

Abstract
With the imminent availability of ultra-high-volume genotyping platforms (on the order of 100,000–1,000,000 genotypes per sample) at a manageable cost, there is growing interest in the possibility of conducting genomewide association studies for a variety of diseases but, so far, little consensus on methods to design and analyze them. In April 2005, an international group of >100 investigators convened at the University of Southern California over the course of 2 days to compare notes on planned or ongoing studies and to debate alternative technologies, study designs, and statistical methods. This report summarizes these discussions in the context of the relevant literature. A broad consensus emerged that the time was now ripe for launching such studies, and several common themes were identified—most notably the considerable efficiency gains of multistage sampling design, specifically those made by testing only a portion of the subjects with a high-density genomewide technology, followed by testing additional subjects and/or additional SNPs at regions identified by this initial scan.
Introduction
A traditional means of discovering disease genes begins with family-based linkage scans, looking for regions of the genome that tend to be transmitted through families in a manner that parallels the transmission of the trait, followed by a variety of fine-mapping techniques. This approach has been highly successful for mapping major genes responsible for Mendelian disorders, in part because the breadth of a linkage signal means that a genomewide scan can be accomplished with a few hundred microsatellite or a few thousand SNP markers. However, finer resolution of the putative risk susceptibility loci through linkage analyses will only be feasible with the availability of sufficient recombination events, requiring large pedigrees (Boehnke 1994), and the utility of the linkage approach for identifying multiple low-penetrance variants involved in common diseases has been questioned.
As an alternative, the past decade has seen a rapid escalation in hypothesis-driven candidate gene association studies or fine-mapping studies exploiting linkage disequilibrium (LD), but these have usually been restricted to a few dozen genes. Recent advances in high-volume genotyping technology have now made it possible to consider using empirical LD patterns to search the genome for risk-associated variants. These studies are based on the premise that “unrelated” individuals are more distantly related than subjects from large pedigrees, thus allowing for sufficient recombination events to have taken place (Nordborg and Taveré 2002). Coupled with the efforts by the International Haplotype Mapping (“HapMap”) Project (Gibbs et al. 2003) to catalog millions of SNPs and haplotypes across diverse populations and to use these to identify subsets of highly informative “tag” SNPs, genomewide association scans involving hundreds of thousands or more markers on thousands of subjects—first suggested a decade ago by Risch and Merikangas (1996)—are now a real possibility.
Numerous research groups are planning or have underway genomewide searches for a range of disorders and the first reports of such studies (using early versions of high-density SNP chips) are just beginning to appear (Ozaki et al. 2002; Klein et al. 2005). These groups are using a variety of population-based and family-based epidemiological designs or model organisms, but so far there has been little general discussion of how best to design and analyze such studies. In April 2005, an international group of 165 investigators met at the University of Southern California for a 2-day workshop to discuss their efforts and consider various methodological problems. This report provides a brief summary of the major themes addressed at this workshop and a review of the relevant background literature. The reader is also referred to several recent review articles (Hirschhorn and Daly 2005; Wang et al. 2005; Palmer and Cardon, in press).
The workshop started with descriptions of some ongoing and planned genomewide association studies. Eleven studies were described and the diseases under study included breast, colorectal, and prostate cancer, type I diabetes, age-related macular degeneration (AMD), Parkinson disease, and systemic lupus erethematosis (SLE). A study in northern Finland is collecting data on a wide range of phenotypes (e.g., birth defects, neurological outcomes, mental illness/personality traits, asthma, cardiovascular events, infections, and diabetes), enabling investigators to study a range of phenotypes simultaneously in the same genomewide scan. Most of the studies were not isolated efforts but were integrated into research programs that usually included candidate-gene association studies and sometimes included linkage studies using affected-pair designs. All of the studies either are using or plan to use some version of a multistage design, and none employ pooling of DNA. They differ in the number of stages, ranging from two to four; in criteria for case selection, with some electing to enhance the initial series with “genetically enriched” cases and some not to; and in the nature of the control group, with some using population-based and some using family-based controls. The number of SNPs currently typed at each stage also differed between studies; although all groups expressed interest in eventually using the 500K panel from Affymetrix (and some are using early-access versions now), they wondered about the actual coverage of that panel. The morning session closed with a description of the research opportunities that derive from studying admixed populations, focusing on Hispanic/Latino populations, and with a discussion of the advantages of studying nonhuman model organisms and of how such studies would complement studies on human populations. More-detailed descriptions of each presentation are provided in appendix A (online only).
The first afternoon began with a series of presentations about genotyping technologies and bioinformatics support. Appendix A (online only) provides some details about the currently available early-release 100–500K SNP platforms from Illumina, Affymetrix, and other companies, as well as the Genetrix bioinformatics suite.
Epidemiologic Study Design
Although the case-control design has become the workhorse of genetic association studies, there has been considerable discussion about its merits relative to cohort studies, nested case-control or case-cohort designs, and family-based designs (Langholz et al. 1999; Witte et al. 1999; Cardon and Bell 2001; Clayton and McKeigue 2001; Thomas and Witte 2002; Cardon and Palmer 2003). Lyle Palmer noted that one of the advantages of the cohort design is that it allows for many disease endpoints to be considered simultaneously using a common set of controls that he dubbed “universal controls” (Palmer and Cardon, in press).
Sobell et al. (1993) first suggested a two-stage design for association studies, which has recently been extended to genomewide scale by Satagopan and colleagues (2002, 2003, 2004), Lowe et al. (2004), and van den Oord and Sullivan (2003). An initial sample of subjects is tested for a dense set of markers, and then an independent sample is tested only on a subset of the most “significant” markers. They describe methods for optimizing the numbers of subjects and significance levels at each stage to maximize power, subject to a constraint on cost and the overall type I error rate. Jaya Satagopan demonstrated that the two-stage design could yield substantial cost savings over a one-stage design with the same test size and power. She also discussed likelihood inference for a quantitative trait locus (QTL), optimizing by selective sampling of subjects with extreme trait values. A related problem for testing single candidate genes was recently considered by Thomas et al. (2004), in which a relatively small sample was used to select tag SNPs, which were then tested in the full study. Both designs use all the data from both samples in the final analysis. Duncan Thomas and Daniel Stram described some extensions of the Satagopan et al. approach for the design of the CFR and MEC genomewide studies. For these studies, it appears that the optimal design typically entails allocating 80%–90% of the costs to the first stage, with a significance level of ∼0.001–0.005 at the first stage. This can be expected to yield ∼500–2,500 loci to be tested at the second stage at a significance level of ∼0.00001. A sample size of ∼2,000 subjects at stage 1 and ∼2,000 at stage 2 would be expected to yield 80% power for detecting a locus with allele frequency 10% conferring a relative risk of 1.3, at an overall (“experimentwise”) type I error rate of 5% and a total cost of ∼$5 million. Extensions required for these calculations involve different costs at the two stages, allowance for the r2 for prediction of unobserved causal SNPs, and use of additional markers at the second stage (see illustrative calculations in appendix A [online only]).
Other design considerations include whether to use a family-based or population-based design and whether to use stratified sampling to enrich one or more of the stages for genetically predisposed cases. The appropriate choice depends in part on whether one intends a search for common polymorphisms having main effects on disease risk or those having modifying effects on other genes or environmental factors, as well as prior beliefs about the “common disease common variant” (Cargill and Daley 2000; Reich and Lander 2001; Lohmueller et al. 2003) versus “multiple rare variant” (Pritchard 2001; Pritchard and Cox 2002; Wang et al. 2003; Fearnhead et al. 2004) hypotheses. Although restriction of the case series to those with a positive family history can be effective at enriching for genetic susceptibility, it risks introducing cryptic relatedness, since cases may share greater kinship with one another than controls, particularly in small regions or population isolates. Selecting cases on the basis of “severity” could also have the counterproductive effect of enriching environmental as well as genetic factors. Genomewide scans could also be used to identify genes that interact with particular environmental agents or other modifying factors already known to play a major role in the etiology of a particular disease. Stratifying on microsatellite instability (MSI) in colorectal tumors or restricting to MSI-stable cases—or stratifying on family history, age, or any of a number of other factors—might lead to greater etiologic homogeneity and improve power for detecting single-gene effects.
The problem of population stratification has been widely debated (Wacholder et al. 2000; Thomas and Witte 2002; Wacholder et al. 2002; Cardon and Palmer 2003; Freedman et al. 2004). David Clayton noted that this can lead to three distinct problems: confounding; cryptic relatedness, resulting in overdispersion of the test statistic; and selection bias. Unlike some other biases, these problems do not become smaller with increasing sample size—on the contrary, the potential inflation of type I error rates will be much larger in studies of the size needed to demonstrate significance at the genomewide scale. Family-based case-control designs offer protection from population stratification, but at the expense of some loss of power from “overmatching” on genotype. The availability of an enormous number of unlinked markers might provide ample opportunity to control for population stratification by the methods of genomic control (Devlin et al. 2001), structured association (Pritchard et al. 2000), or simple logistic regression (Tang et al. 2004) in studies using unrelated controls. Although population stratification will generally cause overdispersion of test statistics (thereby inflating significance levels overall), the significance of any specific test could be either increased or decreased. Thus, the genomic control method, while yielding a test procedure with the correct type I error rate, may suffer from some power loss. It is not known whether the structured association method, which aims to correct each association by stratifying on individual ancestry estimates, would suffer from power loss to the same extent, but it would require a much more computationally intensive analysis.
Population Selection
White populations have hitherto been the primary focus of most association studies, and one of the populations being intensively studied in the HapMap and other genomic variation projects. An open question remains about the “transferability” to other populations of a panel of tag SNPs that has been optimized for whites (Carlson et al. 2003; Mueller et al. 2005). Duncan Thomas described preliminary simulation studies suggesting that a testing procedure that combines a test of overall race-adjusted association and a test of race heterogeneity (each tested at significance level α/2) could yield higher power than either test alone (at level α), even under the hypothesis that the relative risk for an unobserved causal variant was the same across populations. The true relative risk for a causal variant might also be expected to vary across populations because of interactions with other genes or environmental factors with differing prevalence.
Itsik Pe’er discussed the utility of isolated populations for association studies because of their reduced genetic diversity, longer LD, and extreme phenotype frequencies for particular conditions. As an example, he described efforts to construct a haplotype map for the Kosrae population of Micronesia, an isolate for ∼2,000 years with European admixture beginning in the 19th century (Wijsman et al. 2003). These data are based on typing 100,000 SNPs with the 100K GeneChip in 30 parent-child trios. Although the general patterns of allele frequencies and decay of LD were similar to the non-African HapMap data, there was a striking excess of single-copy alleles, with 14 individuals carrying 80% of these rare alleles and a tendency for them to cluster along the genome, suggesting the effects of recent European admixture. This implies a modest improvement in power for single-marker associations and less diversity in long-range haplotypes.
Marker Selection
Eric Jorgenson and John Witte elaborated on the relative merits of “map-based” (i.e., uniformly spaced and or tagSNPs) versus function or “gene-based” (i.e., occurring only in coding, splice site, regulatory regions, and or highly conserved intronic regions) approaches to whole genome association studies (Collins et al. 1997; Tabor et al. 2002; Botstein and Risch 2003; Carlson et al. 2004; Neale and Sham 2004; Palmer and Cardon, in press). They described sample size and cost calculations, concluding that the gene-based approach would be considerably less expensive, because of the reduction in the number of SNPs that must be genotyped and the resulting smaller sample sizes required, but would undoubtedly miss some potentially relevant regions (e.g., enhancers). A map-based approach would also be likely to miss effects of some variation in genes, unless the panel included adequate density of markers within genes (see appendix A [online only] for discussion).
Statistical Analyses
Two main approaches have been advocated for testing gene associations: a “direct” method, based on a simple χ2 test for association, and an “indirect” method, based on associations with haplotypes inferred from unphased multilocus genotypes (Schaid et al. 2002; Zaykin et al. 2002; Stram et al. 2003), the haplotypes being assumed to carry information about possibly unobserved causal variants in the region. In a genomewide context, either approach involves testing an enormous number of hypotheses simultaneously, thereby raising the problem of multiple comparisons. Bonferroni correction is one commonly used approach to address this problem, requiring an extremely small P value (say, 0.05/500,000=10-7) to claim genomewide significance for any particular SNP—or an even smaller P value if multiple subgroups, additional markers, or multiple methods of analysis (e.g., SNPs and haplotypes) are considered. Others have suggested a Bayesian approach, such as the False Positive Report Probability (Wacholder et al. 2004), requiring explicit consideration of the prior probability for each hypothesis under consideration. Under the assumption that there could be many true positive associations, however, the Bonferroni correction is too conservative, and a variety of methods based on the False Discovery Rate have been advocated (Benjamini and Hochberg 1995; Efron and Tibshirani 2002; Sabatti et al. 2003; Storey and Tibshirani 2003).
Two papers have recently proposed analytic approaches for genomewide association studies that go well beyond simple exhaustive testing of all SNPs separately. Lin et al. (2004) proposed exhaustive testing of haplotype associations over all possible windows of segments, using a computationally efficient permutation procedure to assess the significance of these correlated tests. Marchini et al. (2005) proposed exhaustive testing of all possible pairwise gene-gene interactions. Nelson Freimer introduced the idea of haplotype sharing among case-case pairs as an alternative to case-control association; Duncan Thomas provided a formal test of haplotype sharing and showed how this test could be decomposed into principal components representing case-control associations with clusters of similar haplotypes, in the spirit of Tzeng et al. (2003), Nyholt (2004), and Lin and Altman (2004).
It is generally agreed that no amount of association testing in epidemiological studies alone can distinguish between the true-positive and false-positive signals obtained in a multistage genomewide scan (Page et al. 2003). Approaches that could be taken at this stage might include comparative genomics (Sidow 2002; Bejerano et al. 2004), linkage analysis of expression data (Morley et al. 2004), or computational approaches to predicting function (Ng and Henikoff 2003; Taylor and Greene 2003; Livingston et al. 2004; Xi et al. 2004; Zhu et al. 2004), before launching into the labor-intensive and time-consuming process of developing functional tests. Eleazar Eskin illustrated this by incorporating predictions of variation function, using as an example the Chromogranin A gene (CHGA) involved in hypertension. By use of the HAP phasing algorithm (Hinds et al. 2005), six common haplotypes were identified, one of which appeared to be strongly associated with the trait. A combination of comparative genomic analysis and known binding-site analysis identified a specific variant that could be responsible, G462A, shown by in vitro assay to alter reporter expression.
Chiara Sabatti discussed the interpretation of stretches of homozygosity, using data on Costa Rican case-parent trios with Bipolar-1 disorder, typed at ∼3,000 SNPs on chromosome 22. She described a hidden Markov model approach to estimation of the inbreeding coefficient from genomic data (Leutenegger et al. 2003), which showed that all but three of parents had estimated inbreeding coefficients of zero. Another possible explanation for long homozygous stretches is large-scale copy number variation (Iafrate et al. 2004; Sebat et al. 2004). She discussed the applicability of methods used to deal with genomic losses in cancer (Newton et al. 1998) to other types of phenotypes, but she concluded that, without a good model for instability, such techniques were more useful for evaluating the likelihood of seeing particular stretches of large-scale copy number variation than for detecting their existence.
Power
David Clayton showed the sample-size requirements for a single-stage association study using both the direct and indirect approaches (see appendix A [online only]). At a marker density on the order of 1 every 6 kb (500,000 markers), we expect that most associations would be detected indirectly by LD rather than with causal variants directly. Under the assumption that an average of 8 tag SNPs would yield an r2 of 0.8 and with the use of an 8-df test, the sample sizes required for such indirect associations would be slightly less than double those that would be needed for a direct association.
Paul de Bakker summarized a comparison of various SNP selection and analysis methods, using simulations based on the HapMap-ENCODE regions, representative 500-kb regions of the genome with complete ascertainment of common variation in 270 individuals (ENCODE Project Consortium 2004). By nominating all common SNPs as a causal allele, one by one, they generated simulated case-control data sets, from which they computed the power to detect an association under different tagging and testing scenarios. They found that choosing tag SNPs from a 5-kb panel (such as the Phase I HapMap) gave surprisingly good power for common (>5% frequency) causal alleles, and that specified haplotype-based tests further improved genotyping efficiency—a 33% reduction in the number of tag SNPs required, with no loss of power. Additional sliding windows of haplotypes did not help for common causal alleles, once the increase in the number of tests was allowed for, but there was some improvement in power when the causal allele was rare (minor allele frequency [MAF] < 5%).
Daniel Stram described similar simulations, focusing on the power of some very simple analyses of whole genome scans using tag SNPs. Power is determined by the noncentrality parameter, a function of the causal allele frequency, its true relative risk, and the r2 for the prediction of the unobserved causal variant by nearby SNP(s). For relatively small regions, a Bonferroni-adjusted single-SNP analysis is generally more powerful than a multivariate test of association, but, on a genomewide scale, the effective number of “independent” tests is a function of the extent of LD. By determining the block structure, choosing tag SNPs within blocks, conducting multivariate tests within each block, and applying a Bonferroni correction for the number of blocks instead, he showed that this method yielded better power than simply using all SNPs with Bonferroni correction for the number of SNPs.
Genotyping Errors
David Clayton noted that genotyping errors are generally assumed to be nondifferential (i.e., not related to phenotype), leading to some loss of power and bias in relative risk estimates towards the null, but no increase in type I errors (except in case-parent trio designs). However, he pointed out that it may be difficult to ensure that all aspects of DNA processing and analysis are the same for cases and controls, particularly if the ascertainment of these two groups is not concurrent. To test this assumption, he showed data from a study of nonsynonymous SNPs (nsSNPs) and type I diabetes, which revealed that some of the overdispersion of association tests that were not obviously true positives could be explained by questionable allele calls or by those not replicated on another platform, as well as by regional stratification and substructure (see appendix A [online only] for additional details of this analysis). Particularly disturbing were shifts between cases and controls in the point clouds corresponding to each genotype, presumably due to differences in DNA processing. Standard laboratory practice of using blinded samples to determine the parameters for allele calling could thus lead to differential misclassification (with consequent inflation of type I error rates and relative risk estimates biased away from the null). Instead, it would appear that, to minimize such misclassification, it would be necessary to calibrate the software separately for each group.
Derek Gordon showed the effects of nondifferential genotyping error on both family-based and population-based tests of association. For case-parent trio data, even nondifferential errors can inflate type I error rates (Mitchell et al. 2003). To overcome this problem, Gordon et al. (2001) introduced the likelihood-based TDTae (“adjusted for errors”). Similar issues have been addressed for case-control designs using unrelated individuals (Rice and Holmans 2003). Phenotyping errors would be expected to have similar effects. Recent work on the use of double sampling, combining fallible methods on a large sample with a “gold standard” method on a subset, appears to improve power for tests of association (Gordon et al. 2004). Methods that formally incorporate information on accuracy of genotype or haplotype calls into the statistical test of association (Hao and Wang 2004) have some potential for extension to the whole-genome scale.
DNA pooling has been suggested as an efficient procedure for screening many samples for differences in allele frequencies at many loci (Bansal et al. 2002; Sham et al. 2002). Various authors have discussed experimental design for such studies (Barratt et al. 2002; Pfeiffer et al. 2002; Sham et al. 2002), including the number of pools and pool sizes needed for accurate allele frequency determination. The general sense of the participants was that this approach is not sufficiently reliable for use on a genomewide scale at this time, despite its obvious cost appeal.
Conclusions
All genomewide association studies of human populations that have been described above are using or plan to use a multistage design, and none are proposing to use DNA pooling. Studies differ in the number of stages and in the nature of cases and controls selected for each stage. Some of the studies in the United Kingdom and the CFRs employ a strategy that aims to enhance genetically caused cases, while others (e.g., the AMD study) choose not to employ such a strategy. There was general agreement that it is probably helpful to decrease heterogeneity in the case series, either by exclusion of selected subgroups (e.g., exclusion of colorectal cancer cases on the basis of MSI-H tumors or evidence of a germline mutation in a mismatch repair gene) or by a stratified selection that would ensure sufficient sample size in the major strata of interest. In the study of cancer, there is growing recognition of the value of using molecular markers derived from the tumor to define sources of heterogeneity. Similar markers are under development with other diseases. Thus, even for diseases like cancer that are traditionally analyzed as simple dichotomous phenotypes, there are often several dimensions on which to characterize cases; for other diseases, like diabetes, the number of variables needed to fully characterize the phenotype can be very large. A genomewide scan for genomic determinants of gene expression levels, for example, would entail potentially many billion comparisons; several such scans are currently underway.
Advantages and disadvantages of alternative control series, usually discussed in the context of candidate gene studies (e.g., trade-offs between power and control of population stratification), are also relevant to genomewide association studies. Some studies—for example, the CFRs—are in a position to use both types of controls and currently plan to use unrelated controls in stage 1 to enhance power and family-based controls in stage 2. If the main objective of a second stage is to replicate the findings of a first stage, then one needs to be mindful about introducing possible sources of heterogeneity (e.g., by using different types of cases or controls) between stages, which will complicate interpretations of results.
Most studies are not or do not plan to incorporate information on environmental exposures in the early stages of the genomewide studies. This concerned some investigators, since genomewide scans could miss important genetic causes where the effect of the gene is only detectable when information on the relevant environmental exposure(s) is incorporated into the analyses, particularly since common genetic variants for common diseases may plausibly interact with environmental exposures.
Several very general issues were raised in the concluding discussion. Alice Whittemore began by asking (1) Is the technology driving the science? (2) Can we afford the technology? (3) When is an association scan unwarranted? and (4) When it is warranted, how can the epidemiology and biology of the disease drive our choice of design? An example where such an approach might not be warranted is Hodgkin disease, in which the risk to DZ twins is very low and the risk to MZ twins nearly 100% (Mack et al. 1995), suggesting multiple rare variants (Risch 1990), a scenario not amenable to association mapping. Robert Haile asked whether the time was ripe in terms of the technology development, the need for coordination by the many investigators who are likely to be proposing such studies in the near future for a range of diseases and even for different species, and how best to deal with the problems of etiologic heterogeneity and complexity. Nevertheless, there seemed to be a broad consensus that the time was indeed ripe for launching the first generation of genomewide association studies, but that each would require careful justification and coordination among groups studying similar conditions to ensure optimal allocation of the limited resources available for such expensive undertakings.
Acknowledgments
This workshop was supported by the University of Southern California (USC) Center of Excellence in Genomic Sciences (grant 1P50 HG002790), the Southern California Environmental Health Sciences Center (grant 5P30 ES07048), and the USC Keck School of Medicine. Invited speakers included Habib Ahsan (Columbia University), Fernando Arena (National Cancer Institute, National Institutes of Health), Paul de Bakker (Massachusetts General Hospital), Timothy Bishop (Leeds University), Jonathan Buckley (University of Southern California), Graham Casey (Cleveland Clinic Foundation), David Clayton (Cambridge Institute of Medical Research), Mariza de Andrade (Mayo Clinic), David Duggan (TGen), Eleazar Eskin (University of California at San Diego), Nelson Freimer (University of California at Los Angeles), Ellen Goode (Mayo Clinic), Derek Gordon (Rockefeller University), Robert Haile (University of Southern California), Brian Henderson (University of Southern California), John Hopper (University of Melbourne), Eric Jorgenson (University of California at San Francisco), Magnus Nordborg (University of Southern California), Lyle Palmer (University of Western Australia), Itsik Pe’er (Broad Institute), Chiara Sabatti (University of California at Los Angeles), Jaya Satagopan (Memorial Sloan Kettering Cancer Center), Nik Schork (University of California at San Diego), Daniela Seminara (National Cancer Institute, National Institutes of Health), Susan Service (University of California at Los Angeles), Dan Stram (University of Southern California), Simon Tavaré (University of Southern California), Nicole Tedeschi (University of Southern California), David Van Den Berg (University of Southern California), Alice Whittemore (Stanford University), and John Witte (University of California at San Francisco).
Appendix A
Current and Proposed Studies
Human Studies
U.K. Diabetes and Breast Cancer Studies
David Clayton opened the workshop by describing three ongoing or planned whole-genome association studies in the United Kingdom. The first is an ongoing multistage study of type I diabetes, comprising 8,000 cases, which represents 50% of all juvenile onset type I diabetes in Great Britain, and 8,000 controls drawn from the 1958 birth cohort. The first stage involved 1,000 cases, 1,000 controls, and the genotyping of ∼10,000 SNPs by use of the ParAllele/Affymetrix platform and was completed in October, 2004. The second stage recapitulated the first stage, in that 1,000 cases and 1,000 controls were studied with mostly the same SNPs that were used in stage 1. This stage was completed in April, 2005. Stage 3 involves 2,000 cases, 2,000, controls and the genotyping of the 3,000 most significant SNPs identified from stages 1 and 2. Stage 4 is planned to confirm any significant findings in a further set of 4,000 cases, 4,000 controls, and >3,000 case-parent trios.
The second is an ongoing study of breast cancer, again performed using a multistage design. Four hundred “genetically enriched” cases of breast cancer and 400 controls identified from the EPIC-Norfolk cohort are being studied in the first stage, scheduled to be completed by June, 2005. Genotyping involves 200,000 SNPs (Perlegen/Affymetrix platform). SNPs with P<.05 will be genotyped in the second stage in an additional 4,600 cases (East Anglian Registry) and 4,000 controls (EPIC). DNA pooling was considered but was rejected at this point in time.
The third study is the Wellcome Trust Case-Control Consortium, which is planned to start in the near future. This large study merges three separate proposals submitted to the Wellcome Trust in 2003, and will include eight different case groups covering a range of pathologies, including cardiovascular, cancer, autoimmune, and psychiatric outcomes and two control groups (blood donors and subjects derived from the 1958 birth cohort). It is the current plan to genotype 500,000 SNPs, including all known common nonsynonymous SNPs, a dense set of tag SNPs in the MHC region, and tag SNPs for as much as possible of the remaining genome, with the genotyping platform still under consideration. This study will also employ a multistage design, with the details of each stage still under development.
Multiethnic Cohort Study
Brian Henderson described the biological rationale for the ongoing candidate gene association studies in the Multi-Ethnic Cohort (MEC) study focused on steroid hormones (Kolonel et al. 2004). The MEC comprises ∼215,000 individuals from five ethnic groups—African Americans, Hispanics, Japanese Americans, Native Hawaiians, and whites—from Los Angeles and Hawaii, being followed prospectively by linkage to the corresponding SEER registries. A genomewide association study, using a two- or three-stage approach, is planned, to complement the ongoing candidate gene-association studies. In the first stage, ∼2,000 breast, 2,000 prostate, and 1,000 colorectal cancers will each be compared with a common group of 3,000 controls drawn from the MEC, by use of the Affymetrix 500K GeneChip. All ethnic groups will be included at this stage, but additional markers may be needed to improve coverage in African Americans. The initial test will be based on a combination of main effects of each SNP individually, as well as tests of heterogeneity between the five ethnic groups (more on this strategy below). The investigators plan to use HapMap data to predict common SNPs not on the 500K GeneChip, thereby extending the effective coverage and improving power of this initial scan. The follow-up stages of the project will combine two strategies: adding additional markers in the regions suggested at stage 1 and genotyping a roughly equal number of additional subjects on this subset of markers. The optimal way of combining these two strategies is still under discussion.
Breast and Colorectal Cancer Family Registries
Genomewide association studies are also being planned by both the Breast and Colorectal Cancer Family Registries (CFRs). The Colon CFR (Haile et al. 1999; Newcomb et al. 2002) is an NCI-supported consortium initiated in 1997 that established a comprehensive infrastructure for genetic epidemiology studies of colorectal cancer. Six registries, including the Fred Hutchinson Cancer Research Center, the University of Hawaii, the Mayo Clinic, and consortia at Cancer Care Ontario, the University of Southern California, and the University of Melbourne collect risk factor information and biospecimens from families identified through population-based sources and clinics. The registry currently comprises ∼10,000 families, roughly equally divided between population- and clinic-based ascertainment.
Graham Casey described the plans for the genomewide association scan using the Colon CFR resource. The Colon CFR plans to utilize its relatively large sample size of combined clinic-based and population-based study participants who have been screened for known mismatch-repair mutations and selected tumor alterations (e.g., MSI). A two-stage design has been proposed in which a subset of cases (enriched for family history and/or young age at onset) are compared with unrelated age- and sex-matched population controls in the first stage by use of the Affymetrix 500K GeneChip (Satagopan et al. 2002, 2004; Satagopan and Elston 2003). Power in this first stage is expected to be enhanced by the use of unrelated controls (Risch and Merikangas 1996) and what are presumed to be genetically enriched cases (Antoniou and Easton 2003; Thompson et al. 2004). A stratified analysis is planned to address issues of genetic heterogeneity and interactions with nongenetic factors; certain subgroups will be oversampled so that relationships between important molecular characteristics (e.g., MSI), risk factors (e.g., smoking), and family history (very dense families vs. moderately dense families) can be clarified. Approximately 800 cases and 800 controls are expected to be included in the first-stage genome screen.
The second stage of the proposed Colon CFR study design will take advantage of related controls available within the resource to address population stratification concerns in the first stage. Study participants included in the second stage will be similar to those in the first stage with respect to family history and stratifying factors. Second stage controls will be age- and sex-matched unaffected siblings, half-siblings, or cousins. Selections of regions to follow-up for additional, denser SNP analysis in the second stage will be based on a P-value threshold of .008 in the first stage. Additional markers are expected to be used in ∼400 regions of interest, requiring a custom genotyping panel of up to 4,000 SNPs. Approximately 2,000 cases and 2,000 related same-generation controls are expected to be included in the second-stage analysis.
Single-SNP analyses, as well as long-range haplotype analyses (Lin et al. 2004), are planned. Power calculations suggest the sample sizes described above will yield 80% power to detect an allele with a susceptibility prevalence of 20%, conferring a relative risk of 1.5 in the first stage and 1.3 in the second stage (due to the lack of enriched cases in the second stage), 50% relative efficiency in the second stage (due to use of related controls), with a first-stage significance level of .008 and a combined two-stage significance level of .05.
The Breast CFR (John et al. 2004) is structured similarly to the Colon CFR, with contributing registries from Ontario, Northern California, Australia (population based), Utah, Philadelphia, and New York (clinic based), and currently totals 7,156 families. Habib Ahsan described their plans for a genomewide association scan using this resource to identify novel alleles associated with breast cancer risk. Like the Colon CFR, the focus will be on early-onset cases, to enrich the study sample with genetically caused disease. To enhance study efficiency and validity a two-stage design will be employed: (a) a flexible and statistically powerful population-based case-control design, to screen for promising loci in stage I; and (b) a methodologically robust and focused family-based design to confirm the findings in stage II. Two different but complimentary genotyping platforms will be used. In stage I, cases and population controls will be genotyped by the Affymetrix 500K GeneChip and the ParAllele nonsynonymous SNP chip enriched with breast cancer–related candidate SNPs. For stage 2 genotyping, a custom ParAllele chip will be designed by including only the tags for detecting the SNPs identified in stage I and additional SNPs surrounding these loci. The determination of sample sizes for the two stages and the analyses of data will be similar to the plans outlined above for the Colon CFR. The genomewide association scan will be complemented by genomewide linkage and loss-of-heterozygosity scans.
There are several unresolved issues in the design of genomewide association studies using this resource. One issue is whether the inclusion of a third stage in the study design to narrow down the candidate regions would provide benefits over a two-stage approach followed by publication and independent replication. A large variety of analytical issues remain to be addressed related to multiple comparisons, haplotype analysis, and selection of regions for follow-up. Unresolved issues particularly relevant to studies using family-based data sets include the advantages and disadvantages of including parental information in discordant sibling analyses, the interface between genomewide linkage and association scans, and implementation of family-based association testing on a genomewide level.
Prostate Cancer
John Witte described a planned study of prostate cancer. The Prostate Cancer Case-Control Consortium study will include 2,000 cases and 2,000 unrelated controls. The case group will be restricted to men with more-advanced disease at diagnosis (i.e., Gleason score ⩾7 or tumor stage ⩾T2c). Their current plan is very similar to others: use of a two-stage design with the Affymetrix 500K GeneChip in the first stage and a more focused set of markers in the second stage (possibly using the Illumina platform). It remains unclear exactly how many subjects to include in each stage, although initial power calculations suggest an approximately 50–50 split between the two.
Northern Finland Birth Cohorts
Nelson Freimer described a phenome-genome analysis that is currently ongoing among birth cohorts in northern Finland, where a phenome is a comprehensive representation of phenotypes and a phenomic approach is an attempt to bring scale, scope, and standardization to phenotyping. Two birth cohorts are included: the birth cohort of 1966, which has a sample size of 12,000 and currently includes 5,000 DNA samples, and the birth cohort of 1986, which has a sample size of 9,500 and includes 9,000 DNA samples. Data on a wide range of phenotypes (e.g., birth defects, neurological outcomes, mental illness/personality traits, asthma, cardiovascular events, infections, and diabetes) are collected at 24 wk gestation, at birth, at 1 year of age, at 14–15 years of age, and at 31 years of age, with continued data collection planned for the future. In contrast to most genomewide studies that focus on one phenotype (e.g., breast cancer), this design has the advantage that many phenotypes may be studied in association with genotype in a genomewide association. The sample size is large, so power should be high for quantitative traits and the 1986 cohort may be used to replicate findings observed from the 1966 cohort.
Age-Related Macular Degeneration
Three papers (Edwards et al. 2005; Haines et al. 2005; Klein et al. 2005) recently reported an association of age-related macular degeneration (AMD) with a novel locus, Complement Factor H, which appears to confer a population attributable risk of ∼40%. The initial discovery of this association (Klein et al. 2005) was found in a genomewide association scan using the Affymetrix 100K GeneChip. Nicole Tedeschi described an ongoing study of early AMD. Subjects are chosen from within the population-based Los Angeles Latino Eye Study (LALES). Roughly 67% of all LALES participants have donated finger-prick blood samples, from which they have obtained whole-genome amplification product of their DNA. Of these subjects from whom they have amplified DNA, they have chosen all available early AMD phenotype cases (none of which are early age at onset and none of which also have advanced diabetic retinopathy), leaving a total of 285 cases for study. Controls were selected from those subjects from whom they have amplified DNA (excluding any that have advanced diabetic retinopathy or AMD). For each case, two controls were selected, randomly matching on age (by exact year for all possible [77%] and by closest age [± 6 years or less] for 23%), birthplace (the United States, Mexico, or other) and smoking status (smokers vs. nonsmokers). Genotyping is being conducted with amplified DNA, by use of Affymetrix 100K and early access 500K GeneChips.
Parkinson Disease
Mariza de Andrade described a comprehensive approach to the genetics of Parkinson disease that includes a candidate-gene association study, linkage scans, and a two-stage genomewide association scan. In stage 1, 443 case-unaffected sib pairs have been genotyped at 200,000 SNPs (1 per 10 kb, minor allele frequencies >10%). This design yields 80% power to detect odds ratios >2 (at P<.01) under a log-additive model and a causal allele frequency of 10%. In stage 2, 2,000 SNPs selected from stage 1, plus an additional 312 SNPs to serve as a genomic control, were genotyped in 334 case-unrelated control pairs. For this stage, the minimum detectable odds ratio was 1.56 with 80% power.
Systemic Lupus Erythematosus (SLE)
Chaim Jacob described a study being conducted by the SLEGEN Consortium to investigate the genetics of SLE. Their initial study aims to perform a genomewide association study by use of the Affymetrix 500K GeneChip in a multistage design. In stage 1, 400 cases will be chosen randomly from among the probands of >650 European-derived multiplex pedigrees and will be matched with 400 controls on self-reported ethnicity, gender, and geographic origin. Jacob and colleagues are exploring the possibility of exchanging up to half of their controls for genotypes with other studies at the Broad Institute on the 500K Affymetrix GeneChips. Stages 2 and 3 comprise two replication studies. In replicate group A, a total of 750 cases will be selected from the ∼250 remaining probands from the European-derived multiplex families and from the ∼500 SLE cases with the strongest evidence for familial SLE (i.e., of a second SLE-affected relative who satisfies ACR criteria for SLE). In this stage, the investigators will continue to exploit the theoretical advantage that the familial cases provide greater power because they may be enriched for genetic causes of SLE. European-derived controls will be matched by sex and geographic origin. Replicate group B will encompass a thorough consideration of the polymorphisms identified in stages 1 and 2. Another set of 750 independently ascertained SLE cases with matched controls will be evaluated in an attempt to eliminate false positives. The cases and controls will be taken from the remainder of the collected and available European-derived materials, with preference given to complete trios (SLE affected and both parents).
Admixed Populations
Fernando Arena described planned studies of admixture in the Hispanic/Latino populations, which are thought to have West African, Native American, and European ancestry accumulated over the past 5 centuries. The genetic variability of each of the ancestral populations and the varying proportions within the gene pool that contribute to the many Hispanic/Latino groups has resulted in different gene frequencies, which, in interactions with varying environmental factors, generate diverse ethnic identities and cultural backgrounds. With the use of admixture mapping, an understanding of the genetic structure of admixed populations, such as Hispanics/Latinos, can allow us to identify genes that may be responsible for ethnic variations in disease risk (Patterson et al. 2004). Another use is in controlling for confounding of associations between genetic factors and cancer risk in admixed populations. Models of individual and population admixture can be used in planning future studies in those admixed populations to develop efficient study designs and sample size estimates.
Studies have observed a tendency for Hispanics/Latinos to have lower-than-average rates of some chronic illnesses, despite the fact that many of them live in relatively poor social or economic conditions. This tendency has been termed the “Hispanic Paradox” (Markides and Coreil 1986). In fact, Hispanics/Latinos have lower incidence and mortality rates from all major cancers (breast, prostate, lung and bronchus, colon, and rectum) than non-Hispanic whites. In contrast, rates are higher in Hispanics/Latinos for cancers of the stomach, liver, uterine cervix, and gallbladder (O’Brien et al. 2003), as well as other chronic diseases like diabetes.
Preliminary data generated in the United States (Parra et al. 1998, 2001; McKeigue et al. 2000; Pfaff et al. 2001, 2002; Akey et al. 2004) and in countries in Latin America (Carvalho-Silva et al. 1999; Sans 2000; Rodriguez-Delfin et al. 2001; Salzano and Bortolini 2001; Tarazona-Santos et al. 2001; Bortolini et al. 2003; Cifuentes et al. 2004) have provided some initial insight into the geographic pattern of genetic stratification and admixture in the United States and in Central and South American regions. Of the relatively small number of studies that have examined this issue to date, many have suffered from insufficient sample size, limited coverage, and lack of a standardized ancestry-informative panel of DNA markers. Part of the problem stems from an almost complete lack of data on genetic variation in indigenous populations, such as Native Americans in the United States and native populations in other countries. These populations have been neglected in several large-scale studies of genetic variation. Furthermore, most efforts have been isolated, without a systematic and coordinated exchange of knowledge, resources, and data within the scientific community. There is a need to encourage research and to develop the tools necessary to investigate and understand population stratification and admixture in the American continent. Overall, knowledge about genetic structure of populations, together with admixture mapping approaches can help identify genetic factors that contribute to the etiology of complex human traits and to understand how these interact with environmental factors.
Model Organisms
The Workshop also included a brief discussion of the use of nonhuman model systems to complement the ongoing efforts to map genes responsible for human disease. Magnus Nordborg described why the plant Arabidopsis thaliana is, in many ways, ideal for genomewide LD studies. First, because the species is self-fertilizing, it consists of naturally occurring inbred lines, so that genotypes sampled in nature can be replicated in the laboratory, and many different phenotypes can be measured with replication. It is, for example, possible to study the response of a particular genotype to a range of environmental conditions by growing it under all those conditions. Second, inbreeding has led to considerable LD across the genome, with similar decay (25–50 kb) to that seen in humans. Its small genome of 120 Mb means that genomewide scans become affordable, making this model organism particularly attractive for some studies.
Nik Schork described aspects of genomewide studies in mice and argued that genetic background can be a surrogate for the combined effects of many genes too small to be revealed in multiple single-locus analyses. Measures of genetic background can be derived from mouse experiments (especially inbred line crosses), and controlling for genetic background in quantitative trait locus (QTL) is relatively straightforward. Further, in silico mapping can be combined with molecular phenotyping to identify candidate genes. These may then be evaluated further in subsequent experiments with mice and their human homologues in humans.
Genotyping Technologies
David Van Den Berg summarized Illumina’s GoldenGate assay and introduced the whole-genome genotyping (WGG) assay, which forms the basis of Illumina’s higher-density arrays (Gunderson et al. 2005). As proof of principle, Gunderson et al. genotyped several hundred SNPs previously genotyped via the GoldenGate (Fan et al. 2003). The whole-genome amplified products generated as part of the WGG assay were shown to represent >95% of the unamplified loci (r2=0.758). Call rates, accuracy, and concordance were assessed using both monomorphic controls (99.7%, 99.96%, and 99.97%, respectively, for 176 of 186 successful assays) and HapMap quality control SNPs (99.7%, 99.99%, and 99.9%, respectively, for 819 successful SNP assays of 1,500). The first high-density Illumina array (Sentrix Human-1 Genotyping Beadchip, also known as the Infinium 100K) includes >100,000 SNPs selected from dbSNP: 23% are located in transcripts, 48% are within 10 kb of an exon, 15% are in highly conserved regions, and 14% are uniformly spaced.
Multistage designs need to include numerous high-throughput technologies (both in terms of samples and SNPs). David Duggan discussed the niche areas, including genomewide, candidate region/pathway/gene, and individual SNPs. Perlegen’s approach to genomewide association makes use of the LD pattern among 1.5 million SNPs and their tagSNPs (Hinds et al. 2005). The approach makes use of long-range PCR and Affymetrix custom arrays and is 99.54% concordant with the HapMap project data. SNPs for Affymetrix’s early-access 500K GeneChip were selected from >2.7 million Perlegen and dbSNP SNPs and are genotyped using restriction enzyme digestion, ligation of a PCR linker, PCR, and hybridization-based allele discrimination similar to that described previously for the 10K and 100K GeneChips (Matsuzaki et al. 2004a, 2004b). The resulting early-access 500K GeneChip has ∼200,000 SNPs in or around genes, a median intermarker spacing of 3.3 kb, mean 5.4 kb, average heterozygosity of 30%, average MAF of 22%, and >80% of the genome is within 10 kb of a SNP (Greg Marcus, Affymetrix, personal communication). Performance specifications are not yet available but are expected to be similar to those for the 100K: >95% call rate, 99.96% reproducibility, and 99.73% concordance with the HapMap genotypes (Matsuzaki et al. 2004b).
The next-generation high-density arrays from both Affymetrix and Illumina are expected to contain SNPs selected on the basis of empirical data from phases I and II of the HapMap project. A further discussion of the adequacy of genome coverage for these and other LD-based approaches is presented in the following section. Other genomewide technologies described include ParAllele’s 100K Panel and Sequenom’s 75K and 100K SNP panels. Second-stage or candidate region/pathway/gene technologies include ParAllele’s 3K, 5K, and 10K custom SNP panels, Illumina’s GoldenGate, and Sequenom’s MassArray.
Adequacy of Genome Coverage with Existing Technologies
Although the decreased costs of genotyping and availability of genomewide SNP genotyping platforms enable this type of study, it is not clear how adequate the genomic coverage of available products is. Only a fraction of all common SNPs in the human population have been characterized to date (phase I of HapMap has 1 million SNPs of an estimated 10 million common SNPs); thus, association studies rely on the expectation that an undiscovered, disease-associated variant is likely to be correlated with an allele of an assayed SNP. Since marker spacing on the physical map is less important than on an LD map, detailed information about LD between available markers and untyped nearby SNPs is essential. Uniform spacing may mean that, say, 75% of the common SNPs in the genome are captured with r2>0.8, but may leave a substantial part of the genome captured inadequately. One way to address this is to analyze correlations between typed and untyped markers in the HapMap-ENCODE regions, which serve as a “gold standard” data set for common variation (P. I. W. de Bakker, R. Yelensky, I. Pe'er, S. B. Gabriel, D. Daly, and D. Altshuler, unpublished data). However, unless the LD patterns of each gene are empirically determined, missense SNPs might well be missed, because choosing SNPs on the basis of physical proximity does not guarantee that nearby SNPs will be captured (Carlson et al. 2004). Several authors have considered the required density for a map-based approach (Kruglyak 1999; Judson et al. 2002; Carlson et al. 2003, 2004; Goldstein et al. 2003; Wang and Todd 2003; Ke et al. 2004; Hinds et al. 2005), providing estimates ranging from ∼200,000 to >1 million. Lyle Palmer showed that increasing density of markers revealed smaller and smaller haplotype blocks within a region, concluding that guidelines about average numbers of SNPs needed were not particularly helpful. Finally, genecentric approaches assume that we know the location of all human genes. A recent report suggests that there may be twice as many polyadenylated transcribed sequences and an equal number of nonpolyadenylated and nuclear transcripts yet to be identified (Cheng et al. 2005). Moreover, regulatory variants further away from a gene will almost certainly not be surveyed using a genecentric approach. Nonetheless, both gene-based (using PCR-Invader assay of 90,000 SNPs) and map-based (using 100K GeneChips) approaches have been successful in detecting strong associations with common, complex diseases (Ozaki et al. 2002, 2004).
Evidence for Differential Genotyping Error in the U.K. Study of nsSNPs and NIDDM
Examination of the distribution of association tests using the original ParAllele genotyping revealed the expected large number of highly significant associations with SNPs in the HLA region, but some overdispersion of the remaining SNPs ( 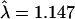 ). Closer examination of the most extreme tests raised some concerns about genotyping errors, and, indeed, the majority of these were not confirmed by reanalysis using Taqman in the original samples and were not significant in additional replication samples. Comparison of the two platforms revealed that the Taqman assay failed to yield a call more frequently in controls, instead yielding them apparently randomly, while ParAllele call failures were differential between cases and controls. More disturbingly, there were major disagreements between called genotypes, but only in cases. These differences seem to be due to shifts in the point clouds corresponding to each genotype between cases and controls, presumably due to differences in DNA processing. Further analysis of overdispersion of the non-HLA association tests showed that the initial estimate could be reduced by restriction to confident allele calls (
). Closer examination of the most extreme tests raised some concerns about genotyping errors, and, indeed, the majority of these were not confirmed by reanalysis using Taqman in the original samples and were not significant in additional replication samples. Comparison of the two platforms revealed that the Taqman assay failed to yield a call more frequently in controls, instead yielding them apparently randomly, while ParAllele call failures were differential between cases and controls. More disturbingly, there were major disagreements between called genotypes, but only in cases. These differences seem to be due to shifts in the point clouds corresponding to each genotype between cases and controls, presumably due to differences in DNA processing. Further analysis of overdispersion of the non-HLA association tests showed that the initial estimate could be reduced by restriction to confident allele calls (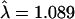 ), further stratification by region (
), further stratification by region (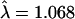 ), adjusting for substructure (
), adjusting for substructure (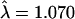 ), and finally by restriction to only the “best” allele calls (
), and finally by restriction to only the “best” allele calls (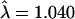 ). Genomic control methods can then be used to assess the significance of these remaining findings.
). Genomic control methods can then be used to assess the significance of these remaining findings.
Informatics Support
Informatics support will be essential for successful management, analysis, visualization, interpretation, and integration of genomewide data. Jonathan Buckley addressed these issues and described a platform that is currently under development. Genetrix will provide flexible tools for SNP selection, based on such criteria as data quality (Hardy-Weinberg equilibrium, call percentage, and allele frequency), location (relative to each other or relative to genes), allele frequency, functional effects of polymorphisms, known disease associations, LD patterns or FST values, and, for case selection, on such factors as data quality, disease severity or subtype, and demographic features. The vast amount of data also presents a challenge for visualization of results in a manner that allows the investigator to organize and highlight key items, while being able to “drill down” on the details of items of particular interest. Perhaps the biggest challenges are interpretation and integration with other biological knowledge including expression, proteomics, methylation, microRNA, and splice variation data, as well as the wealth of information available from Web resources.
Sample Size Requirements
A general sense of the sample size requirements that would be needed for a single-stage association study is provided in table 1 (data presented by David Clayton). At a marker density on the order of 1 every 6 kb (500,000 markers), we expect that most associations would be detected indirectly, by LD, rather than directly, with causal variants. Under the assumption that an average of 8 tag SNPs would yield an r2 of 0.8 and by use of an 8-df test, the sample sizes required for such indirect associations would be slightly less than double those needed for a direct association. These calculations assume a single-stage design and would need to be modified to incorporate a multistage design. Table 2 (data provided by Daniel Stram) provides illustrative calculations for a two-stage design, demonstrating that the vast majority of the costs are incurred at stage one.
Table A1.
Sample-Size Requirements for Case-Control Studies of a Multiplicative Effect[Note]
|
Sample Size Required for Study Type |
||||||
| Direct (1-df Test) with α of |
Indirect (8-df Test) with α of |
|||||
| P and R | 10−4 | 10−5 | 10−6 | 10−4 | 10−5 | 10−6 |
| P=.5 | ||||||
| 1.5 | 670 | 810 | 950 | 1,250 | 1,460 | 1,670 |
| 1.3 | 1,570 | 1,910 | 2,240 | 2,940 | 3,440 | 3,920 |
| P=.25 | ||||||
| 1.5 | 800 | 970 | 1,140 | 1,500 | 1,760 | 2,000 |
| 1.3 | 1,960 | 2,380 | 2,790 | 3,670 | 4,290 | 4,890 |
| P=.01 | ||||||
| 1.5 | 1,560 | 1,890 | 2,220 | 2,920 | 3,420 | 3,890 |
| 1.3 | 3,910 | 4,750 | 5,580 | 7,330 | 8,560 | 9,760 |
| P=.05 | ||||||
| 1.5 | 2,880 | 3,500 | 4,110 | 5,410 | 6,320 | 7,200 |
| 1.3 | 7,290 | 8,860 | 10,410 | 13,680 | 15,990 | 18,224 |
Note.— Shown is the number of cases and controls required for 90% power, given causal allele frequency P and relative risk per copy of causal allele R (under the assumptions that D′=1 and that there are 8 tag SNPs for r2=0.8 with the causal polymorphism).
Table A2.
Illustrative Calculations for a Two-Stage Optimal Design[Note]
| OR and Allele Frequency | N1 (Cases + Controls) | N2 (Cases + Controls) | Total No. of Cases | α1 | β1 | Cost of Stage I | Total Cost |
| 1.3: | |||||||
| 5% | 16,179 | 17,159 | 16,700 | .0015 | .96 | $16,179,000 | $18,431,000 |
| 10% | 8,745 | 9,327 | 9,036 | .0015 | .96 | $8,745,000 | $9,696,000 |
| 20% | 5,156 | 5,557 | 5,356 | .0015 | .96 | $5,156,000 | $5,885,000 |
| 1.5: | |||||||
| 5% | 6,085 | 6,300 | 6,192 | .0015 | .96 | $6,085,000 | $6,912,000 |
| 10% | 3,338 | 3,489 | 3,413 | .0015 | .96 | $3,337,000 | $3,796,000 |
| 20% | 2,024 | 2,165 | 2,095 | .0015 | .96 | $2,024,000 | $2,166,000 |
| 2.0: | |||||||
| 5% | 1,682 | 1,682 | 1,682 | .0015 | .96 | $1,682,000 | $1,903,000 |
| 10% | 954 | 963 | 959 | .0015 | .96 | $954,000 | $1,081,000 |
| 20% | 616 | 640 | 628 | .0015 | .96 | $616,000 | $700,000 |
Note.— Illustrative calculations for a two-stage optimal design required to yield a power of 90% at a genomewide significance level of 0.05 (SNPs specific overall significance level of 5×10-8), under the following assumptions: costs per genotype of $0.002 at stage I and $0.035 at stage II, four additional markers tested at stage II for each marker identified at stage I, maximum pairwise r2=0.6 at stage I and multivariate r2=0.9 at stage II, and a 1:1 case:control ratio.
Supplemental References
- Akey JM, Eberle MA, Rieder MJ, Carlson CS, Shriver MD, Nickerson DA, Kruglyak L (2004) Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol 2:e286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou AC, Easton DF (2003) Polygenic inheritance of breast cancer: implications for design of association studies. Genet Epidemiol 25:190–202 [DOI] [PubMed] [Google Scholar]
- Bortolini MC, Salzano FM, Thomas MG, Stuart S, Nasanen SP, Bau CH, Hutz MH, Layrisse Z, Petzl-Erler ML, Tsuneto LT, Hill K, Hurtado AM, Castro-de-Guerra D, Torres MM, Groot H, Michalski R, Nymadawa P, Bedoya G, Bradman N, Labuda D, Ruiz-Linares A (2003) Y-chromosome evidence for differing ancient demographic histories in the Americas. Am J Hum Genet 73:524–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Rieder MJ, Smith JD, Kruglyak L, Nickerson DA (2003) Additional SNPs and linkage-disequilibrium analyses are necessary for whole-genome association studies in humans. Nat Genet 33:518–521 [DOI] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA (2004) Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet 74:106–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho-Silva DR, Santos FR, Hutz MH, Salzano FM, Pena SD (1999) Divergent human Y-chromosome microsatellite evolution rates. J Mol Evol 49:204–214 [DOI] [PubMed] [Google Scholar]
- Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G, Sementchenko V, Piccolboni A, Bekiranov S, Bailey DK, Ganesh M, Ghosh S, Bell I, Gerhard DS, Gingeras TR (2005) Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308:1149–1154 [DOI] [PubMed] [Google Scholar]
- Cifuentes L, Morales R, Sepulveda D, Jorquera H, Acuna M (2004) DYS19 and DYS199 loci in a Chilean population of mixed ancestry. Am J Phys Anthropol 125:85–89 [DOI] [PubMed] [Google Scholar]
- Edwards AO, Ritter I, Robert, Abel KJ, Manning A, Panhuysen C, Farrer LA (2005) Complement factor H polymorphism and age-related macular degeneration. Science 308:421–424 [DOI] [PubMed] [Google Scholar]
- Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, Hansen M, et al (2003) Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol 68:69–78 [DOI] [PubMed] [Google Scholar]
- Goldstein DB, Ahmadi KR, Weale ME, Wood NW (2003) Genome scans and candidate gene approaches in the study of common diseases and variable drug responses. Trends Genet 19:615–622 [DOI] [PubMed] [Google Scholar]
- Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS (2005) A genome-wide scalable SNP genotyping assay using microarray technology. Nat Genet 37:549–554 [DOI] [PubMed] [Google Scholar]
- Haile R, Siegmund K, Gauderman W, Thomas D (1999) Study design issues in the development of the University of Southern California consortium’s colorectal cancer registry. Monogr Natl Cancer Inst 26:89–93 [DOI] [PubMed] [Google Scholar]
- Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA (2005) Complement factor H variant increases the risk of age-related macular degeneration. Science 308:419–421 [DOI] [PubMed] [Google Scholar]
- Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, Ballinger DG, Frazer KA, Cox DR (2005) Whole-genome patterns of common DNA variation in three human populations. Science 307:1072–1079 [DOI] [PubMed] [Google Scholar]
- John EM, Hopper JL, Beck JC, Knight JA, Neuhausen SL, Senie RT, Ziogas A, Andrulis IL, Anton-Culver H, Boyd N, Buys SS, Daly MB, O’Malley FP, Santella RM, Southey MC, Venne VL, Venter DJ, West DW, Whittemore AS, Seminara D (2004) The Breast Cancer Family Registry: an infrastructure for cooperative multinational, interdisciplinary and translational studies of the genetic epidemiology of breast cancer. Breast Cancer Res 6:R375–R389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson R, Salisbury B, Schneider J, Windemuth A, Stephens JC (2002) How many SNPs does a genome-wide haplotype map require? Pharmacogenomics 3:379–391 [DOI] [PubMed] [Google Scholar]
- Ke X, Hunt S, Tapper W, Lawrence R, Stavrides G, Ghori J, Whittaker P, Collins A, Morris AP, Bentley D, Cardon LR, Deloukas P (2004) The impact of SNP density on fine-scale patterns of linkage disequilibrium. Hum Mol Genet 13:577–588 [DOI] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai J-Y, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308:385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolonel LN, Altshuler D, Henderson BE (2004) The multiethnic cohort study: exploring genes, lifestyle and cancer risk. Nat Rev Cancer 4:519–527 [DOI] [PubMed] [Google Scholar]
- Kruglyak L (1999) Prospects for whole-genome linkage disequilibrium mapping. Nat Genet 22:139–144 [DOI] [PubMed] [Google Scholar]
- Lin S, Chakravarti A, Cutler DJ (2004) Exhaustive allelic transmission disequilibrium tests as a new approach to genome-wide association studies. Nat Genet 36:1181–1188 [DOI] [PubMed] [Google Scholar]
- Markides KS, Coreil J (1986) The health of Hispanics in the southwestern United States: an epidemiologic paradox. Public Health Rep 101:253–265 [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki H, Dong S, Loi H, Di X, Liu G, Hubbell E, Law J, Berntsen T, Chadha M, Hui H, Yang G, Kennedy GC, Webster TA, Cawley S, Walsh PS, Jones KW, Fodor SP, Mei R (2004a) Genotyping over 100,000 SNPs on a pair of oligonucleotide arrays. Nat Methods 1:109–111 [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Loi H, Dong S, Tsai YY, Fang J, Law J, Di X, Liu WM, Yang G, Liu G, Huang J, Kennedy GC, Ryder TB, Marcus GA, Walsh PS, Shriver MD, Puck JM, Jones KW, Mei R (2004b) Parallel genotyping of over 10,000 SNPs using a one-primer assay on a high-density oligonucleotide array. Genome Res 14:414–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeigue PM, Carpenter JR, Parra EJ, Shriver MD (2000) Estimation of admixture and detection of linkage in admixed populations by a Bayesian approach: application to African-American populations. Ann Hum Genet 64:171–186 [DOI] [PubMed] [Google Scholar]
- Newcomb P, Haile R, Anton-Culver H, Gallinger S, Hopper J, Jass J, Le Marchand L, Lindor N, Potter J, Seminara D (2002) The Colorectal Cancer Family Registry: 1998–2002 (Abstract D205). Cancer Epidemiol Biomarkers Prev 11:1222s [DOI] [PubMed] [Google Scholar]
- O’Brien K, Cokkinides V, Jemal A, Cardinez CJ, Murray T, Samuels A, Ward E, Thun MJ (2003) Cancer statistics for Hispanics, 2003. CA Cancer J Clin 53:208–226 [DOI] [PubMed] [Google Scholar]
- Ozaki K, Inoue K, Sato H, Iida A, Ohnishi Y, Sekine A, Odashiro K, Nobuyoshi M, Hori M, Nakamura Y, Tanaka T (2004) Functional variation in LGALS2 confers risk of myocardial infarction and regulates lymphotoxin-alpha secretion in vitro. Nature 429:72–75 [DOI] [PubMed] [Google Scholar]
- Ozaki K, Ohnishi Y, Iida A, Sekine A, Yamada R, Tsunoda T, Sato H, Hori M, Nakamura Y, Tanaka T (2002) Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat Genet 32:650–654 [DOI] [PubMed] [Google Scholar]
- Parra EJ, Kittles RA, Argyropoulos G, Pfaff CL, Hiester K, Bonilla C, Sylvester N, Parrish-Gause D, Garvey WT, Jin L, McKeigue PM, Kamboh MI, Ferrell RE, Pollitzer WS, Shriver MD (2001) Ancestral proportions and admixture dynamics in geographically defined African Americans living in South Carolina. Am J Phys Anthropol 114:18–29 [DOI] [PubMed] [Google Scholar]
- Parra EJ, Marcini A, Akey J, Martinson J, Batzer MA, Cooper R, Forrester T, Allison DB, Deka R, Ferrell RE, Shriver MD (1998) Estimating African American admixture proportions by use of population-specific alleles. Am J Hum Genet 63:1839–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Hattangadi N, Lane B, Lohmueller KE, Hafler DA, Oksenberg JR, Hauser SL, Smith MW, O’Brien SJ, Altshuler D, Daly MJ, Reich D (2004) Methods for high-density admixture mapping of disease genes. Am J Hum Genet 74:979–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff CL, Kittles RA, Shriver MD (2002) Adjusting for population structure in admixed populations. Genet Epidemiol 22:196–201 [DOI] [PubMed] [Google Scholar]
- Pfaff CL, Parra EJ, Bonilla C, Hiester K, McKeigue PM, Kamboh MI, Hutchinson RG, Ferrell RE, Boerwinkle E, Shriver MD (2001) Population structure in admixed populations: effect of admixture dynamics on the pattern of linkage disequilibrium. Am J Hum Genet 68:198–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Delfin LA, Rubin-de-Celis VE, Zago MA (2001) Genetic diversity in an Andean population from Peru and regional migration patterns of Amerindians in South America: data from Y chromosome and mitochondrial DNA. Hum Hered 51:97–106 [DOI] [PubMed] [Google Scholar]
- Salzano FM, Bortolini MC (2001) The evolution and genetics of Latin American populations. Cambridge University Press, Cambridge [Google Scholar]
- Sans M (2000) Admixture studies in Latin America: from the 20th to the 21st century. Hum Biol 72:155–177 [PubMed] [Google Scholar]
- Tarazona-Santos E, Carvalho-Silva DR, Pettener D, Luiselli D, De Stefano GF, Labarga CM, Rickards O, Tyler-Smith C, Pena SD, Santos FR (2001) Genetic differentiation in South Amerindians is related to environmental and cultural diversity: evidence from the Y chromosome. Am J Hum Genet 68:1485–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson D, Witte JS, Slattery M, Goldgar D (2004) Increased power for case-control studies of single nucleotide polymorphisms through incorporation of family history and genetic constraints. Genet Epidemiol 27:215–224 [DOI] [PubMed] [Google Scholar]
- Wang WY, Todd JA (2003) The usefulness of different density SNP maps for disease association studies of common variants. Hum Mol Genet 12:3145–3149 [DOI] [PubMed] [Google Scholar]
Web Resources
The URLs for data presented herein are as follows:
- Affymetrix, http://www.affymetrix.com/index.affx
- Epicenter Software, http://www.epicentersoftware.com/genetrix.php (for Genetrix)
- Illumina, http://www.illumina.com
- ParAllele BioScience, http://www.parallelebio.com (for 100K panel)
- Sequenom, http://www.sequenom.com (for 75K and 100K panels)
References
- Bansal A, van den Boom D, Kammerer S, Honisch C, Adam G, Cantor CR, Kleyn P, Braun A (2002) Association testing by DNA pooling: an effective initial screen. Proc Natl Acad Sci USA 99:16871–16874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barratt BJ, Payne F, Rance HE, Nutland S, Todd JA, Clayton DG (2002) Identification of the sources of error in allele frequency estimations from pooled DNA indicates an optimal experimental design. Ann Hum Genet 66:393–405 [DOI] [PubMed] [Google Scholar]
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D (2004) Ultraconserved elements in the human genome. Science 304:1321–1325 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300 [Google Scholar]
- Boehnke M (1994) Limits of resolution of genetic linkage studies: implications for the positional cloning of human disease genes. Am J Hum Genet 55:379–390 [PMC free article] [PubMed] [Google Scholar]
- Botstein D, Risch N (2003) Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet 33:228–237 [DOI] [PubMed] [Google Scholar]
- Cardon LR, Bell JI (2001) Association study designs for complex diseases. Nat Rev Genet 2:91–99 [DOI] [PubMed] [Google Scholar]
- Cardon LR, Palmer LJ (2003) Population stratification and spurious allelic association. Lancet 361:598–604 [DOI] [PubMed] [Google Scholar]
- Cargill M, Daley GQ (2000) Mining for SNPs: putting the common variants—common disease hypothesis to the test. Pharmacogenomics 1:27–37 [DOI] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Kruglyak L, Nickerson DA (2004) Mapping complex disease loci in whole-genome association studies. Nature 429:446–452 [DOI] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Rieder MJ, Smith JD, Kruglyak L, Nickerson DA (2003) Additional SNPs and linkage-disequilibrium analyses are necessary for whole-genome association studies in humans. Nat Genet 33:518–521 [DOI] [PubMed] [Google Scholar]
- Clayton DG, McKeigue PM (2001) Epidemiological methods for studying genes and environmental factors in complex diseases. Lancet 358:1357–1360 [DOI] [PubMed] [Google Scholar]
- Collins FS, Guyer MS, Charkravarti A (1997) Variations on a theme: cataloging human DNA sequence variation. Science 278:1580–1581 [DOI] [PubMed] [Google Scholar]
- Devlin B, Roeder K, Wasserman L (2001) Genomic control, a new approach to genetic-based association studies. Theor Pop Biol 60:155–160 [DOI] [PubMed] [Google Scholar]
- Efron B, Tibshirani R (2002) Empirical Bayes methods and false discovery rates for microarrays. Genet Epidemiol 23:70–86 [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2004) The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 306:636–640 [DOI] [PubMed] [Google Scholar]
- Fearnhead NS, Wilding JL, Winney B, Tonks S, Bartlett S, Bicknell DC, Tomlinson IP, Mortensen NJ, Bodmer WF (2004) Multiple rare variants in different genes account for multifactorial inherited susceptibility to colorectal adenomas. Proc Natl Acad Sci USA 101:15992–15997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman ML, Reich D, Penney KL, McDonald GJ, Mignault AA, Patterson N, Gabriel SB, Topol EJ, Smoller JW, Pato CN, Pato MT, Petryshen TL, Kolonel LN, Lander ES, Sklar P, Henderson B, Hirschhorn JN, Altshuler D (2004) Assessing the impact of population stratification on genetic association studies. Nat Genet 36:388–393 [DOI] [PubMed] [Google Scholar]
- Gibbs RA, Belmont JW, Hardenbol P, Willis TD, Yu F, Yang H, Ch’ang LY, et al (2003) The International HapMap Project. Nature 426:789–796 [DOI] [PubMed] [Google Scholar]
- Gordon D, Heath SC, Liu X, Ott J (2001) A transmission/disequilibrium test that allows for genotyping errors in the analysis of single-nucleotide polymorphism data. Am J Hum Genet 69:371–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Yang Y, Haynes C, Finch SJ, Mendell NR, Brown AM, Haroutunian V (2004) Increasing power for tests of genetic association in the presence of phenotype and/or genotype error by use of double-sampling. Stat Appl Genet Mol Biol 3:1–35 [DOI] [PubMed] [Google Scholar]
- Hao K, Wang X (2004) Incorporating individual error rate into association test of unmatched case-control design. Hum Hered 58:154–163 [DOI] [PubMed] [Google Scholar]
- Hinds DA, Stuve LL, Nilsen GB, Halperin E, Eskin E, Ballinger DG, Frazer KA, Cox DR (2005) Whole-genome patterns of common DNA variation in three human populations. Science 307:1072–1079 [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN, Daly MJ (2005) Genome-wide association studies for common disease and complex traits. Nat Rev Genet 6:95–108 [DOI] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C (2004) Detection of large-scale variation in the human genome. Nat Genet 36:949–951 [DOI] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai J-Y, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308:385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langholz B, Rothman N, Wacholder S, Thomas D (1999) Cohort studies for characterizing measured genes. Monogr Natl Cancer Inst 26:39–42 [DOI] [PubMed] [Google Scholar]
- Leutenegger AL, Prum B, Genin E, Verny C, Lemainque A, Clerget-Darpoux F, Thompson EA (2003) Estimation of the inbreeding coefficient through use of genomic data. Am J Hum Genet 73:516–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Altman RB (2004) Finding haplotype tagging SNPs by use of principal components analysis. Am J Hum Genet 75:850–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Chakravarti A, Cutler DJ (2004) Exhaustive allelic transmission disequilibrium tests as a new approach to genome-wide association studies. Nat Genet 36:1181–1188 [DOI] [PubMed] [Google Scholar]
- Livingston RJ, von Niederhausern A, Jegga AG, Crawford DC, Carlson CS, Rieder MJ, Gowrisankar S, Aronow BJ, Weiss RB, Nickerson DA (2004) Pattern of sequence variation across 213 environmental response genes. Genome Res 14:1821–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, Pearce CL, Pike MC, Lander ES, Hirschhorn JN (2003) Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 33:177–182 [DOI] [PubMed] [Google Scholar]
- Lowe CE, Cooper JD, Chapman JM, Barratt BJ, Twells RC, Green EA, Savage DA, Guja C, Ionescu-Tirgoviste C, Tuomilehto-Wolf E, Tuomilehto J, Todd JA, Clayton DG (2004) Cost-effective analysis of candidate genes using htSNPs: a staged approach. Genes Immun 5:301–305 [DOI] [PubMed] [Google Scholar]
- Mack TM, Cozen W, Shibata DK, Weiss LM, Nathwani BN, Hernandez AM, Taylor CR, Hamilton AS, Deapen DM, Rappaport EB (1995) Concordance for Hodgkin’s disease in identical twins suggesting genetic susceptibility to the young-adult form of the disease. N Engl J Med 332:413–418 [DOI] [PubMed] [Google Scholar]
- Marchini J, Donnelly P, Cardon LR (2005) Genome-wide strategies for detecting multiple loci that influence complex diseases. Nat Genet 37:413–417 [DOI] [PubMed] [Google Scholar]
- Mitchell AA, Cutler DJ, Chakravarti A (2003) Undetected genotyping errors cause apparent overtransmission of common alleles in the transmission/disequilibrium test. Am J Hum Genet 72:598–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS, Cheung VG (2004) Genetic analysis of genome-wide variation in human gene expression. Nature 430:743–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller JC, Lohmussaar E, Magi R, Remm M, Bettecken T, Lichtner P, Biskup S, Illig T, Pfeufer A, Luedemann J, Schreiber S, Pramstaller P, Pichler I, Romeo G, Gaddi A, Testa A, Wichmann HE, Metspalu A, Meitinger T (2005) Linkage disequilibrium patterns and tagSNP transferability among European populations. Am J Hum Genet 76:387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Sham PC (2004) The future of association studies: gene-based analysis and replication. Am J Hum Genet 75:353–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton MA, Gould MN, Reznikoff CA, Haag JD (1998) On the statistical analysis of allelic-loss data. Stat Med 17:1425–1445 [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31:3812–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M, Taveré S (2002) Linkage disequilibrium: what history has to tell us. Trends Genet 18:83–90 [DOI] [PubMed] [Google Scholar]
- Nyholt DR (2004) A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet 74:765–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki K, Ohnishi Y, Iida A, Sekine A, Yamada R, Tsunoda T, Sato H, Hori M, Nakamura Y, Tanaka T (2002) Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat Genet 32:650–654 [DOI] [PubMed] [Google Scholar]
- Page GP, George V, Go RC, Page PZ, Allison DB (2003) “Are we there yet?”: deciding when one has demonstrated specific genetic causation in complex diseases and quantitative traits. Am J Hum Genet 73:711–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LJ, Cardon LR. Shaking the tree: mapping complex disease genes using linkage disequilibrium. Lancet (in press) [DOI] [PubMed] [Google Scholar]
- Pfeiffer RM, Rutter JL, Gail MH, Struewing J, Gastwirth JL (2002) Efficiency of DNA pooling to estimate joint allele frequencies and measure linkage disequilibrium. Genet Epidemiol 22:94–102 [DOI] [PubMed] [Google Scholar]
- Pritchard JK (2001) Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet 69:124–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Cox NJ (2002) The allelic architecture of human disease genes: common disease-common variant…or not? Hum Mol Genet 11:2417–2423 [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67:170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Lander ES (2001) On the allelic spectrum of human disease. Trends Genet 17:502–510 [DOI] [PubMed] [Google Scholar]
- Rice KM, Holmans P (2003) Allowing for genotyping error in analysis of unmatched case-control studies. Ann Hum Genet 67:165–174 [DOI] [PubMed] [Google Scholar]
- Risch N (1990) Linkage strategies for genetically complex traits. I. Multilocus models. Am J Hum Genet 46:222–228 [PMC free article] [PubMed] [Google Scholar]
- Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1616–1617 [DOI] [PubMed] [Google Scholar]
- Sabatti C, Service S, Freimer N (2003) False discovery rate in linkage and association genome screens for complex disorders. Genetics 164:829–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satagopan JM, Elston RC (2003) Optimal two-stage genotyping in population-based association studies. Genet Epidemiol 25:149–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satagopan JM, Venkatraman ES, Begg CB (2004) Two-stage designs for gene-disease association studies with sample size constraints. Biometrics 60:589–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satagopan JM, Verbel DA, Venkatraman ES, Offit KE, Begg CB (2002) Two-stage designs for gene-disease association studies. Biometrics 58:163–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA (2002) Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet 70:425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M (2004) Large-scale copy number polymorphism in the human genome. Science 305:525–528 [DOI] [PubMed] [Google Scholar]
- Sham P, Bader JS, Craig I, O’Donovan M, Owen M (2002) DNA pooling: a tool for large-scale association studies. Nat Rev Genet 3:862–871 [DOI] [PubMed] [Google Scholar]
- Sidow A (2002) Sequence first. Ask questions later. Cell 111:13–16 [DOI] [PubMed] [Google Scholar]
- Sobell JL, Heston LL, Sommer SS (1993) Novel association approach for determining the genetic predisposition to schizophrenia: case-control resource and testing of a candidate gene. Am J Med Genet 48:28–35 [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100:9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stram DO, Pearce CL, Bretsky P, Freedman M, Hirschhorn JN, Altshuler D, Kolonel LN, Henderson BE, Thomas DC (2003) Modeling and E-M estimation of haplotype-specific relative risks from genotype data for a case-control study of unrelated individuals. Hum Hered 55:179–190 [DOI] [PubMed] [Google Scholar]
- Tabor HK, Risch NJ, Myers RM (2002) Opinion: candidate-gene approaches for studying complex genetic traits: practical considerations. Nat Rev Genet 3:391–397 [DOI] [PubMed] [Google Scholar]
- Tang H, Quertermous T, Rodriguez B, Kardia SL, Zhu X, Brown A, Pankow JS, Province MA, Hunt SC, Boerwinkle E, Schork NJ, Risch NJ (2004) Genetic structure, self-identified race/ethnicity, and confounding in case-control association studies. Am J Hum Genet 76:268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor NE, Greene EA (2003) PARSESNP: A tool for the analysis of nucleotide polymorphisms. Nucleic Acids Res 31:3808–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DC, Witte JS (2002) Point: Population stratification: A problem for case-control studies of candidate gene associations? Cancer Epidemiol Biomarkers Prev 11:505–512 [PubMed] [Google Scholar]
- Thomas DC, Xie R, Gebregziabher M (2004) Two-stage sampling designs for gene association studies. Genet Epidemiol 27:401–414 [DOI] [PubMed] [Google Scholar]
- Tzeng JY, Devlin B, Wasserman L, Roeder K (2003) On the identification of disease mutations by the analysis of haplotype similarity and goodness of fit. Am J Hum Genet 72:891–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Oord EJ, Sullivan PF (2003) A framework for controlling false discovery rates and minimizing the amount of genotyping in the search for disease mutations. Hum Hered 56:188–199 [DOI] [PubMed] [Google Scholar]
- Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N (2004) Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst 96:434–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacholder S, Rothman N, Caporaso N (2000) Population stratification in epidemiologic studies of common genetic variants and cancer: quantification of bias. J Natl Cancer Inst 92:1151–8 [DOI] [PubMed] [Google Scholar]
- Wacholder S, Rothman N, Caporaso N (2002) Counterpoint: Bias from population stratification is not a major threat to the validity of conclusions from epidemiologic studies of common polymorphisms and cancer. Cancer Epidemiol Biomarkers Prev 11:513–520 [PubMed] [Google Scholar]
- Wang WY, Cordell HJ, Todd JA (2003) Association mapping of complex diseases in linked regions: estimation of genetic effects and feasibility of testing rare variants. Genet Epidemiol 24:36–43 [DOI] [PubMed] [Google Scholar]
- Wang WYS, Barratt BJ, Clayton DG, Todd JA (2005) Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet 6:109–118 [DOI] [PubMed] [Google Scholar]
- Wijsman EM, Rosenthal EA, Hall D, Blundell ML, Sobin C, Heath SC, Williams R, Brownstein MJ, Gogos JA, Karayiorgou M (2003) Genome-wide scan in a large complex pedigree with predominantly male schizophrenics from the island of Kosrae: evidence for linkage to chromosome 2q. Mol Psychiatry 8:695–705, 643 [DOI] [PubMed] [Google Scholar]
- Witte JS, Gauderman WJ, Thomas DC (1999) Asymptotic bias and efficiency in case-control studies of candidate genes and gene-environment interactions: Basic family designs. Am J Epidemiol 148:693–705 [DOI] [PubMed] [Google Scholar]
- Xi T, Jones IM, Mohrenweiser HW (2004) Many amino acid substitution variants identified in DNA repair genes during human population screenings are predicted to impact protein function. Genomics 83:970–979 [DOI] [PubMed] [Google Scholar]
- Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG (2002) Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered 53:79–91 [DOI] [PubMed] [Google Scholar]
- Zhu Y, Spitz MR, Amos CI, Lin J, Schabath MB, Wu X (2004) An evolutionary perspective on single-nucleotide polymorphism screening in molecular cancer epidemiology. Cancer Res 64:2251–2257 [DOI] [PubMed] [Google Scholar]