (1980) A plasma porphyrin fluorescence marker for variegate porphyria. Arch Dermatol 116:543–547 [PubMed] [Google Scholar]
- Puy H, Deybach JC, Lamoril J, Robreau AM, Da Silva V, Gouya L, Grandchamp B, Nordmann Y (1997) Molecular epidemiology and diagnosis of PBG deaminase gene defects in acute intermittent porphyria. Am J Hum Genet 60:1373–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AG, Puy H, Dailey TA, Morgan RR, Whatley SD, Dailey HA, Martasek P, Nordmann Y, Deybach JC, Elder GH (1998). Molecular characterisation of homozygous variegate porphyria. Hum Mol Genet 7:1921–1925 [DOI] [PubMed] [Google Scholar]
- Rosipal R, Lamoril J, Puy H, Da Silva V, Gouya L, De Rooij FWM, Te Velde K, Nordmann Y, Martasek P, Deybach JC (1999) Systematic analysis of coproporphyrinogen oxidase gene defects in hereditary coproporphyria and mutation update. Hum Mut 13:44–53 [DOI] [PubMed] [Google Scholar]
- Schreiber WE, Zhang X, Senz J, Jamani A (1997) Hereditary coproporphyria: exon screening by heteroduplex analysis detects two novel mutations in the coproporphyrinogen oxidase gene. Hum Mutat 10:196–200 [DOI] [PubMed] [Google Scholar]
- Susa S, Daimon M, Yamamori I, Kondo M, Yamatani K, Sasaki H, Kato T (1998) A novel mutation of coproporphyrinogen oxidase (CPO) gene in a Japanese family. J Hum Genet 43:182–184 [DOI] [PubMed] [Google Scholar]
- Taketani S, Kohno H, Furukawa T, Yoshinaga T, Tokunaga R (1994) Molecular cloning, sequencing and expression of cDNA encoding human coproporphyrinogen oxidase. Biochim Biophys Acta 1183:547–549 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) Clustal W improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whatley SD, Puy H, Morgan RR, Robreau AM, Roberts AG, Nordmann Y, Elder GH, Deybach JC (1999a) Variegate porphyria in Western Europe: identification of PPOX gene mutations in 104 families, extent allelic heterogeneity, and absence of correlation between phenotype and type of mutation. Am J Hum Genet 65:984–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whatley SD, Woolf JR, Elder GH (1999b) Comparison of complementary and genomic DNA sequencing for the detection of mutations in the HMBS gene in British patients with acute intermittent porphyria: identification of twenty-five novel mutations. Hum Genet 104:505–510 [DOI] [PubMed] [Google Scholar]