

Abstract
Nemaline myopathy (NM) is a clinically and genetically heterogeneous disorder characterized by muscle weakness and the presence of nemaline bodies (rods) in skeletal muscle. Disease-causing mutations have been reported in five genes, each encoding a protein component of the sarcomeric thin filament. Recently, we identified mutations in the muscle α-skeletal-actin gene (ACTA1) in a subset of patients with NM. In the present study, we evaluated a new series of 35 patients with NM. We identified five novel missense mutations in ACTA1, which suggested that mutations in muscle α-skeletal actin account for the disease in ∼15% of patients with NM. The mutations appeared de novo and represent new dominant mutations. One proband subsequently had two affected children, a result consistent with autosomal dominant transmission. The seven patients exhibited marked clinical variability, ranging from severe congenital-onset weakness, with death from respiratory failure during the 1st year of life, to a mild childhood-onset myopathy, with survival into adulthood. There was marked variation in both age at onset and clinical severity in the three affected members of one family. Common pathological features included abnormal fiber type differentiation, glycogen accumulation, myofibrillar disruption, and “whorling” of actin thin filaments. The percentage of fibers with rods did not correlate with clinical severity; however, the severe, lethal phenotype was associated with both severe, generalized disorganization of sarcomeric structure and abnormal localization of sarcomeric actin. The marked variability, in clinical phenotype, among patients with different mutations in ACTA1 suggests that both the site of the mutation and the nature of the amino acid change have differential effects on thin-filament formation and protein-protein interactions. The intrafamilial variability suggests that α-actin genotype is not the sole determinant of phenotype.
Introduction
Nemaline myopathy (NM) is a genetic muscle disorder characterized clinically by muscle weakness and hypotonia and pathologically by the presence of rod-shaped structures (nemaline bodies) in muscle fibers of affected patients. Clinically, the disorder is heterogeneous, ranging from death, in utero, associated with fetal akinesia (Lammens et al. 1997), to a mild skeletal myopathy presenting during childhood or adulthood (North et al. 1997). The weakness typically affects proximal musculature, with variable involvement of the facial, bulbar, and respiratory muscles. Cardiac muscle is usually spared. Nemaline bodies (rods) are clearly detectable at the light-microscopy (LM) level, and, ultrastructurally, they originate from the Z-disks of the sarcomeres. One of the main components of rods is α-actinin 2, an actin-binding protein that localizes to the Z-disk (Blanchard et al. 1989). Other pathological features of NM include abnormal fiber type differentiation and fiber atrophy and/or hypertrophy (Volpe et al. 1982; Miike et al. 1986; North et al. 1997). In a subset of patients with NM, mutations have been identified in five different genes—those for α-tropomyosin slow (TPM3) (Laing et al. 1995), nebulin (NEB) (Pelin et al. 1999), β-tropomyosin (TPM2) (Donner et al. 2000), troponin T1 (TNNT1) (Johnston et al. 2000), and muscle α-skeletal actin (ACTA1 [MIM 102610]) (Nowak et al. 1999). There are additional cases of NM that do not link to any of the five identified loci, suggesting further genetic heterogeneity.
In 1999, we published the first identified cases of NM due to mutations in muscle α-skeletal actin (Nowak et al. 1999). In skeletal muscle, actin function depends on the balance between monomeric G-actin (globular) and filamentous F-actin, a polymer of G-actin subunits. F-actin, together with tropomyosin, troponin, and nebulin, forms the thin filaments of the skeletal-muscle contractile apparatus. F-actin directly interacts with myosin to generate contractile force (Adelstein and Eisenberg 1980).
The frequency of actin NM and the mechanism by which mutations in ACTA1 result in the clinical and pathological features of NM are unknown. Here we report a new series of patients with ACTA1 mutations who are derived from a large Australian cohort. We define the frequency and range of clinical and pathological phenotypes associated with mutations in muscle α-skeletal actin, and we provide insights into the pathogenetic mechanisms underlying the disease process.
Subjects and Methods
Ascertainment of Subjects
The Australian cohort of 72 patients with NM was ascertained through records of the neurology and pathology services in each state. The diagnosis of NM was based on European Neuromuscular Centre (ENMC) criteria (Wallgren-Pettersson and Laing 2000).
Mutation Analysis
DNA for ACTA1 mutation analysis was available from 35 patients. All patients were screened for mutations in ACTA1, by sequencing of PCR products amplified from exons 2–7 of genomic DNA. DNA samples isolated from ⩾100 normal individuals served as controls. When available, parental DNA samples also were screened. The methodology was per Nowak et al. (1999), except for the use of a different reverse primer (5RB-5′-tgcccgccgactccatacctgg- 3′) for exon 5 and a different forward primer (6FB-5′-ccagccctccttcatcggtgag-3′) for exon 6, to avoid a newly identified polymorphism present in intron 5 on ∼15% of chromosomes.
Histochemistry and Immunohistochemistry for LM
Cryostat sections (8.0 μm) were stained by means of the modified Gomori trichrome method described by Engel and Cunningham (1963). We performed indirect immunofluorescence, using mouse monoclonal (mAb) anti–skeletal slow myosin (identifies type 1 fibers) (1:250; Chemicon International), mouse mAb anti–skeletal fast myosin (identifies type 2 fibers) (1:200; Sigma), mouse mAb anti–neonatal skeletal myosin (1:100; Novocastra), affinity-purified rabbit polyclonal anti–α-actinin 2 (1:500) and anti–α-actinin 3 (1:50; North and Beggs 1996), mouse mAb anti–sarcomeric tropomyosin (1:400; Sigma), and mouse mAb anti-nebulin (1:400; Sigma). To determine the expression of α–skeletal actin and other actin isoforms, we used mouse mAb anti–sarcomeric actin (1:250; Sigma), mouse mAb anti–α-cardiac actin (1:10; American Research Products), mouse mAb anti–α-smooth-muscle actin (1:100; Sigma), mouse mAb anti–β-cytoplasmic actin (1:200; Sigma), and sheep anti–γ-cytoplasmic actin (1:200; Dr. J. C. Bulinski). Rhodamine-phalloidin was used to identify filamentous actin (1:400; Sigma). We used either affiniPure goat anti-mouse and anti-rabbit CY3- or fluoroscein-conjugated secondary antibodies (1:250 and 1:200, respectively; Jackson ImmunoResearch Laboratories) and Alexa488-conjugated goat anti-mouse (1:100; Molecular Probes) for confocal images. All studies were repeated on at least two occasions, to confirm initial findings. All results were compared with results for muscle tissue with normal histology, from age-matched controls.
Stained serial sections were examined under an Olympus BX50 microscope, and images were recorded by either a SPOT Colour Digital Imaging Camera or Scanning Laser Confocal Microscopy (Leica SP2). Analysis of fiber typing and fiber diameters was performed on photographs of serial sections stained with anti–skeletal slow and anti–skeletal fast muscle myosin heavy-chain antibodies.
RNA Slot-Blot Analysis
RNA was isolated from 40 frozen muscle sections (5 μm), from patient 2 and from an age- and sex-matched control. RNA was resuspended in 20 μl of DEPC-MilliQ water, and equal volumes were loaded, in duplicate, onto a nylon Hybond-N+ transfer membrane (Amersham) (Kingston 1997). The blots were probed with [γ-32P] end-labeled oligonucleotides complementary to the 3′ UTR of the α–skeletal-actin (gaagattcgtcgtcctgagaagtcgcgtgctggaggtggagtgtg) and α-cardiac-actin (aataccgtcatcctgactggaaggtagatggagagagaaggcatc) transcripts. The blots were stripped and rehybridized with a human β-spectrin–specific oligonucleotide probe (ggtctctgcgcgtcccgactccgccgcgcccgccagccccacctg). Levels of transcripts were quantified by densitometric analysis (ImageQuant, version 4.0; Molecular Dynamics).
Results
Mutation Analysis
Five different missense ACTA1 mutations were identified in the five probands (patients 1–5) (fig. 1); these represent new dominant mutations, on the basis of the absence of family history and of sequencing of genomic DNA from both parents of patients 1–3 and from the mother and two unaffected children of patient 5. Patients 6 and 7 inherited the missense mutation (Asn115Ser) in ACTA1 from their mother (patient 5). All the mutations gave rise to SSCP variants that were not found in DNA from ⩾100 controls, and all mutated amino acid residues were highly conserved in known actins (fig. 2) (Sheterline et al. 1999). Prenatal diagnosis for ACTA1 mutation was performed for the family of patient 1 during a subsequent pregnancy; neither the parents nor the fetus carried the ACTA1 mutation identified in the proband.
Figure 1.

Position of mutated residues in α-skeletal actin. Five different missense mutations were identified in seven patients with NM. These mutations' respective locations within the actin molecule are represented by a schematic model of the three-dimensional structure of actin (Kabsch et al. 1990, p. 37 [reprinted by permission from Nature]). Patient 1 (I357L) (orange) shows lethal severe congenital; patient 2 (R183G) (pink), lethal severe congenital; patient 3 (G268C) (yellow), childhood onset; patient 4 (I136M) (blue), typical congenital. Also represented is the autosomal dominant family (N115S) (purple).
Figure 2.

Comparison of muscle α-skeletal-actin protein sequences in various species and in the five mutated residues in the patients. All mutated residues in α-skeletal actin are highly conserved across the species shown. An asterisk (*) denotes conserved amino acid, and a dash (–) denotes a deleted amino acid. The amino acid numbering nomenclature corresponds to that of the human muscle α-skeletal-actin protein sequence.
Clinical Features
Patient 1
This female infant was born, at term, with severe hypotonia, minimal spontaneous movements of the arms and legs, sparse facial movements, and fractures of both femurs. Both the parents and a 3-year-old sibling were clinically normal. The patient failed to achieve any motor milestones and required a gastrostomy tube for feeding. Results of both cranial magnetic-resonance imaging (MRI) and echocardiography were normal. The patient died of respiratory failure at age 6 mo.
Patient 2
This male infant was born, at term, with severe hypotonia, reduced muscle bulk, lack of antigravity movements, and facial diplegia. Decreased fetal movements were noted during the last few weeks of pregnancy. Both the parents and two siblings were clinically normal. The patient failed to achieve any motor milestones and had both difficulty in swallowing and poor respiratory effort. Results of electrocardiography, echocardiography, and cranial MRI were normal. The patient died of respiratory failure at age 13 mo.
Patient 3
This male patient, the first child of healthy parents, had no problems during the neonatal period. At age 5 years, he presented with both inability to run and frequent falls; he had poor muscle bulk, pes cavus, and bilateral foot drop, suggestive of a peripheral neuropathy. Electromyography showed a myopathic pattern, and results of nerve conduction studies were normal. The patient had no feeding or respiratory difficulties, and echocardiography showed normal cardiac function. At the time of the present study, he was 10 years old and had slowly progressive weakness, with involvement of proximal muscles.
Patient 4
This male patient was weak and floppy from birth and had feeding difficulties, with a poor suck. Both the parents and four siblings were clinically normal. At age 2 years, the patient walked with assistance and had recurrent lower-respiratory-tract infections. His weakness has been nonprogressive. At the time of the present study, he was 45 years old and, although he had poor muscle bulk and mild proximal weakness, was physically active and regularly engaged in long-distance competitive cycling; he had a weak cough and frequent respiratory infections. Echocardiography showed normal cardiac function.
Family A
Family A has autosomal dominant NM. Patient 5 was the third child of clinically unaffected normal parents with no family history of neuromuscular disease. Both the affected mother (patient 5, who was 35 years old at the time of the present study) and the youngest child (patient 7, who was 4 years old at the time of the present study) display clinical features characteristic of typical congenital NM, with onset of feeding difficulties, recurrent lower-respiratory-tract infections, hypotonia, facial diplegia, and proximal weakness during the first weeks of life, as well as delayed motor milestones. At age 3 years, patient 7 underwent a sleep study, which demonstrated mild nocturnal hypoventilation without significant oxygen desaturation. The weakness has been nonprogressive, with improvement in respiratory status over time. Because of the family history of NM, a pediatric neurologist evaluated patient 6 (the daughter of patient 5) at age 4 years, and no abnormality was detected; however, beginning at age 6 years, she exhibited mild proximal weakness, with frequent falls, and developed progressive scoliosis requiring surgery at age 14 years. She has a slowly progressive disease course. At the time of the present study, she was 19 years old and had recurrent respiratory infections with a weak cough and was able to run only short distances. In all family members, echocardiography showed normal cardiac function.
LM and Electron Microscopy (EM)
Nemaline bodies were present on Gomori trichrome stain in biopsy samples from patients 1–6 (patient 7 has not been biopsied) (fig. 3) and were either scattered within the cytoplasm or clustered under the sarcolemma (fig. 3). There was marked interpatient variability in the percentage of fibers with rods, the proportion of individual fibers occupied by rods, and the extent of myofibrillar disruption in the area surrounding the rods (as shown by EM). Rods were present in 98%–100% of fibers in patients 1, 4, and 6, whereas patients 2, 3, and 5 had, respectively, 50%, 14%, and 35% of fibers containing rods (table 1).
Figure 3.

Gomori trichrome staining of muscle-biopsy sections. Gomori trichrome treatment results in deep-purple staining of nemaline bodies (indicated by arrows), which were observed in muscle-biopsy sections from all patients. Stained sections are shown for patients 1 (severe congenital [a]), 2 (severe congenital [b]), and 6 (childhood onset [c]).
Table 1.
Clinical, Pathological, and Mutational Data on Patients with Actin NM
|
Age at |
Fiber Type(%) |
Fiber Status(%) |
||||||||||
| Patient (Family) | Clinical Classificationa | Mode of Inheritanceb | Time of Study | Biopsy | Slow Myosin | Fast Myosin | Fibers with Rods(%) | Atrophied | Hypertrophied | Actin Mutation | Amino Acid Substitution | Binding Sites/Other Functions |
| 1 | Severe congenital | Sporadic | (Died at 6 mo) | 6 wk | ∼99 | 40–50 | 99 | 5–10 | 0 | ATC→CTC | Ile357Leu | α-Actinin,c tropomyosind |
| 2 | Severe congenital | Sporadic | (Died at 13 mo) | 5 wk | 50 | 50 | 50 | 5 | 0 | CGC→GGC | Arg183Gly | DNase Ie |
| 3 | Childhood onset | Sporadic | 10 years | 5 years | 100 | 10–15 | 14 | 5 | 5 | GGT→TGT | Gly268Cys | Filament stabilizationf |
| 4 | Typical congenital | Sporadic | 45 years | 27 years | 90 | 10 | 98 | 0 | 90 | ATC→ATG | Ile136Met | ? |
| 5 (A) | Typical congenital | AD | 35 years | 34 years | 100 | 20 | 35 | 30 | 0 | AAC→AGC | Asn115Ser | α-Actinin,g filaminh |
| 6 (A) | Childhood onset | AD | 19 years | 17 years | 100 | ∼1 | 100 | ∼10 | 0 | AAC→AGC | Asn115Ser | α-Actinin,g filaminh |
| 7 (A)i | Typical congenital | AD | 4 years | … | … | … | … | … | … | AAC→AGC | Asn115Ser | α-Actinin,g filaminh |
In general, fiber atrophy was not a dominant histopathological feature. However, EM revealed dramatically atrophied fibers that were not visible by LM in the severely affected patients (i.e., patients 1 and 2) (see fig. 4). Interestingly, in patient 4 (i.e., the cyclist), 90% of fibers were hypertrophied (mean deltoid-fiber size ∼102 μm, compared with an age-matched normal mean of ∼55 μm) (Polgar et al. 1973).
Figure 4.

EM of muscle samples from patients 1 (severe congenital NM [a and b]) and 6 (childhood onset NM [c and d]). Nemaline bodies are present in all patients and are associated with disruption of the sarcomeric register. Rods appear either as extensions of sarcomeric Z-lines, in random array without obvious attachment to Z-lines (often in areas devoid of sarcomeres) or in large clusters localized at the sarcolemma or intermyofibrillar spaces (d). Glycogen accumulation (long arrow in panel a) and an increase in intermyofibrillar spaces (arrow in panel d) are common. Additional features include intranuclear rods (short arrow in panel a) and dramatically atrophied fibers (arrow in panel b) in patient 1, as well as whorling of thin filaments in patient 6 (arrow in panel c).
In all patients, there were abnormal glycogen accumulations at the EM level, with abundant glycogen granules free in both the sarcoplasm and intermyofibrillar spaces of most fibers (fig. 4). By EM, we observed ultrastructural changes that correlated with clinical severity. In the two patients with severe congenital NM (i.e., patients 1 and 2), areas containing sarcomeres were disordered in appearance, and nemaline bodies occupied a large proportion of fibers, often in areas completely devoid of sarcomeric structural units. Patient 1 also had occasional central nuclei and intranuclear rods. We have observed intranuclear rods in a total of five infants with severe congenital NM, but only patient 1 was found to have a mutation in α-skeletal actin (authors' unpublished data). In contrast, the mildly affected patients had fibers containing large areas of normal sarcomeric register. Patient 6 also had many fibers with both a “moth-eaten” appearance and “whorling” of thin filaments (fig. 4), features that we have not observed in an age-matched disease control without an actin mutation.
Immunohistochemistry
Fiber typing
Staining with slow myosin demonstrated a marked type I fiber predominance (90%–100%) in five of the six patients, with coexpression of both slow- and fast-myosin isoforms in a subset of fibers in patients 1, 3, and 5 (table 1). α-Actinin 3 is a Z-line protein expressed only in type II (i.e., fast) fibers (North and Beggs 1996). Patients 1, 3, and 4 (in whom there was 90%–100% type I fiber predominance) showed abnormal expression of α-actinin 3 in type I (i.e., slow-myosin positive) fibers and/or in a subset of fibers expressing both slow- and fast-myosin isoforms. Patients 5 and 6 did not express detectable levels of α-actinin 3, because of homozygosity for a stop codon in exon 16 of ACTN3, present in ∼18% of the normal population (North et al. 1999). Patient 2 had normal type II fiber–restricted α-actinin 3 expression.
Actin
Immunocytochemical staining of muscle-biopsy sections from controls, by use of an anti–α-skeletal-actin antibody (α-sr-1, which also recognizes α-cardiac actin) demonstrated a homogenous “honeycomb” pattern of staining in a meshlike network surrounding individual myofibrils. Unexpectedly, this antibody did not give the striated staining pattern that would be expected if it stained the sarcomeric thin filaments. To unmask the actin epitope in thin filaments, we performed a variety of staining protocols, including acetone fixation and glutaraldehyde fixation, detergent treatment, antigen unmasking by heating, and the use of high-salt buffers. None of these protocols revealed a striated staining pattern when α-sr-1 was used (data not shown). Other laboratories doing research on muscle tissue have obtained similar results with α-sr-1 (C. Sewry and H. Jungbluth, personal communication). Therefore, α-sr-1 appears to recognize α-skeletal actin at the periphery of myofibrils, rather than an epitope within sarcomeric thin filaments.
α-Sarcomeric-actin staining of muscle sections from patients 3–6 revealed a uniform honeycomb staining pattern when α-sr-1 was used, similar to that observed in age-matched controls (fig. 5). However, in patients 1 and 2 (who has lethal severe congenital NM), large areas of many fibers showed a complete absence of α-skeletal-actin immunoreactivity, with abnormal aggregations of actin in a disordered distribution across the fiber (fig. 5f). This staining pattern was not observed in an age-matched patient with severe congenital NM who does not have an actin mutation (data not shown). α-sr-1 did not stain the rods. However, staining with phalloidin demonstrated a striated staining pattern and also intensely stained the rod bodies (fig. 5), suggesting that filamentous actin is a constituent of rods in these patients.
Figure 5.

α-Actinin 2, sarcomeric-actin, and phalloidin staining of muscle-biopsy sections from patients 1 and 6. Immunolabeling with α-actinin 2 both demonstrated intense positive staining of Z-lines in all the patients and intensely stained the rods (arrowheads in panels e and i). Muscle sections labeled with a sarcomeric-actin antibody reveal a meshlike honeycomb staining pattern in the control (b) and in mildly affected patients (as shown for patient 6, in panel j). The two severely affected patients showed abnormal localization of actin, with large areas devoid of staining (as shown for patient 1, in panel f). Double labeling with α-actinin 2 and α-skeletal actin (g) demonstrated that areas devoid of sarcomeric actin in patients 1 and 2 contained α-actinin 2–reactive striations. Unlike the sarcomeric-actin antibody, phalloidin demonstrated a striated staining pattern and intensely stained the rods (arrowheads in panels h and l).
Sarcomeric proteins
In all patients, immunohistochemistry using an antibody that recognizes α-actinin 2 resulted in intense striated labeling of muscle sections and also positively stained the rod bodies (fig. 5). Double labeling with α-actinin 2 and α-skeletal actin (fig. 5g) demonstrated that areas devoid of sarcomeric actin in patients 1 and 2 contained α-actinin 2–reactive striations. Immunostaining with antibodies that recognize tropomyosin and nebulin produced intense striated labeling of thin filaments but did not stain rods. In patients 1 and 2, there was striking interfiber variability in the intensity of staining, both for tropomyosin and for nebulin, a feature not observed in mildly affected patients.
Possible Compensation by Other Actin Isoforms
α-Cardiac actin is the major actin isoform expressed in developing and regenerating skeletal muscle. We hypothesized that α-cardiac actin may compensate for mutant α-skeletal actin, and we sought to determine expression levels in patients with NM and in controls. Immunostaining of muscle sections by an antibody that specifically recognizes α-cardiac actin produced intense striated labeling of 10%–20% of fibers in patients 1 and 2, 1%–5% of fibers in patient 3, and occasional or absent staining in patients 4–6. For patients 3–6, these results did not differ significantly from those observed in age-matched controls. The age-matched control for patients 1 and 2 showed only 1%–5% of fibers staining for α-cardiac actin. However, α-cardiac-actin staining was positive in 10%–20% of fibers in an age-matched patient with severe congenital NM but without an actin mutation, suggesting that there is an increased proportion of regenerative fibers as part of the severe disease phenotype, rather than specific compensatory up-regulation of α-cardiac actin (data not shown). To confirm these results, we performed slot-blot analysis of mRNA encoding α-skeletal actin and α-cardiac actin, in muscle-biopsy sections from patient 2 and from an age-matched control (fig. 6). In both samples, the α-skeletal actin:α-cardiac actin ratio was 4–5:1 and was not suggestive of transcriptional up-regulation of α-cardiac actin in patient 2.
Figure 6.
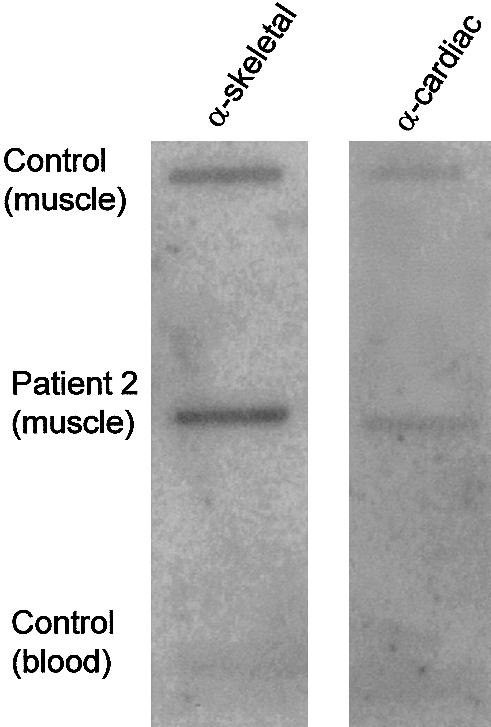
RNA slot-blot analysis of α-skeletal-actin and α-cardiac-actin transcripts. RNA from patient 2 and from an age-matched control was blotted, in duplicate, onto membranes. Membranes were hybridized with oligonucleotide probes complementary to the 3′ UTR of the α-skeletal-actin or α-cardiac-actin transcripts, were washed, and then were exposed to film. The α-skeletal actin:α-cardiac actin ratio was ∼4–5:1 in both the patient and the control, on the basis of densitometric analysis. Specificity of oligonucleotide labeling was confirmed by an absence of hybridization to RNA isolated from blood, and equal loading between duplicate samples was verified by reprobing with an oligonucleotide complementary to β-spectrin.
To determine whether there was altered expression of other actin isoforms in these patients, we stained muscle sections with antibodies specific to α-smooth-muscle actin, β-cytoplasmic actin, and γ-cytoplasmic actin. We did not detect any significant difference between samples from the patients and those from controls (data not shown).
Discussion
In this study, we have identified five novel α-skeletal-actin mutations in a series of 35 patients with NM, suggesting that ACTA1 mutations may account for ∼15% of NM cases. The actin mutations in the five sporadic cases represent new dominant mutations. One proband subsequently had two affected children, indicating a dominant inheritance pattern. The marked variability, in clinical phenotype, among patients with different mutations in muscle α-skeletal actin suggests that the site of the mutation and the nature of the amino acid change have differential effects on thin-filament formation or on protein-protein interactions. For example, patient 1 (who has severe congenital NM) has a mutation at residue 357 (Ile357Leu), which is an active site for competitive interactions with α-actinin (Mimura and Asano 1987) and tropomyosin (Moir and Levine 1986). Patient 2 (who has severe congenital NM) has a mutation at residue 183 (Arg183Gly), which is involved in interactions with DNase I and in ionic interactions with other parts of actin (Kabsch et al. 1990). This residue, Arg183Cys, also was mutated in a patient with a similar clinical phenotype who was reported by Nowak et al. (1999). The family with autosomal dominant inheritance has a mutation at position 115 (Asn115Ser), a region also implicated in α-actinin 2 binding (Lebart et al. 1993). The variation in both age at onset and clinical severity in this family suggests that ACTA1 genotype is not the sole determinant of phenotype and that modifying genetic loci or stochastic factors may influence the clinical presentation.
The pathogenesis of muscle weakness and hypotonia in patients with actin NM is unknown, although disruption of actin structure and interactions is likely to impair myofiber contractility. The percentage of fibers containing rods did not correlate with disease severity. However, the proportion of fibers occupied by rods, the size of the rod clusters, the organization of sarcomeric actin, and the degree of myofibrillar disruption appear to correlate with clinical severity. For example, the two patients with the lethal severe congenital form of NM had both markedly abnormal localization of sarcomeric actin and generalized disorganization of sarcomeric structure, with many of the fibers' regions being completely devoid of ordered sarcomeric register. Numerous fibers in the severely affected patients were almost completely occupied by rods, and there were many atrophied, necrotic rod-filled fibers not visible at the LM level. In contrast, there was both minimal sarcomeric disruption by rods and comparatively normal ultrastructural appearance in patients with a milder phenotype. Accumulation of glycogen was evident in all patients and may reflect a decrease in efficiency of energy utilization. Other features—such as abnormalities of sarcomeric-actin staining, whorling of thin filaments (seen in patient 6), and an excess of thin filaments (Goebel et al. 1997)—may be specific to actin myopathy and assist in guiding a mutational analysis.
Type I fiber predominance and altered expression of fiber type–specific genes was a common histopathological feature. Coexpression of myosin isoforms and expression of fast fiber–specific proteins, such as α-actinin 3, in fibers expressing only slow myosin suggests, as part of the disease process, either fiber type conversion or an abnormality in the maturation of fiber typing. Compensatory fiber hypertrophy was a striking feature in patient 4, who remains physically active and cycles regularly. Although anecdotal, this observation suggests that patients with actin NM may benefit from regular low-impact exercise.
There was abnormal expression of tropomyosin and nebulin in the patients with severe congenital NM. Both proteins are closely associated with actin in the thin filament, and it is likely that the ACTA1 mutations affect nebulin and tropomyosin distribution. Transfection of normal and mutant cytoplasmic actin genes into myoblasts results in isoform-specific changes in both the expression and organization of tropomyosin (Schevzov et al. 1993).
In striated muscle, there is coexpression of two sarcomeric actins—α-skeletal actin and α-cardiac actin—that are highly conserved and that, in their amino acid sequences, differ by only four residues. Cardiac actin is thought to be the major actin isoform present in embryonic skeletal muscle, but it is rapidly replaced, during the postnatal period, by α-skeletal actin (Collins et al. 1997). The expression of cardiac actin in human fetal skeletal muscle may explain why the two patients with severe congenital NM survived to term but exhibited both severe hypotonia and absence of antigravity movements from birth. In human fetal heart, α-cardiac actin is the prevalent isoform, whereas α-skeletal actin is predominant (∼60%) in the adult (Boheler et al. 1991). Although mutations in cardiac actin result in dilated cardiomyopathy (Olson et al. 1998), none of the patients with NM with identified mutations in α-skeletal actin have clinically detectable abnormalities of cardiac-muscle contractility (by echocardiography). Although we cannot exclude subclinical or histological abnormalities of cardiac muscle, the high level of cardiovascular fitness achieved by patient 4 (the competitive long-distance cyclist) suggests that expression of wild-type cardiac actin and the presence of one normal ACTA1 allele) may be sufficient for normal cardiac-muscle function.
A disease phenotype resulting from a new dominant mutation could be the result either of a functional dominant-negative effect exerted by the mutant gene or of haploinsufficiency. Mice heterozygous for a cardiac-actin knockout are indistinguishable from their wild-type littermates, suggesting that haploinsufficiency does not result in a disease phenotype (Kumar et al. 1997). Our two patients with lethal severe congenital NM had abnormal localization of muscle α-skeletal actin, and filamentous actin was present in the rods. Taken together, these results suggest that the mutant actin isoform exerts a dominant-negative effect, rather than causing a deficiency in steady-state levels of wild-type actin.
Acknowledgments
We wish to thank the European Neuromuscular Centre Nemaline Myopathy Consortium, particularly Drs. Carina Wallgren-Pettersson and Alan Beggs, for their valued discussions of this work, and we thank Dr. Ted Wills for his assistance with the EM. We are indebted to all the families who participated in this study. This study was supported by grants from the National Health and Medical Research Council and from the Children’s Hospital Fund and by a Murdoch University Research Scholarship.
Electronic-Database Information
The accession number and URL for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for ACTA1 [MIM 102610])
References
- Adelstein RS, Eisenberg E (1980) Regulation and kinetics of the actin-myosin-ATP interaction. Annu Rev Biochem 49:921–956 [DOI] [PubMed] [Google Scholar]
- Blanchard A, Ohanian V, Critchley D (1989) The structure and function of α-actinin. J Muscle Res Cell Motil 10:280–289 [DOI] [PubMed] [Google Scholar]
- Boheler KR, Carrier L, de la Bastie D, Allen PD, Komajda M, Mercadier J-J, Schwartz K (1991) Skeletal actin mRNA increases in the human heart during ontogenic development and is the major isoform of control and failing adult hearts. J Clin Invest 88:323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins T, Joya JE, Arkell RM, Ferguson V, Hardeman EC (1997) Reappearance of the minor α-sarcomeric actins in postnatal muscle. Am J Physiol 273:C1801–C1810 [DOI] [PubMed] [Google Scholar]
- Donner K, Ollikainen M, Pelin K, Gronholm M, Carpen O, Wallgren-Pettersson C, Ridanpaa M (2000) Mutations in the β-tropomyosin (TPM2) gene in rare cases of autosomal dominant nemaline myopathy. World Muscle Society abstract. Neuromuscul Disord 10:342–343 [Google Scholar]
- Engel WK, Cunningham GG (1963) Rapid examination of muscle tissue: an improved trichrome method for fresh frozen biopsy sections. Neurology 13:919–923 [DOI] [PubMed] [Google Scholar]
- Goebel HH, Anderson JR, Hubner C, Oexle K, Warlo I (1997) Congenital myopathy with excess of thin myofilaments. Neuromuscul Disord 7:160–168 [DOI] [PubMed] [Google Scholar]
- Holmes KC, Popp D, Gebhard W, Kabsch W (1990) Atomic model of the actin filament. Nature 347:44–49 [DOI] [PubMed] [Google Scholar]
- Johnston JJ, Kelley RI, Crawford TO, Morton DH, Agarwala R, Koch T, Schäffer AA, Francomano CA, Biesecker LG (2000) A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am J Hum Genet 67:814–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC (1990) Atomic structure of the actin:DNase I complex. Nature 347:37–44 [DOI] [PubMed] [Google Scholar]
- Kingston RE (1997) Preparation and analysis of RNA. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (eds) Current protocols in molecular biology. John Wiley & Sons, New York, pp 4.0.1–4.10.11 [Google Scholar]
- Kumar A, Crawford K, Close L, Madison M, Lorenz J, Doetschman T, Pawlowski S, Duffy J, Neumann J, Robbins J, Boivin GP, O’Toole BA, Lessard JL (1997) Rescue of cardiac α-actin-deficient mice by enteric smooth muscle γ-actin. Proc Natl Acad Sci USA 94:4406–4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing NG, Wilton SD, Akkari PA, Dorosz S, Boundy K, Kneebone C, Blumbergs P, White S, Watkins H, Love DR, Haan E (1995) A mutation in the α tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy. Nat Genet 9:75–79 [DOI] [PubMed] [Google Scholar]
- Lammens M, Moerman P, Fryns JP, Lemmens F, van de Kamp GM, Goemans N, Dom R (1997) Fetal akinesia sequence caused by nemaline myopathy. Neuropediatrics 28:116–119 [DOI] [PubMed] [Google Scholar]
- Lebart MC, Mejean C, Roustan C, Benyamin Y (1993) Further characterization of the α-actinin-actin interface and comparison with filamin-binding sites on actin. J Biol Chem 268:5642–5648 [PubMed] [Google Scholar]
- Mejean C, Lebart MC, Boyer M, Roustan C, Benyamin Y (1992) Localization and identification of actin structures involved in the filamin-actin interaction. Eur J Biochem 209:555–562 [DOI] [PubMed] [Google Scholar]
- Miike T, Ohtani Y, Tamari H, Ishitsu T, Une Y (1986) Muscle fiber type transformation in nemaline myopathy and congenital fiber type disproportion. Brain Dev 8:526–532 [DOI] [PubMed] [Google Scholar]
- Mimura N, Asano A (1987) Further characterization of a conserved actin-binding 27-kDa fragment of actinogelin and α-actinins and mapping of their binding sites on the actin molecule by chemical cross-linking. J Biol Chem 262:4717–4723 [PubMed] [Google Scholar]
- Moir AJ, Levine BA (1986) Protein cognitive sites on the surface of actin: a proton NMR study. J Inorg Biochem 28:271–278 [DOI] [PubMed] [Google Scholar]
- North KN, Beggs AH (1996) Deficiency of a skeletal muscle isoform of α-actinin (alpha-actinin-3) in merosin-positive congenital muscular dystrophy. Neuromuscul Disord 6:229–235 [DOI] [PubMed] [Google Scholar]
- North KN, Laing NG, Wallgren-Pettersson C (1997) Nemaline myopathy: current concepts. The ENMC International Consortium and Nemaline Myopathy. J Med Genet 34:705–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- North KN, Yang N, Wattanasirichaigoon D, Mills M, Easteal S, Beggs AH (1999) A common nonsense mutation results in α-actinin-3 deficiency in the general population. Nat Genet 21:353–354 [DOI] [PubMed] [Google Scholar]
- Nowak KJ, Wattanasirichaigoon D, Goebel HH, Wilce M, Pelin K, Donner K, Jacob RL, Hubner C, Oexle K, Anderson JR, Verity CM, North KN, Iannaccone ST, Muller CR, Nurnberg P, Muntoni F, Sewry C, Hughes I, Sutphen R, Lacson AG, Swoboda KJ, Vigneron J, Wallgren-Pettersson C, Beggs AH, Laing NG (1999) Mutations in the skeletal muscle α-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet 23:208–212 [DOI] [PubMed] [Google Scholar]
- Olson TM, Michels VV, Thibodeau SN, Tai YS, Keating MT (1998) Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 280:750–752 [DOI] [PubMed] [Google Scholar]
- Pelin K, Hilpela P, Donner K, Sewry C, Akkari PA, Wilton SD, Wattanasirichaigoon D, Bang M-L, Centner T, Hanefeld F, Odent S, Fardeau M, Urtizberea AJ, Muntoni F, Dubowitz V, Beggs AH, Laing NG, Labeit S, de la Chapelle A, Wallgren-Pettersson C (1999) Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc Natl Acad Sci USA 96:2305–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgar J, Johnson MA, Weightman D, Appleton D (1973) Data on fiber size in thirty-six human muscles: an autopsy study. J Neurol Sci 19:307–318 [DOI] [PubMed] [Google Scholar]
- Schevzov G, Lloyd C, Hailstones D, Gunning P (1993) Differential regulation of tropomyosin isoform organization and gene expression in response to altered actin gene expression. J Cell Biol 121:811–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheterline P, Clayton J, Sparrow JC (1999) Actin 1-272. Oxford Univeristy Press, Oxford [Google Scholar]
- Volpe P, Damiani E, Margreth A, Pellegrini G, Scarlato G (1982) Fast to slow change of myosin in nemaline myopathy: electrophoretic and immunologic evidence. Neurology 32:37–41 [DOI] [PubMed] [Google Scholar]
- Wallgren-Pettersson C, Laing NG (2000) Report of the 70th ENMC International Workshop: nemaline myopathy, 11–13 June 1999, Naarden, The Netherlands. Neuromuscul Disord 10:299–306 [DOI] [PubMed] [Google Scholar]