

Abstract
François-Neetens fleck corneal dystrophy (CFD) is a rare, autosomal dominant corneal dystrophy characterized by numerous small white flecks scattered in all layers of the stroma. Linkage analysis localized CFD to a 24-cM (18-Mb) interval of chromosome 2q35 flanked by D2S2289 and D2S126 and containing PIP5K3. PIP5K3 is a member of the phosphoinositide 3-kinase family and regulates the sorting and traffic of peripheral endosomes that contain lysosomally directed fluid phase cargo, by controlling the morphogenesis and function of multivesicular bodies. Sequencing analysis disclosed missense, frameshift, and/or protein-truncating mutations in 8 of 10 families with CFD that were studied, including 2256delA, 2274delCT, 2709C→T (R851X), 3120C→T (Q988X), IVS19-1G→C, 3246G→T (E1030X), 3270C→T (R1038X), and 3466A→G (K1103R). The histological and clinical characteristics of patients with CFD are consistent with biochemical studies of PIP5K3 that indicate a role in endosomal sorting.
Introduction
François-Neetens fleck corneal dystrophy (CFD [MIM 121850]) is an autosomal dominant corneal dystrophy characterized by numerous small white flecks scattered in all layers of the stroma. François and Neetens described this disorder for the first time in 1957, calling it “hérédodystrophie mouchetée” (François and Neetens 1957). Typically, the stroma in between the flecks is clear, and the endothelium, epithelium, Bowman layer, and Descemet membrane are normal. Except for an occasional patient with minor photophobia, patients are usually asymptomatic and have normal vision; the disease is most often found at routine examination. The corneal flecks appear to not progress significantly throughout life (Patten et al. 1976; Akova et al. 1994). In vivo confocal microscopy examination has made the clinical diagnosis more straightforward, with CFD evident by small bright deposits around keratocyte nuclei throughout the stroma.
The flecks in CFD may appear as early as at 2 years of age or even at birth, but generally they are reported not to progress significantly throughout life. However, individual 15 in family 50003 showed no signs of CFD on slit-lamp examination at age 2 years, but, at age 6 years, it was observed that two stromal lesions in the left eye and one in the right eye had developed (Jiao et al. 2003). Although CFD is thought to be rare, specific incidence figures are difficult to obtain. It has been suggested that, because of its subtle presentation, CFD might be much more common than has been appreciated in the literature (Streeten and Falls 1961; Birndorf and Ginsberg 1972; Patten et al. 1976; Goldberg et al. 1977).
On histological examination, the corneal flecks appear to correspond to abnormal keratocytes swollen with membrane-limited intracytoplasmic vesicles containing complex lipids and acid mucopolysaccharides. There are no extracellular abnormalities (Nicholson et al. 1977; Purcell et al. 1977), and laboratory screening has shown no systemic metabolic abnormalities (Patten et al. 1976). Thus, it was thought that this dystrophy might represent a dominantly inherited metabolic disorder confined to the cornea (Nicholson et al. 1977). CFD was previously mapped to a 27.9-cM region of chromosome 2q35 flanked by D2S117 and D2S126, with the highest LOD scores shown at D2S2289 (Zmax=4.46; θ=0), D2S325 (Zmax=3.28; θ=0), D2S317 (Zmax=3.1; θ=0), D2S143 (Zmax=3.8; θ=0.03), and D2S2382 (Zmax=5.0; θ=0). Multipoint analysis confirmed linkage to the region between D2S117 and D2S126, with a maximum multipoint LOD score of 5.0 located midway between D2S2289 and D2S325. Analysis of CFD in the same families with the assumption of 90% penetrance increased the maximum LOD score to 6.28 at D2S157 (Jiao et al. 2003).
The present study was performed with 10 families, including the original 4 families studied by Jiao et al. (2003), and refined the linked region to a 24-cM interval flanked by D2S2289 and D2S126 and containing ∼18 Mb. Sequencing of numerous candidate genes in this region in affected members from 8 of 10 families showed mutations in PIP5K3, which encodes a widely expressed 2,089-aa phosphoinositide 3-kinase family member active in post-Golgi vesicular sorting.
Material and Methods
Patients and Clinical Findings
Ten unrelated families of European origin that contain 69 individuals, including 32 affected individuals, 27 unaffected individuals, and 10 unaffected spouses, were enrolled in this study. Families 50001 and 50007 are of mixed European origin (American), family 50002 is of Italian origin, family 50004 is of Yugoslavian origin, and the remaining families are of Swiss origin. Control individuals were of mixed European origin (from the United States, Italy, and Switzerland). The diagnosis of CFD was based on the presence of typical small white flecks throughout the corneal stroma. Both affected and unaffected family members underwent a complete ophthalmological examination. In affected individuals, the corneal epithelium and endothelial layer showed no abnormalities. The corneal stroma showed the presence of typical hyperreflective dots throughout but was otherwise normal. Confocal microscopy findings for family 5005 also showed hyperreflective inclusions associated with the basal nerves (Frueh and Bohnke 1999). Detailed ocular, medical, and family histories were obtained for each available family member. This project was approved by the institutional review board of the National Eye Institute and by the Ethics Committee of the Faculty of Medicine, University of Lausanne. The patients admitted to this study gave their informed consent, and the study adhered to the tenets of the Declaration of Helsinki.
Genotyping and Linkage Analyses
DNA was extracted either directly from blood or from transformed lymphoblastoid cell lines, by use of standard phenol-chloroform protocols (Smith et al. 1989). Two-point linkage analysis was performed using the FASTLINK version (Schaffer et al. 1994) of MLINK from the LINKAGE program package (Lathrop and Lalouel 1984; Cottingham et al. 1993). Maximum LOD scores were calculated using ILINK. CFD was initially analyzed as a fully penetrant, autosomal dominant trait. The gene frequency of CFD was set at 0.0001, and an equal allele frequency was used for all markers.
Candidate-Gene Analysis
Candidate genes were chosen in the 24-cM CFD-linked interval flanked by D2S2289 and D2S126 on chromosome 2q35, estimated to contain >300 genes or potential genes. To confirm that the candidate genes are expressed in the human cornea, human corneal total RNA was extracted with an RNeasy Fibrous Tissue Mini kit (QIAGEN), corneal cDNA was made using RT-PCR with the use of SuperScriptTM III First-Strand Synthesis System (Invitrogen), and direct PCR amplification of the cDNA was performed. Expression of PIP5K3 was tested by PCR analysis of corneal cDNA with the use of exon 1 forward and exon 3 reverse primers (table 1).
Table 1.
PCR Primers for 41 Exons and cDNA of PIP5K3 (Primers for DHPLC)
|
Primer Sequence |
|||
| Primers for | Forward(5′→3′) | Reverse(5′→3′) | AnnealingTemperature(°C) |
| Exon1 | TCGATGCTGTTTGGAATCAA | GAGCCACTGGAGACAAAG GA | 55 |
| Exon 2 | TCATCTACT TCC TTCTCATTGCTG | CCTGCTATAAACAACCTAGGAAACA | 55 |
| Exon 3 | TAACCGGGTGGGAGAACATA | ATGGCATGATCCCCATAAGC | 55 |
| Exon 4 | TTTTGACCTTTCGTATT CCATA | TGAAAATTGGCTTAACACTATCC | 55 |
| Exon 5 | GATGAACTGTTGACCCAAGGA | CTGAAAGCACTCAAAGGTCAGA | 55 |
| Exon 6 | TGTTAAAGCGCCTCATTGAA | TTCAGCTTTTAGAGCTACGGAAG | 55 |
| Exon 7 | TCCTTTACCAAATTTCTCCCAAT | CAGTCACATGAACATTCTCTATGC | 55 |
| Exon 8 | CACAGGAAAACATCTGAAAAGG | TCAGATTGGAGGAGGGTTAAG | 55 |
| Exon 9 | CAAGGGCCATTTGAAATTCT | TTCCCACTGATGAAGAAAGACT | 55 |
| Exon 10 | TTTATAGTGTGACAGCCTCAACA | TTTCCATTTAGTTGGAGCCATAA | 55 |
| Exon 11 | CCTTTCCTCCAATACCCTGA | ACCCACACCAAATACCCTCA | 55 |
| Exon 12 | TGTGTGTGGGCGTATTCTCT | GCACCTTCTCTGTGCTAATCAA | 55 |
| Exon 13 | TGTTCTTGCCTTTTTACACCAA | CCCTTTTTAAGAGGAATGATGC | 55 |
| Exon 14 | TCTGTGAGCCGTAATTGAAATG | CAATCAGTCCTGCCTCATCA | 55 |
| Exon 15 | TTGATCGTTGGTTGAAAGGA | CTGGGAAATCTTCCCTGAA | 55 |
| Exon 16 | TGAAGAATTTTGCATTGTTCTGA | GAGTGACAGAGGTTTTGGTAAGC | 55 |
| Exon 17 | TGTCCAGATACTGCATTTTACCA | TCTTGTATACCTACCATACGTGCAT | 55 |
| Exon 18 | GGTTTCAGTGGGCATATATTGA | TGCCTATCCTCACTTACCCACTA | 55 |
| Exon 19a | TGTACTTCTGGATTGCCACCT | CATCACAGGGCAGAGACTCA | 55 |
| Exon 19b | GGGCTGTCCAAGAGCAGTA | CAGTGTCATCCTGTAGAGGGTCT | 61 |
| Exon 19c | ACTCCCTGTGGATGACCAAC | TCCCTGAGCAGCTGTTTCTT | 55 |
| Exon 19d | TTGCAGAGCAGGTTTACTGG | TGCCTTTGAGGAGTCATTCA | 55 |
| Exon 20 | TGGAGGCTTTACAAATAGAGTGG | GCTGGAATTCTGTTCTTATGTGG | 55 |
| Exon 21 | GATTGGGAGTGAAAAAGATGC | TGCACTATGAAAGTGAGGATCA | 55 |
| Exon 22 | CACCGTGCTGTAGCTGAAGA | TGAACATACAGCACTGGAAGG | 55 |
| Exon 23 | CTGGGTGACAGAGCGAGACT | TAAATGGGGAATCCCACAAA | 55 |
| Exon 24 | GAAACTTTT CCTCTTATGCCTCA | CCAAGCATGCATAGCATACAA | 55 |
| Exon 25 | TTTCAATTACATTTTGCATGTAGC | CAAGGAACAGAAACAACTTCAGG | 55 |
| Exon 26 | GCAGTTTCTTTTGGGGTATGTC | GAGGAAAGGAGAGTGACAGGAA | 55 |
| Exon 27 | ACAGCACTCAAGGGGCTAGA | CAAAATGGAAAACCCTACTGGA | 55 |
| Exon 28 | TGGGTCCTGAGACCAAAAAG | TTCCCCTAATAGCAGCCTTC | 55 |
| Exon 29 | GGAAGTAGGCATATGAATGAGCA | AAACAGAAAGCAAGGCATGAA | 55 |
| Exon 30 | TCCGAAAGGAAAAGAACCAA | TGAAGCGAGCAACTAAGGAAA | 55 |
| Exon 31a | TCTGCATACATTTTGGCTTTC | CATTTTGCTTTATGTGAAAGATTCC | 55 |
| Exon 31a | GTGACTATTGCAGAACTCTCTG | GAACAAGCTATCACGACTAAAG | 55 |
| Exon 32 | TGATTGCTTTTGTCAGTTAACCA | TGAGTGCCACTATCTGAATCCT | 55 |
| Exon 33 | TGTAGGTGGGCTTAGGTAGGAA | TCATAAGTAGTAAATCATTGCCAGA | 55 |
| Exon 34 | GCATTACTCAGATTGATTAGCTTCA | GCCCCAGTTTCAGCACTAAG | 55 |
| Exon 35 | GGAAAAAGGAAAGGACAAAATG | TTCATTTCAAGAACTGGGACA | 55 |
| Exon 36 | AACATGCAAAATGCCATCAT | TTCCTGCTGCATTTTCTGAG | 55 |
| Exon 37 | AGGATCTTTGGAATGATGTGTT | CAAATTACCCAGCCTCAGGT | 55 |
| Exon 38 | TTGCTGCTGTACTCACTTCTGA | CCAAAGGTACAAAAGGTTCCA | 55 |
| Exon 39 | AATCATTTGTGGCAACCTGT | TTGACTCTGACTTCAATCATGAAA | 55 |
| Exon 40 | ACCCTGAGAACAGCACTTGG | CACCCAATATTTCCCCACCT | 55 |
| Exon 41 | CTTTCCTGCCCACTCAGACT | GGCATACAGTGGTGAACAC | 55 |
| cDNA | GAGCCCCCTTTTAAATCAGC | CCTCCAAAGGTAGGTTCTGC | 55 |
An extra set of primers was used for DHPLC screening of exon 31.
Denaturing High-Pressure Liquid Chromatography (DHPLC) and Sequence Analysis
Coding exons and adjacent intronic sequences of PIP5K3 were amplified from genomic DNA by use of the primers shown in table 1. PCR amplification of each of the 41 exons was performed in 20-μl reactions involving 2.5 U of AmpliTaq Gold (ABI), 2 μl of 10× AmpliTaq Gold Buffer, 1.9 mM MgCl2, 0.25 mM dNTPs, 0.25 mM of primers, and 40 g of genomic DNA. PCR cycling conditions consisted of an initial 10-min denaturation step at 95°C for 5 min; 32 cycles at 94°C for 40 s, 55°C for 30 s (61°C for exon 19b), and 72°C for 40 s; and a final elongation step at 72°C for 7 min. PCR products were screened by DHPLC on a WAVE system (Transgenomics). Buffer A contained 0.1 M triethylammonium acetate (TEAA [Transgenomics]). Buffer B contained 0.1 M TEAA and 25% acetonitrile, HPLC grade (Sigma-Aldrich). The flow rate was set at 0.9 ml/min, and the buffer B gradient increased by 2% per min for 4 min. The optimum temperature was determined for each DNA fragment by Wavemaker software (Transgenomics). When multiple melting domains were established, each domain was analyzed at the appropriate temperature. Initial buffer B concentrations and temperatures for each fragment are listed in table 2. PCR products were purified using Millipore Montage PCRμ96 plates or QIAquick PCR purification kits. The PCR templates were sequenced using ABI Dye Terminator, version 1 or 3, in a final reaction volume of 10 μl. Products of the sequencing reactions were purified using the Edge Biosystems Performa TM DTR gel-filtration system and were run on a 3100 ABI genetic analyzer. Sequences were aligned using the Seqman program of the DNASTAR package (DNASTAR) or Chromas, version 2.23 (Technelysium). The reference genomic sequence is available from NCBI (accession number NM_015040).
Table 2.
Conditions for DHPLC of PIP5K3 Exons
| Exon | Size(bp) | Temperature(s)(°C) | Initial % of Buffer B |
| 1 | 327 | 56; 58.5 | 55.5 |
| 2 | 300 | 56; 60; 62 | 54.7 |
| 3 | 300 | 56.1; 59; 61 | 54.7 |
| 4 | 397 | 54.1; 57; 60 | 57 |
| 5 | 386 | 55.1; 57.1; 60 | 56.8 |
| 6 | 286 | 55.5; 56.3; 58 | 54.3 |
| 7 | 374 | 53.3; 58 | 56.5 |
| 8 | 393 | 54.5; 58; 59.5 | 56.9 |
| 9 | 310 | 54.6; 59 | 55 |
| 10 | 342 | 54.8; 57.5; 59; 61 | 55.8 |
| 11 | 370 | 59.3; 61.3 | 56.5 |
| 12 | 250 | 56.6; 59 | 53 |
| 13 | 360 | 53.1; 60 | 56.2 |
| 14 | 351 | 54.5; 58; 60 | 56 |
| 15 | 398 | 53.2; 57; 58.5 | 57 |
| 16 | 456 | 51.5; 54.2; 55 | 58 |
| 17 | 275 | 52.8; 54.8; 59.5 | 53.9 |
| 18 | 432 | 52.3; 54.5; 59 | 57.6 |
| 19a | 375 | 57.3; 62.3 | 56.6 |
| 19b | 372 | 60.1; 61.3 | 56.5 |
| 19c | 384 | 57.5 | 56.7 |
| 19d | 442 | 58.7 | 57.8 |
| 20 | 245 | 59.2; 60.2 | 52.8 |
| 21 | 398 | 54.8; 57.5; 59; 61 | 57 |
| 22 | 482 | 54.3; 58.8 | 58.3 |
| 23 | 399 | 54.4; 57.4; 61 | 57 |
| 24 | 347 | 52.6; 54.2; 57.6 | 56 |
| 25 | 349 | 53.1; 54.5 | 56 |
| 26 | 366 | 54.8; 58.5; 59.8 | 56.4 |
| 27 | 278 | 53.5; 59.5 | 54 |
| 28 | 396 | 53.6; 55.7; 58 | 57 |
| 29 | 371 | 54.4; 61 | 56.5 |
| 30 | 272 | 54.4; 56.5; 58.5 | 53.8 |
| 31a | 438 | 53; 55; 60 | 57.7 |
| 32 | 360 | 53.2; 57.5 | 56.2 |
| 33 | 331 | 52.5; 54.5; 58 | 55.6 |
| 34 | 398 | 53.5; 61 | 57 |
| 35 | 248 | 54.7; 56.7 | 53 |
| 36 | 318 | 56.1; 60; 62 | 55.2 |
| 37 | 299 | 54.2; 56.2; 61 | 54.7 |
| 38 | 354 | 53.5; 57.4 | 56.1 |
| 39 | 364 | 55.9; 57.4 | 56.3 |
| 40 | 398 | 53.2; 55.2 | 57 |
| 41 | 359 | 57; 59 | 56.2 |
An extra set of primers was used for DHPLC screening of exon 31.
Protein Analysis
Structural motifs of the lipoprotein lipid attachment site were localized in the phosphatidylinositol-4-phosphate 5-kinase type III sequence (PIP5K3) by use of PROSITE. The search for spectrin sequence repeats was performed using rapid automatic detection and alignment of repeats (RADAR), and a search for structural motifs was performed using the simple modular architecture research tool (SMART), the Superfamily 1.65 HMM library and genome assignment service, the NIH homology alert service Whales, and the protein families database (PFAM) (see Web Resources). Sequence alignment was performed using the GCG-Lite+ ClustalW 1.74 multiple sequence alignment program (see Web Resources).
The chaperone-like domain structure of PIP5K3 was derived by homology modeling, with the crystal coordinates of protein database (PDB) file 1a6d as the structural template (Abola et al. 1987). For each model, amino acid sequences were aligned using the method of Needleman and Wunsch (1970). The three-dimensional structure was built using the program Look, version 3.5.2 (Lee and Subbiah 1991; Lee 1994), followed by 500 cycles of nonbound energy minimization. The conformation of mutant proteins was refined by 500 cycles of self-consistent ensemble optimization (Lee 1994). The geometry of the predicted structures was tested with the Procheck program (Laskowski et al. 1993).
Results
Patient Ascertainment
Individuals were diagnosed as affected on the basis of the typical appearance of corneal flecks on slit-lamp examination and, in some individuals, in vivo confocal microscopy (fig. 1). Of the 10 families investigated here, 4 (families 50001, 50002, 50003, and 50004) were previously studied by Jiao et al. (2003) and are described more fully in that study. Additional individuals and families 50005, 50006, 50008, 50009, and 50010 showed classical CFD and were asymptomatic, whereas affected individuals from family 50007 showed photophobia, which was severe enough in individual 5 (see pedigree in fig. 2) to result in blepharospasm and facial spasm, requiring multiple botulinum toxin injections to control the spasms. Family 50005 and one affected individual from family 50006 were reported elsewhere (Frueh and Bohnke 1999).
Figure 1.

Slit-lamp image (A) and confocal microscopy image (B) of the cornea in a patient with CFD
Figure 2.

Pedigrees of families, with haplotypes for markers in the CFD region of chromosome 2 in the families used for linkage analysis. Blackened bars indicate the risk haplotype inherited by all affected individuals in a family.
Linkage Analysis
Two-point linkage analysis suggested that the CFD locus is linked to a 12-cM region flanked by D2S2289 and D2S143 (table 3). This region includes markers D2S325 (Zmax=4.64; θ=0) and D2S317 (Zmax=3.67; θ=0). Significant LOD scores were also seen with other markers in the region, including D2S2289 (Zmax=4.7; θ=0.03), D2S143 (Zmax=4.83; θ=0.05), D2S2382 (Zmax=5.27; θ=0.3), and D2S2249 (Zmax=4.54; θ=0.04), although these markers showed obligate recombinants.
Table 3.
Two-Point Linkage Results for Markers in the CFD Region of Chromosome 2q35
|
Position |
LOD Score at θ = |
||||||||||
| Marker | cM | Mb | 0 | .01 | .05 | 0.1 | .02 | .03 | .04 | Zmax | 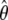 |
| D2S364 | 192.90 | 182.86 | −∞ | −1.32 | 1.14 | 1.89 | 2.05 | 1.57 | .82 | 2.05 | .2 |
| D2S117 | 201.40 | 195.44 | −∞ | −3.90 | −1.22 | −.21 | .48 | .53 | .30 | .53 | .3 |
| D2S311 | 203.10 | 197.39 | −∞ | 1.75 | 2.87 | 3.04 | 2.60 | 1.75 | .75 | 3 | .07 |
| D2S2289 | 205.40 | 203.49 | −∞ | 4.42 | 4.74 | 4.53 | 3.66 | 2.48 | 1.16 | 4.7 | .03 |
| D2S325 | 210.90 | 208.10 | 4.64 | 4.60 | 4.36 | 3.99 | 3.11 | 2.08 | .98 | 4.64 | 0 |
| D2S157 | 212.60 | 211.03 | 1.32 | 1.55 | 1.91 | 2.00 | 1.79 | 1.31 | .69 | 2 | .1 |
| D2S317 | 215.10 | 213.33 | 3.67 | 3.61 | 3.38 | 3.05 | 2.29 | 1.45 | .63 | 3.67 | 0 |
| D2S143 | 217.00 | 214.70 | −∞ | 3.87 | 4.83 | 4.82 | 4.02 | 2.75 | 1.25 | 4.83 | .05 |
| D2S2382 | 220.70 | 216.87 | −∞ | 5.01 | 5.25 | 4.94 | 3.90 | 2.57 | 1.12 | 5.27 | .3 |
| D2S2249 | 223.10 | 218.54 | −∞ | 4.18 | 4.51 | 4.30 | 3.46 | 2.34 | 1.06 | 4.54 | .04 |
| D2S2359 | 225.60 | 220.77 | −∞ | −1.16 | .49 | 1.05 | 1.18 | .85 | .40 | 1.18 | .2 |
| D2S126 | 228.80 | 221.84 | −∞ | −1.75 | 1.26 | 2.13 | 2.26 | 1.65 | .77 | 2.26 | .2 |
| D2S396 | 240.20 | 230.51 | −∞ | −6.20 | −2.25 | −.79 | .20 | .32 | .12 | .32 | .3 |
Haplotype analysis confirmed the critical interval (fig. 2). A recombination event in affected individual 7 of family 50007 set the centromeric boundary at D2S2289, and other centromeric obligate recombination events took place between D2S117 and D2S311 in individual 10 of family 50001, individual 16 of family 50003, and individual 7 of family 50004. Telomeric obligate recombination events took place between D2S126 and D2S396 in affected individuals 12 and 16 of family 50003, and the recombination in affected individual 14 of family 50003 set the telomeric boundary at D2S126. In addition, unaffected individual 9 of family 50001 showed a recombination event at D2S2359, and unaffected individual 7 of family 50001 showed an obligate recombination at D2S143. Thus, the CFD locus is linked to chromosome 2 between D2S2289 and D2S126, and—on the basis of recombination events in unaffected individuals in whom the gene might not, in fact, be penetrant—the locus is probably between D2S2289 and D2S143. In family 50003, unaffected individuals 3 and 18 share part of the risk haplotype, although individuals 1, 2, and 4 (through whom individuals 3 and 18 are connected to the affected individuals in this family) were not available for genotyping and examination.
DHPLC and Mutation Analysis
The interval contains PIP5K3, which has 41 coding exons spanning >89 kb and encodes a 2,098-aa protein. PCR amplification and DHPLC, followed by direct sequencing of the putative exons and intron-exon boundaries, showed mutations in 8 of 10 families studied (fig. 3). Mutations were confirmed in all available affected family members.
Figure 3.

Sequence analysis for representative affected individuals from families with CFD. HTZ = heterozygous.
Screening of exon 16 revealed two significant sequence variations. Family 50001 showed mutation 2256delA, which causes a shift of the reading frame and the generation of an early stop codon after 6 aa, removing the final 1,392 aa. Family 50004 showed 2274delCT, which also causes a shift of the ORF and an early truncation after 5 aa. Five other families exhibit mutations in exon 19. Nonsense mutation 2962C→T (Q988X) was observed in family 50003; 2709C→T (R851X) in family 50005; 3246G→T (E1030X) in family 50008; and 3270C→T (R1038X) in family 50009. Family 50010 displays the only missense mutation observed to date. Nucleotide A at position 3466 was mutated to G, changing the lysine codon at position 1103 to arginine. In family 50007, the splice-acceptor site of exon 20 shows an IVS19-1 G→C transversion. This is predicted to cause skipping of exon 20, with a resultant frame shift and an early truncation after 10 additional amino acids. All these mutations were observed in a heterozygous state and were not observed in 100 control individuals (200 control individuals for mutation K1103R) of similar (European) ethnic background. In two families, 50002 and 50006, the complete screening of the coding regions of PIP5K3 revealed neither point mutations nor small insertions and deletions. In family 50002, a single base insertion (IVS15-82insA) was observed in intron 15, as well as the 13-bp deletion IVS15+11→24. Both these sequence variants are observed in control individuals in a homozygous state and are not considered pathogenic.
PIP5K3 Molecular Organization
A schematic diagram of the PIP5K3 protein structure is shown in figure 4A. Homology to several structural fragments from the PDB was found. First, homology to a zinc finger–containing phosphoinositide kinase (FYVE [PDB: 1dvp]) is located at positions 150–219 (E value = 5.9e−18), with eight cysteines, at positions 160, 164, 167, 180, 183, 188, 191, and 192, that are potentially able to coordinate two zinc atoms. Second, a structural motif shows similarity to a DEP domain (Disheveled, EGL-10, plextrin homology domains of ∼70 aa that are present in numerous signaling proteins [PDB: 1fsh]) of unknown function found in secreted proteins (positions 365–440; E value = 8.3e−27). A search with Superfamily 1.65 localizes a cytosolic chaperone CCTγ apical domain–like motif (PDB: 1gml) at position 677 through 843 of the PIP5K3 protein (E value = 1.3e−27), with a sequence identity of 29.8% and a similarity of 58.5%. The 1791–2085 region of the last C-terminal fragment is similar to that of the common kinase core found in the type IIβ phosphatidylinositol-4-phosphate 5-kinase (PDB: 1bo1) (Rao et al. 1998). The location of these four motifs agrees with that published previously (Shisheva et al. 2001). In addition, a small β-sheet “winged helix” DNA/RNA–binding motif at positions 348–489 (E value = 3.8e−18) and two spectrin repeats at positions 1490–1538 and 1679–1729 were found. Spectrin repeats form a three-helix bundle (PDB: 1quu) and are found in several proteins involved in cytoskeletal structure.
Figure 4.

Schematic diagram of PIP5K3, showing the locations and effect of the dominant mutations. A, Domain structure of PIP5K3. FYVE represents the FYVE zinc-finger domain; DEP represents the Disheveled, EGL-10, plextrin homology domain; Cpn60-TCP1 represents the chaperone-like Cpn-60-TCP1 domain; SPEC represents the two spectrin repeats; and PIPKc represents the domain with phosphatidylinositol-4-phosphate 5-kinase activity. Red arrows show the positions of the truncation mutations associated with CFD, and the blue arrow shows the missense mutation K1103R. B, Molecular structure of the Cpn60-TCP1 domain of PIP5K3, as predicted by homology modeling with the thermosome structure (PDB: 1a6d) as a structural template. The α helices (cylinders) and β strands (ribbons) are shown. C, Effect of dominant mutations in position 711 or 705 on the structure of the Cpn60-TCP1 domain.
A search using the SMART method shows homology to the archaeal chaperonin thermosome structure (PDB: 1a6d), with an E value of 7.0e−14. The sequence of this protein is 36% identical to the CCTγ apical domain (Ditzel et al. 1998), containing ∼170 overlapping residues. ClustalW 1.74 sequence alignment of the thermosome 1a6d and PIP5K3 (residues 462–1026) sequences have shown 20.2% identity and 50% similarity, suggesting that the structure of thermosome is suitable for use as a template for building an atomic model of this sequence fragment. The model of the three-dimensional structure of the domain similar to Cpn60-TCP1 was obtained using the thermosome 1a6d structure, as shown in figure 4B.
CFD nonsense mutations interrupt the PIP4K3 protein sequence at positions 705, 711, 951, 988, 1030, 1038, and 1217. All these mutations affect the C-terminal part of the PIP5K3 sequence, eliminating both the spectrin repeats and the PIPKc sequence. In addition, the two mutations at positions 705 and 711 remove almost half of the C-terminal domain (fig. 4C), destroying the polar surface of the central cavity of the protein domain possibly involved in protein folding (Ditzel et al. 1998), and are predicted to eliminate the chaperone-like domain of the protein.
Discussion
The CFD locus has been mapped in five unrelated families of European origin to a 24-cM (18-Mb) region of chromosome 2q35 flanked by D2S2289 and D2S126, and CFD has been associated with mutations in PIP5K3, a protein which is important for post-Golgi vesicle processing. Mutations were identified in 8 of 10 families in the present study. All but one of these are protein-truncating mutations, predicted to cause termination of the peptide chain before the PIP5K3 structure is formed and to result in loss of activity.
No mutation was identified in family 50002 or 50006. Affected individuals in both of these families have typical CFD and could not be differentiated clinically from affected individuals from other families in the study. Although family 50006 is small and was not included in the linkage study, family 50002 is linked to the PIP5K3 region, with a LOD score of 1.74, which is consistent with involvement of PIP5K3 or a nearby gene, although unaffected individual 3 also inherited the risk haplotype specific for this family. This suggests, but does not prove, that affected individuals in family 50002 may have an intronic or noncoding regulatory mutation that has not yet been identified. Although these will be further investigated as additional families are studied, identification of noncoding mutations can be difficult and may require functional studies of splicing or promoter activity.
A K1103R missense mutation was observed in family 50010. Since most mutations reported here are nonsense mutations, the causative role of this variant may be questionable. Several lines of evidence suggest that K1103R is a mutation. This variant segregated in all affected individuals and was not observed in >400 control chromosomes. A search of dbSNP indicated a total of 15 SNPs in the coding region of PIP5K3, 8 of which are nonsynonymous. Position 1103 is not among them. In addition, K1103 is conserved in various species as divergent as mouse, Xenopus, and Fugu. Confirmation of the causative role of this mutation awaits functional assays or studies of additional families with the same base change.
PIP5K3, a large gene comprising 41 exons that span >89 kb, encodes a predicted 2,098-aa protein. The PIP5K3 protein is a member of the phosphoinositide 3-kinase family, which comprises dual-specificity enzymes possessing an intrinsic protein kinase activity inseparable from a lipid kinase activity. These can generate and relay protein phosphorylation signals to regulate the formation and intracellular location of lipid products (Sbrissa et al. 2000). Both maintenance of mammalian cell morphology and endocytic membrane homeostasis require enzymatically active phosphoinositide 5-kinase PIKfyve (Ikonomov et al. 2001; Shisheva et al. 2001). Cultured mammalian cells transiently transfected with a kinase-deficient point mutant, PIKfyve K1831E, show dilation of PIKfyve-containing vesicles and progressive accumulation of multiple swollen lucent endosomal vacuoles, initially in the perinuclear cytoplasm and later in the cell periphery. This indicates that active PIKfyve enzyme must be present in a distinct set of late endocytic membranes for normal cell morphology (Ikonomov et al. 2001). The formation of enormously dilated late endocytic structures upon transfection of enzymatically inactive PIKfyve suggests a role for PIP5K3 in the endosome-to-trans-Golgi network (Ikonomov et al. 2003b).
It has been suggested that PIP5K3 selectively regulates the sorting and traffic of peripheral endosomes containing lysosomally directed fluid phase cargo by controlling the morphogenesis and function of multivesicular bodies (Ikonomov et al. 2003a). This role is entirely consistent with the clinical and histological findings for CFD, in which the corneal flecks appear to be abnormally swollen keratocytes with membrane-limited intracytoplasmic vacuoles containing complex lipids and glycosaminoglycans (Nicholson et al. 1977). Although the clinical findings for CFD are confined to the cornea, only limited studies of other tissues have been done, and the presence of a subset of abnormal cells in most tissues might not result in clinical symptoms.
In summary, the present study describes both the mapping of the CFD locus in 10 unrelated families of European origin to a 24-cM (18-Mb) region of chromosome 2q35 flanked by D2S2289 and D2S126 and the association of CFD with mutations in the PIP5K3 protein, which is important for post-Golgi vesicle processing. Mutations were identified in 8 of 10 families studied. All but one of these mutations are predicted to result in truncation of the PIP5K3 protein before the protein structure is formed, resulting in the loss of activity. Enzymatic studies of the PIP5K3 enzyme in affected individuals should further elucidate the role of this enzyme in CFD and in normal cellular metabolism.
Acknowledgments
We thank the members of the families, for their participation in this study, and Céline Agosti, Tatiana Favez, and Sylviane Métrailler, for their technical assistance. We acknowledge support from the Swiss National Science Foundation (grant 32-065250.01).
Web Resources
The accession number and URLs for data presented herein are as follows:
- GCG-Lite+ ClustalW 1.74 multiple sequence alignment program, http://www.ebi.ac.uk/clustalw/
- NCBI, http://www.ncbi.nlm.nih.gov/ (for reference genomic sequence [accession number NM_015040])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CFD) [PubMed]
- PFAM, http://www.sanger.ac.uk/Software/Pfam/
- ScanProsite, tool to search the PROSITE database, http://us.expasy.org/tools/scanprosite/
- RADAR, http://www.ebi.ac.uk/Radar/
- SMART at European Laboratory of Molecular Biology, http://smart.embl-heidelberg.de/
- Superfamily 1.65, HMM library and genome assignment service, http://supfam.mrc-lmb.cam.ac.uk/
References
- Abola E, Bernstein FC, Bryant SH, Koetzle TF, Weng J (1987) Protein data bank. In: Allen FH, Bergerhoff G, Sievers R (eds) Crystallographic databases—information content, software systems, scientific applications. Data Comission of the International Union of Crystallography, Cambridge, United Kingdom, pp 107–132 [Google Scholar]
- Akova YA, Unlu N, Duman S (1994) Fleck dystrophy of the cornea: a report of cases from three generations of a family. Eur J Ophthalmol 4:123–125 [DOI] [PubMed] [Google Scholar]
- Birndorf LA, Ginsberg SP (1972) Hereditary fleck dystrophy associated with decreased corneal sensitivity. Am J Ophthalmol 73:670–672 [DOI] [PubMed] [Google Scholar]
- Cottingham RW, Idury RM, Schaffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263 [PMC free article] [PubMed] [Google Scholar]
- Ditzel L, Lowe J, Stock D, Stetter KO, Huber H, Huber R, Steinbacher S (1998) Crystal structure of the thermosome, the archaeal chaperonin and homolog of CCT. Cell 93:125–138 [DOI] [PubMed] [Google Scholar]
- François J, Neetens A (1957) Nouvelle dystrophie hérédofamiliale du parenchyme corneen (hérédo-dystrophie mouchetée). Bull Soc Belge Ophtalmol 114:641–646 [PubMed] [Google Scholar]
- Frueh BE, Bohnke M (1999) In vivo confocal microscopy of fleck dystrophy. Cornea 18:658–660 [DOI] [PubMed] [Google Scholar]
- Goldberg MF, Krimmer B, Sugar J, Sewell J, Wong P (1977) Variable expression in flecked (speckled) dystrophy of the cornea. Ann Ophthalmol 9:889–896 [PubMed] [Google Scholar]
- Ikonomov OC, Sbrissa D, Foti M, Carpentier JL, Shisheva A (2003a) PIKfyve controls fluid phase endocytosis but not recycling/degradation of endocytosed receptors or sorting of procathepsin D by regulating multivesicular body morphogenesis. Mol Biol Cell 14:4581–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomov OC, Sbrissa D, Mlak K, Deeb R, Fligger J, Soans A, Finley RL Jr, Shisheva A (2003b) Active PIKfyve associates with and promotes the membrane attachment of the late endosome-to-trans-Golgi network transport factor Rab9 effector p40. J Biol Chem 278:50863–50871 [DOI] [PubMed] [Google Scholar]
- Ikonomov OC, Sbrissa D, Shisheva A (2001) Mammalian cell morphology and endocytic membrane homeostasis require enzymatically active phosphoinositide 5-kinase PIKfyve. J Biol Chem 276:26141–26147 [DOI] [PubMed] [Google Scholar]
- Jiao X, Munier FL, Schorderet DF, Zografos L, Smith J, Rubin B, Hejtmancik JF (2003) Genetic linkage of Francois-Neetens fleck (mouchetée) corneal dystrophy to chromosome 2q35. Hum Genet 112:593–599 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26:283–291 [Google Scholar]
- Lathrop GM, Lalouel JM (1984) Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet 36:460–465 [PMC free article] [PubMed] [Google Scholar]
- Lee C (1994) Predicting protein mutant energetics by self-consistent ensemble optimization. J Mol Biol 236:918–939 [DOI] [PubMed] [Google Scholar]
- Lee C, Subbiah S (1991) Prediction of protein side-chain conformation by packing optimization. J Mol Biol 217:373–388 [DOI] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 48:443–453 [DOI] [PubMed] [Google Scholar]
- Nicholson DH, Green WR, Cross HE, Kenyon KR, Massof D (1977) A clinical and histopathological study of Francois-Neetens speckled corneal dystrophy. Am J Ophthalmol 83:554–560 [DOI] [PubMed] [Google Scholar]
- Patten JT, Hyndiuk RA, Donaldson DD, Herman SJ, Ostler HB (1976) Fleck (mouchetee) dystrophy of the cornea. Ann Ophthalmol 8:25–32 [PubMed] [Google Scholar]
- Purcell JJ Jr, Krachmer JH, Weingeist TA (1977) Fleck corneal dystrophy. Arch Ophthalmol 95:440–444 [DOI] [PubMed] [Google Scholar]
- Rao VD, Misra S, Boronenkov IV, Anderson RA, Hurley JH (1998) Structure of type IIβ phosphatidylinositol phosphate kinase: a protein kinase fold flattened for interfacial phosphorylation. Cell 94:829–839 [DOI] [PubMed] [Google Scholar]
- Sbrissa D, Ikonomov OC, Shisheva A (2000) PIKfyve lipid kinase is a protein kinase: downregulation of 5′-phosphoinositide product formation by autophosphorylation. Biochemistry 39:15980–15989 [DOI] [PubMed] [Google Scholar]
- Schaffer AA, Gupta SK, Shriram K, Cottingham RW (1994) Avoiding recomputation in genetic linkage analysis. Hum Hered 44:225–237 [DOI] [PubMed] [Google Scholar]
- Shisheva A, Rusin B, Ikonomov OC, DeMarco C, Sbrissa D (2001) Localization and insulin-regulated relocation of phosphoinositide 5-kinase PIKfyve in 3T3-L1 adipocytes. J Biol Chem 276:11859–11869 [DOI] [PubMed] [Google Scholar]
- Smith RJH, Holcomb JD, Daiger SP, Caskey CT, Pelias MZ, Alford BR, Fontenot DD, Hejtmancik JF (1989) Exclusion of Usher syndrome gene from much of chromosome 4. Cytogenet Cell Genet 50:102–106 [DOI] [PubMed] [Google Scholar]
- Streeten BW, Falls HF (1961) Hereditary fleck dystrophy of the cornea. Am J Ophthalmol 51:275–281 [Google Scholar]