

Abstract
During the past 20 years, cystathionine β-synthase (CBS) deficiency has been detected in the former Czechoslovakia with a calculated frequency of 1:349,000. The clinical manifestation was typical of homocystinuria, and about half of the 21 patients were not responsive to pyridoxine. Twelve distinct mutations were detected in 30 independent homocystinuric alleles. One half of the alleles carried either the c.833 T→C or the IVS11−2A→C mutation; the remaining alleles contained private mutations. The abundance of five mutant mRNAs with premature stop codons was analyzed by PCR-RFLP. Two mRNAs, c.828_931ins104 (IVS7+1G→A) and c.1226 G→A, were severely reduced in the cytoplasm as a result of nonsense-mediated decay. In contrast, the other three mRNAs—c.19_20insC, c.28_29delG, and c.210_235del26 (IVS1−1G→C)—were stable. Native western blot analysis of 14 mutant fibroblast lines showed a paucity of CBS antigen, which was detectable only in aggregates. Five mutations—A114V (c.341C→T), A155T (c.463G→A), E176K (c.526G→A), I278T (c.833T→C), and W409_G453del (IVS11−2A→C)—were expressed in Escherichia coli. All five mutant proteins formed substantially more aggregates than did the wild-type CBS, and no aggregates contained heme. These data suggest that abnormal folding, impaired heme binding, and aggregation of mutant CBS polypeptides may be common pathogenic mechanisms in CBS deficiency.
Homocystinuria due to cystathionine β-synthase (CBS) deficiency (MIM 236200) is an autosomal recessive trait that phenotypically resembles Marfan syndrome. Unless diagnosed by neonatal screening and treated appropriately, homocystinuria manifests, usually in infancy and childhood, with disturbances in connective tissue, vasculature, and the central nervous system. Since the first identification of mutations in the CBS mRNA (Kozich and Kraus 1992), >350 homocystinuric alleles with >100 distinct mutations have been reported worldwide (Kraus et al. 1999; authors' Web site). Some of these mutant alleles are present in all populations (e.g., the c.833 T→C mutation conferring pyridoxine responsiveness), but others are confined to specific populations. The vast majority of homocystinuric alleles all over the world carry private mutations (Kraus et al. 1999). Although several mutations have been expressed in Escherichia coli (Kozich and Kraus 1992; de Franchis et al. 1994; Dawson et al. 1997; Kluijtmans et al. 1999) or yeast (Shan and Kruger 1998), detailed reports on pathogenicity of individual mutations in homocystinuria are lacking. In the present report, we describe the mutation analysis of a group of Slavic patients with homocystinuria, with a focus on the consequences of mutations at the RNA and protein levels.
CBS deficiency is the most common disorder of sulfur amino acid metabolism, with an incidence of 1:57,000–1:1,000,000 (Mudd et al. 2001). In the region of the former Czechoslovakia, homocystinuria has been ascertained only by the selective biochemical screening. With a correction for diagnosis delay, the calculated frequency of detection of homocystinuria during the past 20 years is 1:349,000 (95% confidence interval 1:208,000–1:641,000). The clinical phenotype of all 21 Czech and Slovak patients with CBS deficiency was without any peculiar findings and corresponded to the previous report of a series of 629 patients (Mudd et al. 1985). The number of pyridoxine responders equaled that of nonresponders, which is similar to that in other populations in Europe (Mudd et al. 1985).
We analyzed the underlying molecular causes of CBS deficiency in all cases where fibroblasts and/or blood were available. Mutations were detected by sequencing both the cDNA (Kozich and Kraus 1992) and genomic DNA (see Primers and Conditions Web site). In addition, all mutations were verified in genomic DNA by use of independent PCR-RFLP assays. Among the Czech and Slovak patients with CBS deficiency, we detected 12 distinct mutant alleles (see table 1). About one-half of the 30 independent homocystinuric alleles carried private mutations, while the other half was represented equally by alleles that bear the c.833T→C or the IVS11−2A→C mutation.
Table 1.
Overview of Mutant Alleles[Note]
| No. of Alleles | cDNA | Genomic DNA | Restriction- Site Changea | Observed mRNA Changeb | Deduced AminoAcid Change | Activity in E. colic | Decreased or PresentCBS Immunodetection in E. coli | Referencesd |
| 7 | c.833 T→C | g.9122 T→C | BsrI+ | I278T | 0–1.5 | Decreased | 1 | |
| 7 | IVS11−2A→C | g.13217 A→C | MspI+ | c.1224_1358del135 | W409_G453del | 0 | Decreased | 1 |
| 3 | c.19_20insC | g.19_20insC | Mwo I−e | P6fsX36 | ND | ND | ||
| 2f | [c.430 G→A; c.463G→A] | [g.5929G→A; g.6510 G→A] | TaqI−; Sau96I+ | [E144K; A155T] | <1.0; 0.0 | Present | 2, 3 | |
| 2 | c.494 G→A | g.6541 G→A | BsoFI− | C165Y | 1.3 | ND | 4 | |
| 2 | c.526 G→A | g.6573 G→A | BstXI+ | E176K | 0 | Present | 2 | |
| 2 | c.1226 G→A | g.13221 G→A | BslI− | NMD | W409X | ∼200 | Present | 5 |
| 1 | c.28_29delG | g.28_29delG | BslI−e | E9fs, 81X | ND | ND | ||
| 1 | c.194 A→G | g.194 A→G | MaeIII+ | H65R | ND | ND | ||
| 1 | IVS1−1G→C | g.3576 G→C | BslI−e | c.210_235del26 | A69fsX94 | ND | ND | |
| 1 | c.341 C→T | g.5840 C→T | MaeII+ | A114V | 0–54.5 | Decreased/present | 2, 6 | |
| 1f | [IVS7+1 G→A; IVS11+39del99] | [g.8297 G→A; 13000_13098del99] | None | c.737_828del92,g NMD; c.828_931ins104,g NMD | D245fsX298g; R276fsX295g | ND | ND |
Note.— ND = not determined.
− = mutation abolishes restriction site; + = mutation forms a novel restriction site.
NMD = nonsense-mediated decay.
Catalytic activity of mutant CBS in E. coli extracts, expressed as percent of wild-type CBS.
Sources of protein information are as follows: 1, Kozich and Kraus 1992; 2, this study; 3, Dawson et al. 1997; 4, Kluijtmans et al. 1999; 5, Shan and Kruger 1998; and 6, de Franchis et al. 1994.
Restriction sites formed using mismatched PCR primer (see Primers and Conditions Web site).
Allele carrying two mutations in cis.
Mutation IVS7+1G→A yields two mutant mRNA isoforms depending on the use of an alternative splice site; these isoforms are present only in the nuclear RNA preparation (data not shown).
The high prevalence of the c.833T→C allele, which confers pyridoxine responsiveness, was not surprising, because this transition is clearly the most prevalent pathogenic CBS mutation in whites (Kraus et al. 1999). On the other hand, the IVS11−2A→C mutation seems to be present only in central Europe; it has been observed in patients of East German, Austrian, and Turkish origin (Koch et al. 1994), but it has never been detected in numerous homocystinuric alleles of people from western and southern Europe.
Five mRNAs predicted premature termination codons at various exons: c.19_20insC predicted a premature termination codon in exon 1; c.28_29delG and c.210_235del26 (IVS1−1G→C) in exon 2; 828_931ins104 (IVS7+1G→A) in exon 8; and c.1226G→A (W409X) in exon 12. Given that the naturally occurring stop codon in the wild-type CBS mRNA is located in exon 16, mRNAs that carry any of these five mutations would presumably be subject to nonsense-mediated decay (Nagy and Maquat 1998; Frischmeyer and Dietz 1999). Because all patients with these mutations were heterozygous for a neutral synonymous mutation—c.699C→T (Y233Y)—we analyzed the abundance of CBS mRNAs carrying premature stop codon, using a simple PCR-RFLP method (see fig. 1). Both genomic DNA and cDNA were amplified by PCR, and the PCR products were digested with RsaI and electrophoresed on 4% Metaphor agarose, as described elsewhere (Kraus et al. 1993).
Figure 1.

The relative abundance of mRNAs that carry premature termination codon. Top, Analysis of genomic DNA. Restriction-site analysis shows fragments of 122 bp, in the case of a T allele in position 699, and of 92 bp, if the C allele is present. The undigested 304-bp PCR products that contain a control RsaI are not shown. Lanes 1 and 9, Molecular-weight markers. Lane 2, Patient 4 [IVS7+1G→A; c.699C]/[IVS11−2A→C; and c.699T]. Lane 3, Patient 6 [c.1226G→A; c.699T ]/[IVS11−2A→C; c.699C]. Lane 4, Patient 9 [c.19_20insC; c.699C]/[IVS11−2A→C; c.699T]. Lane 5, Patient 10 [IVS1−1G→C; c.699C]/[c.28_29delG; c.699T]. Lane 6, Wild-type control c.699C/C. Lane 7, Wild-type control c.699T/T. Lane 8, Wild-type control c.699C/T. Bottom, RT-PCR/RFLP analysis of total fibroblast RNA. The presence of T and C in position 699 is signified by the 280-bp and 248-bp fragments, respectively. The uncut PCR products of 392 bp that contain a control RsaI are not shown. The lanes contain samples identical to those in top panel. The absence of a fragment signifying the C allele in lane 2 demonstrates that the mRNA that carries the c.828_931ins104 (IVS7+1G→A) is not present in the RNA preparation. Similarly, the absence of the T allele in lane 3 shows the virtual absence of the mRNA molecules carrying the c.1226G→A (W409X).
Surprisingly, only two of five mutants were severely depleted below the detection limit of the RT-PCR–RFLP method (fig. 1, bottom). The loss of heterozygosity for the c.699C→T synonymous mutation suggests that the mRNAs carrying the mutations 828_931ins104 (IVS7+1G→A) or c.1226G→A (W409X) are degraded by nonsense-mediated decay. Our result may explain why the c.1226G→A (W409X) mutation was pathogenic in patients 6 and 18 of the series reported here, whereas the protein that is truncated at or near W409 and that is expressed in bacteria or yeast has an increased activity (Kery et al. 1997; Shan and Kruger 1998). We postulate that the pathogenicity of the c.1226G→A (W409X) mutation in humans results from a nonsense-mediated decay of the mutant mRNA before translation.
In contrast, the mRNA molecules that bear the mutations c.19_20insC, c.28_29delG, and c.210_235del26 (IVS1−1G→C) were abundant and easily detectable by the RT-PCR method, suggesting that their stability was not severely compromised. The c.19_20insC mutation is an extra C inserted into a sequence of six cytidines, and it probably originates from a slipped mispairing at the replication fork (Cooper et al. 1995). The c.19_20insC mutation leads to a frameshift with a premature termination codon in exon 1 and predicts instability of the mRNA resulting from nonsense-mediated decay. Patients who are homozygous for this mutation would be expected to be true null mutants, with no mRNA and no translated CBS protein. However, fibroblasts derived from patient 3, who is homozygous for the c.19_20insC mutation, contained an appreciable amount of CBS mRNA, which allowed mutation analysis (data not shown). In addition, the fibroblasts of patient 3 contained residual CBS activity that was 2.5% of control values (see table 2), although the native western blot analysis did not detect any normal CBS tetramers (fig. 2). It is conceivable that (1) RNA polymerase slippage may have produced some mRNA molecules carrying six cytidines, instead of the seven cytidines contained in the genomic DNA, and (2) translation of these wild-type mRNA molecules yielded small amounts of normal CBS polypeptide. To test whether RNA polymerase slippage may have been operational, we amplified and cloned the 5′ portion of mRNA, using RT-PCR with total RNA preparations. Among the 100 colonies screened for the presence of the c.19_20insC, we observed 1 wild-type clone (which was devoid of mutation) and 99 mutant clones. We examined whether this result was caused by the in vitro infidelity of DNA polymerase during PCR. We subsequently amplified, by PCR, and cloned a part of exon 1, using genomic DNA of this patient; all 100 tested colonies contained only the c.19_20insC, suggesting that PCR did not artificially produce the wild-type allele in the course of cDNA analysis. However, it is also possible that the observed residual activity may have originated from mosaicism for cells carrying both six and seven cytidines, or from translational frameshifting (Horsburgh et al. 1996; Farabaugh 2000). Taken together, our data suggest that RNA polymerase slippage and other mechanisms may function in vivo and thus modify the primary genetic defect in patients with the c.19_20insC mutation.
Table 2.
Description of Patients
|
Genotype |
|||||||
| PatientNumbera | Originb | Sex | Current Age(Years) | PyridoxineResponsec | Allele 1 | Allele 2 | CBS Activityd |
| 1 | CZ | F | 6 | − | c.494 G→A | c.494 G→A | .46 |
| 2 | CZ | M | 11 | − | IVS11−2 A→C | [c.430 G→A; c.463 G→A] | .00 |
| 3 | SK | F | 14 | − | c.19_20insC | c.19_20insC | .46 |
| 4 | CZ | M | 15 | − | IVS11−2 A→C | [IVS7+1 G→A; IVS11+39del99] | .00 |
| 5 | SK | F | 16 | − | Unknown | Unknown | NA |
| 6 | SK | M | 19 | − | IVS11−2 A→C | c.1226 G→A | .10 |
| 7 | SK | M | 20 | − | c.526 G→A | c.526 G→A | .03 |
| 8* | CZ | M | 20 | − | IVS1−1G→C | c.28_29delG | NA |
| 9 | SK | F | 21 | − | IVS11−2A→C | c.19_20insC | .33 |
| 10* | CZ | M | 22e | − | IVS1−1G→C | c.28_29delG | .03 |
| 11 | CZ | M | 33 | − | c.194 A→G | Unknown | .02 |
| 12 | SK | M | 19 | ± | IVS11−2 A→C | Unknown | NA |
| 13† | SK | M | 24 | ± | IVS11−2 A→C | c.833 T→C | .03 |
| 14 | CZ | M | 25 | ± | Unknown | Unknown | NA |
| 15† | SK | F | 25 | ± | IVS11−2 A→C | c.833 T→C | .00 |
| 16‡ | CZ | M | 15 | + | c.833 T→C | [c.430 G→A; c.463 G→A] | NA |
| 17‡ | CZ | F | 17 | + | c.833 T→C | [c.430 G→A; c.463 G→A] | .10 |
| 18 | CZ | M | 21 | + | c.341 C→T | c.1226 G→A | .13 |
| 19 | CZ | F | 26 | + | c.833 T→C | IVS11−2 A→C | .05 |
| 20 | CZ | M | 33 | + | c.833 T→C | c.833 T→C | .23 |
| 21 | CZ | M | 47 | + | c.833 T→C | c.833 T→C | .43 |
Asterisks (*), daggers (†), and double daggers (‡) indicate sib pairs.
CZ = Czech; SK = Slovak.
+ = positive response; ± = partial response; − = no response.
CBS activity in cultured skin fibroblasts expressed in nanomoles of cystathionine per milligram of total protein per hour. Control range 10–40 nmol/mg/h. NA = not applicable.
Patient 10 died at the age of 22 years because of pulmonary embolism.
Figure 2.

Western blot analysis of fibroblast extracts (native PAGE). The normally assembled tetramer is present only in control fibroblasts (10 μg of protein per lane) and is not detectable in any of the mutant cell lines (100 μg of protein per lane). Lane numbers correspond to patient numbers in table 2. Lanes 1, 2, 3, 4, 6 7, 9, 10, and 11 show results in patients who were not responsive to pyridoxine. Lane N shows results in a negative sample, mutant cell line 599 devoid of any CBS antigen (Skovby et al. 1984); lanes 13, 17, 19, and 20 show results in patients who were fully or partially responsive to pyridoxine. Lane C1 shows results in control fibroblasts 2047 with the sample frozen and thawed twice; lane C2 shows results in control fibroblasts 2047 analyzed in a separate experiment. The extract for lane C2 was prepared immediately before loading, with addition of β-mercaptoethanol and protease inhibitors.
Frameshifting of the c.28_29delG and c.210_235del26 (IVS1−1G→C) alleles predicts a premature termination of translation in exon 2 at amino acid residues 81 and 94, respectively. Again, the positions of the premature stop codons are consistent with the rules of nonsense-mediated decay. However, the mRNAs carrying these mutations are clearly abundant in total RNA preparations and are easily detectable by RT-PCR. Because both these mutant alleles carry a downstream methionine (either a newly formed one at position 10, in the case of mutation c.28_29delG, or a naturally occurring one at position 89, in the case of mutation c.210_235del26 [IVS1−1G→C]), we propose that the mutant mRNA molecules may have been stabilized by translation reinitiation (Zhang and Maquat 1997). Unfortunately, the sensitivity of the western blotting for CBS and the specificity of the in vitro translation assay are insufficient to test this hypothesis.
The CBS deficiency was confirmed in cultured skin fibroblasts of 16 patients, in whom a range (0%–2.5% of control values) of the residual activity was present (see table 2). To determine whether the decreased CBS activity originates from an impaired catalytic activity or from the depletion of CBS polypeptides, we performed western blot analysis in fibroblast extracts of 14 cell lines. SDS-PAGE, followed by western blot analysis, repeatedly showed very small amounts of a normal-sized 63-kD CBS in fibroblasts derived from patient 1; none of the other 13 cell lines contained detectable CBS subunits (data not shown). A more sensitive nondenaturing PAGE, followed by western blot analysis, was used to determine the degree of assembly of CBS subunits that are responsible for the observed residual enzyme activity. Cell lysates of cultured skin fibroblasts were prepared from frozen pellets, using the PEE-W1 lysis buffer (Kraus 1987). For western blot analysis, 10–100 μg of total protein was electrophoresed on 7.5% native polyacrylamide gel at 5 V/cm, followed by a transfer onto polyvinylidene difluoride membrane (Immobilon-P, Millipore). CBS subunits were detected by use of anti-CBS antibody (dilution 1:2,000 in 3% BSA in 1×PBS), followed by secondary anti-rabbit IgG conjugated with horseradish peroxidase (Jackson ImmunoResearch) diluted 1:30,000 in 5% nonfat dry milk dissolved in 1×PBS with 0.2% Tween 20, and they were visualized with the ECL detection kit (Amersham Pharmacia Biotech) and fluorography with XOmat AR film (Kodak). The slowly migrating complexes are marked as aggregates. Figure 2 (a representative example of three repeated determinations) shows that none of the mutant cell lines contained any detectable amount of the CBS tetramer. The majority of mutant fibroblast extracts contained minute amounts of large immunoreactive complexes that were slow to emerge from the loading well and slow to migrate on the gel. In normal fibroblasts, these multimeric aggregates have been observed in small amounts in repeatedly frozen and thawed samples (see fig. 2, lane C1) but not in freshly prepared extracts (fig. 2, lane C2). The aggregates are indeed formed by CBS polypeptides—as demonstrated by their absence in extracts from cell line 599 (fig. 2, lane N), which had been previously shown to be devoid of any CBS antigen (Skovby et al. 1984). The data from western blotting under nondenaturing conditions suggest that (1) the steady-state amounts of CBS polypeptides produced from 10 distinct mutant alleles are severely decreased in vivo, (2) the mutants do not form tetramers, and (3) the mutants aggregate.
To verify the hypothesis that the mutant CBS subunits are misfolded and aggregated, we also expressed five mutations in E. coli and performed western blot analysis under denaturing and native conditions. The procedure was the same as that in figure 2, with the following modifications: lysates from E. coli cell pellets were prepared using sonication in potassium phosphate buffer, samples were tested on 9% SDS-PAGE, and affinity-purified CBS antibody was used (Kozich and Kraus 1992). The size and abundance of mutant subunits expressed in E. coli were compared with those of the wild-type enzyme; the wild-type human CBS has the calculated and apparent molecular weight of 60.6 kD and 63 kD, respectively; the shorter band, with deletion of exon 12, has a calculated molecular weight of 55.6 kD. In contrast to human fibroblasts, the A114V (c.341C→T), A155T (c.463G→A), and E176K (c.526G→A) mutants were easily detectable on SDS-PAGE (see fig. 3) in amounts comparable to those in the wild type, a result that demonstrated their stability in E. coli extracts. The steady-state amounts of the I278T (c.833C→T) and of the W409_G453del (IVS11−2A→C) mutants are decreased, compared with wild type and with the other three mutants, a result that suggests a decreased stability in the bacterial expression system.
Figure 3.
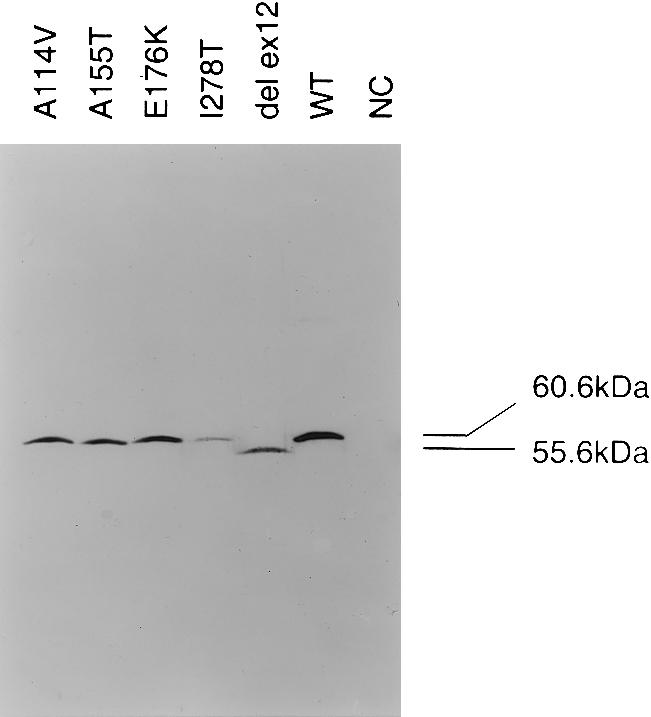
Western blot analysis of mutants expressed in E. coli (SDS-PAGE). Lanes (50 μg of total protein each): A114V (c.341C→T); A155T (c.463G→A); E176K (c.526G→A); I278T (c.833T→C); del ex 12 (W409_G453del [IVS11−2A→C]) mutants; WT indicates wild-type pHCS3; NC indicates negative control (E. coli transformed with pKK 388.1 plasmid without the human CBS insert).
Analysis of crude extracts from bacteria under native conditions was performed to confirm the observations made in fibroblast extracts. The procedure was the same as that shown in figure 3, with the exception of the use of nondenaturing 9% PAGE. Western blot analysis using CBS-specific antibody showed that, under native conditions, the wild-type CBS protein in the crude extract is present in its normal tetrameric form and that a small portion of the wild-type enzyme also forms multimers. In contrast, all five CBS mutants are present in the crude extract, almost exclusively as aggregates with high molecular weights (fig. 4, left). A small amount of a tetramer was detected only in the case of the A114V (c.341C→T) mutant, a result consistent with the mutant's high residual activity, and traces of tetramer were observed in E176K (c.526G→A) at the detection limit of the procedure.
Figure 4.

Oligomeric status and heme content of CBS mutants expressed in E. coli (native PAGE). Left, Immunodetection. The correctly assembled tetramers, marked as “4-mer,” are present in the wild-type lane and, in greatly diminished amounts, in A114V (c.341C→T) and E176K (c.526G→A) mutants. Aggregates of low mobility are present in all extracts as a smear. Lanes (each containing 50 μg of total protein) are as follows: A114V (c.341C→T); A155T (c.463G→A); E176K (c.526G→A); I278T (c.833T→C); W409_G453del (IVS11−2A→C) mutants cloned in pHCS3; WT50%, wild type pHCS3, 25 μg of total protein per lane; WT, wild type pHCS3, 50 μg; NC, negative control, E. coli transformed with pKK 388.1 plasmid without human CBS insert. Right, Heme staining. Non-CBS staining reaction of E. coli heme proteins was observed in all samples, including the negative control (top and bottom bands in all lanes). The protein loading is the same as in the panel at left. Heme bound to CBS is detectable only in the tetramers, both in the wild type and in the A114V (c.341C→T) mutant, but it is absent in the aggregates.
To limit the possibility that aggregates form as an artifact in vitro, we always repeated the nondenaturing western blot analyses at least twice with freshly prepared fibroblast and bacterial extracts, and, on several occasions, we added β-mercaptoethanol and protease inhibitors. These repeated experiments (data not shown) yielded identical results that were indistinguishable from those shown in figures 2 and 4. The ability to detect consistently the normally assembled tetramer in control samples and the presence of aggregates in mutant samples suggest that the in vitro data reflect reasonably well the subunit assembly within cells and are not artifacts.
We have reported elsewhere that heme may be necessary for binding of pyridoxal 5′-phosphate to CBS (Kery et al. 1994), and we postulated that heme may be necessary for correct CBS folding (Kery et al. 1999). We have also reported that the G307S (c.919G→A) mutant expressed in E. coli does not contain heme (Kery et al. 1995). These data suggest that heme may play a role in mutant subunit assembly. We therefore analyzed the heme content in five mutants expressed in E. coli. Bacterial crude extracts from both mutant and wild-type cells were electrophoresed under native conditions, and western blot gels were stained and examined for the presence of heme (Vargas et al. 1993). The experiments demonstrated that only the tetramers of wild-type cell or the A114V (c.341C→T) mutant contain heme, but aggregates of both mutant and wild-type CBS are devoid of this ligand (see fig. 4, right panel). This observation suggests that the inability to bind heme may prevent correct folding and subsequent tetramer formation of both mutant and, to a lesser extent, normal CBS subunits. Conversely, it may be the inability to retain the heme molecule that makes the mutants prone to misfolding and aggregation. Further studies are needed to explore each of these possibilities.
To conclude, we hypothesize that the abnormal folding of mutant CBS that is associated with impaired heme binding alters tertiary structure and exposes hydrophobic domains, most likely in the COOH terminal region. The abnormally folded molecules may be either forced to degradation by proteasome (Kirschner 1999) or may form aggregates. In human cells, these aggregates may be sequestered into aggresomes (Johnston et al. 1998), which are resistant to proteolytic degradation. The net result of these processes within the cells thus may be severe depletion of the abnormally assembled polypeptide complexes, as well as of mutant tetramers. We postulate that, as with other genetic defects (Bross et al. 1999), mutant CBS misfolding and aggregation may be the primary defect in a significant proportion of patients with homocystinuria.
Acknowledgments
The authors would like to thank Drs. J. Zeman, M. Orendáč, and V. Bzdúch, for providing patient samples; Dr. Zorka Novotná and Ms. Tanya Reed, for excellent tissue-culture work; and Ms. Eva Richterová and Ms. Terezie Zezuláková, for expert technical help. Drs. Lynne E. Maquat, Philip Farabaugh, Maria Chloupková, and Ken Maclean were of great help in discussing and critically reading the manuscript. This work was supported by a grant from the Czech Grant Agency (302/97/0476) and from the National Institutes of Health (Fogarty International Center grant R03 TW00989 and NIH-P01-HD08315 to J.P.K.). V.K. was also supported by a project of Charles University First Faculty of Medicine (VZ 111100003).
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Authors' Web site, http://www.uchsc.edu/sm/cbs (for an updated list of mutations)
- Genbank, http://www.ncbi.nlm.nih.gov/Genbank (for human CBS cDNA [accession number L19501] and genomic DNA [accession number AF042836])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CBS deficiency [MIM 236200])
- Primers and Conditions, http://www.lf1.cuni.cz/~mjano/protocols.html (for list of PCR primers and conditions for amplification of all 23 CBS exons from genomic DNA)
References
- Bross P, Corydon TJ, Andresen BS, Jorgensen MM, Bolund L, Gregersen N (1999) Protein misfolding and degradation in genetic diseases. Hum Mutat 14:186–198 [DOI] [PubMed] [Google Scholar]
- Cooper DN, Krawczak M, Antonarakis SE (1995) The nature and mechanisms of human gene mutation. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. 7th ed. McGraw-Hill, New York, pp 259–292 [Google Scholar]
- Dawson PA, Cox AJ, Emmerson BT, Dudman NP, Kraus JP, Gordon RB (1997) Characterisation of five missense mutations in the cystathionine beta-synthase gene from three patients with B6-nonresponsive homocystinuria. Eur J Hum Genet 5:15–21 [PubMed] [Google Scholar]
- de Franchis R, Kozich V, McInnes RR, Kraus JP (1994) Identical genotypes in siblings with different homocystinuric phenotypes: identification of three mutations in cystathionine beta-synthase using an improved bacterial expression system. Hum Mol Genet 3:1103–1108 [DOI] [PubMed] [Google Scholar]
- Farabaugh PJ (2000) Translational frameshifting: implications for the mechanism of translational frame maintenance. Prog Nucleic Acid Res Mol Biol 64:131–170 [DOI] [PubMed] [Google Scholar]
- Frischmeyer PA, Dietz HC (1999) Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet 8:1893–1900 [DOI] [PubMed] [Google Scholar]
- Horsburgh BC, Kollmus H, Hauser H, Coen DM (1996) Translational recoding induced by G-rich mRNA sequences that form unusual structures. Cell 86:949–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143:1883–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kery V, Bukovska G, Kraus JP (1994) Transsulfuration depends on heme in addition to pyridoxal 5′-phosphate: cystathionine beta-synthase is a heme protein. J Biol Chem 269:25283–288 [PubMed] [Google Scholar]
- Kery V, Elleder D, Kraus JP (1995) Delta-aminolevulinate increases heme saturation and yield of human cystathionine beta-synthase expressed in Escherichia coli. Arch Biochem Biophys 316:24–29 [DOI] [PubMed] [Google Scholar]
- Kery V, Poneleit L, Elleder D, Straubhaar JR, Teisinger J, Kraus JP (1997) The carboxy terminus of human cystathionine β-synthase is involved in tetramer formation and activation of the enzyme by S-adenosyl-l-methionine. Faseb J 11:1613 [Google Scholar]
- Kery V, Poneleit L, Meyer JD, Manning MC, Kraus JP (1999) Binding of pyridoxal 5′-phosphate to the heme protein human cystathionine beta-synthase. Biochemistry 38:2716–2724 [DOI] [PubMed] [Google Scholar]
- Kirschner M (1999) Intracellular proteolysis. Trends Cell Biol 9:M42–45 [PubMed] [Google Scholar]
- Kluijtmans LA, Boers GH, Kraus JP, van den Heuvel LP, Cruysberg JR, Trijbels FJ, Blom HJ (1999) The molecular basis of cystathionine beta-synthase deficiency in Dutch patients with homocystinuria: effect of CBS genotype on biochemical and clinical phenotype and on response to treatment. Am J Hum Genet 65:59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch HG, Ullrich K, Deufel T, Harms E (1994) A highly prevalent splice site mutation in the cystathionine-β-synthase gene is associated with poor vitamin B6 response in homocystinuria. Pediatr Res 36:21A [Google Scholar]
- Kozich V, Kraus JP (1992) Screening for mutations by expressing patient cDNA segments in E. coli: homocystinuria due to cystathionine beta-synthase deficiency. Hum Mutat 1:113–123 [DOI] [PubMed] [Google Scholar]
- Kraus JP (1987) Cystathionine beta-synthase (human). Methods Enzymol 143:388–394 [DOI] [PubMed] [Google Scholar]
- Kraus JP, Janosik M, Kozich V, Mandell R, Shih V, Sperandeo MP, Sebastio G, de Franchis R, Andria G, Kluijtmans LA, Blom H, Boers GH, Gordon RB, Kamoun P, Tsai MY, Kruger WD, Koch HG, Ohura T, Gaustadnes M (1999) Cystathionine beta-synthase mutations in homocystinuria. Hum Mutat 13:362–375 [DOI] [PubMed] [Google Scholar]
- Kraus JP, Le K, Swaroop M, Ohura T, Tahara T, Rosenberg LE, Roper MD, Kozich V (1993) Human cystathionine beta-synthase cDNA: sequence, alternative splicing and expression in cultured cells. Hum Mol Genet 2:1633–1638 [DOI] [PubMed] [Google Scholar]
- Mudd SH, Levy HL, Kraus JP (2001) Disorders of transsulfuration. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B (eds) The metabolic and molecular bases of inherited disease. 8th ed. McGraw-Hill, New York, pp 2007–2056 [Google Scholar]
- Mudd SH, Skovby F, Levy HL, Pettigrew KD, Wilcken B, Pyeritz RE, Andria G, Boers GH, Bromberg IL, Cerone R, Fowler B, Grobe H, Schmidt H, Schweitzer L (1985) The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet 37:1–31 [PMC free article] [PubMed] [Google Scholar]
- Nagy E, Maquat LE (1998) A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 23:198–199 [DOI] [PubMed] [Google Scholar]
- Shan X, Kruger WD (1998) Correction of disease-causing CBS mutations in yeast. Nat Genet 19:91–93 [DOI] [PubMed] [Google Scholar]
- Skovby F, Kraus JP, Rosenberg LE (1984) Homocystinuria: biogenesis of cystathionine beta-synthase subunits in cultured fibroblasts and in an in vitro translation system programmed with fibroblast messenger RNA. Am J Hum Genet 36:452–459 [PMC free article] [PubMed] [Google Scholar]
- Vargas C, McEwan AG, Downie JA (1993) Detection of c-type cytochromes using enhanced chemiluminescence. Anal Biochem 209:323–326 [DOI] [PubMed] [Google Scholar]
- Zhang J, Maquat LE (1997) Evidence that translation reinitiation abrogates nonsense-mediated mRNA decay in mammalian cells. Embo J 16:826–833 [DOI] [PMC free article] [PubMed] [Google Scholar]