

Abstract
Chronic lymphocytic leukemia (CLL) and other B-cell lymphoproliferative disorders (LPDs) show clear evidence of familial aggregation, but the inherited basis is largely unknown. To identify a susceptibility gene for CLL, we conducted a genomewide linkage analysis of 115 pedigrees, using a high-density single-nucleotide polymorphism (SNP) array containing 11,560 markers. Multipoint linkage analyses were undertaken using both nonparametric (model-free) and parametric (model-based) methods. Our results confirm that the presence of high linkage disequilibrium (LD) between SNP markers can lead to inflated nonparametric linkage (NPL) and LOD scores. After the removal of high-LD SNPs, we obtained a maximum NPL of 3.14 (P=.0008) on chromosome 11p11. The same genomic position also yielded the highest multipoint heterogeneity LOD (HLOD) score under both dominant (HLOD 1.95) and recessive (HLOD 2.78) models. In addition, four other chromosomal positions (5q22-23, 6p22, 10q25, and 14q32) displayed HLOD scores >1.15 (which corresponds to a nominal P value <.01). None of the regions coincided with areas of common chromosomal abnormalities frequently observed for CLL. These findings strengthen the argument for an inherited predisposition to CLL and related B-cell LPDs.
Introduction
Leukemia affects 1%–2% of the population of Western countries (Ries et al. 2003). B-cell chronic lymphocytic leukemia (CLL [MIM 151400]) is the most common form of leukemia (∼30% of all cases) (Ries et al. 2003). Family and epidemiological studies have now provided strong support for the existence of inherited susceptibility to CLL (reviewed by Sellick et al. [2004a]) and other B-cell lymphoproliferative disorders (LPDs), such as non-Hodgkin lymphoma (NHL [MIM 605027]) and Hodgkin lymphoma (HL [MIM 236000]). In the literature, >50 families have been described that show distinct clustering of CLL, and many striking multiple-case families provide very strong evidence of an increased familial risk (reviewed by Houlston et al. [2003]). Although no formal modeling of the inheritance of CLL-LPD in families has been undertaken, a number of large pedigrees described in the literature are compatible with the existence of dominantly acting alleles with pleiotropic effects (Schweitzer et al. 1973; Gunz et al. 1975). Case-control and cohort studies that have systematically estimated the familial risk of CLL and other LPDs (Cartwright et al. 1987; Linet et al. 1989; Pottern et al. 1991; Radovanovic et al. 1994; Goldin et al. 2004) have shown that all B-cell LPDs display site-specific elevated familial risks—particularly for CLL, for which risks are increased sevenfold in first-degree relatives of patients (Goldin et al. 2004). Furthermore, these studies have also demonstrated that familial associations exist between the different types of B-cell LPDs, with risks of NHL and HL showing twofold increases in relatives of patients with CLL (Goldin et al. 2004).
In 1997, we formed an International CLL Linkage Consortium (ICLLC) to collect biological samples and data from families with CLL and other B-cell LPDs, to identify predisposition genes for these diseases. Here, we report the results of a genomewide linkage search of 115 families segregating CLL with or without additional B-cell LPD cases ascertained through the consortium. Since increased genetic-marker density in whole-genome scans has been shown to increase the linkage information content (IC) (Schaid et al. 2004), we conducted our genomewide scan using the recently introduced Affymetrix Mapping 10Kv1 array, which contains ∼11,000 SNP markers.
Methods
Ascertainment and Collection of Families
Through hematologists in the United Kingdom, Norway, Israel, Italy, Ireland, Germany, Portugal, The Netherlands, and Australia who participate in the ICLLC, 115 families with two or more affected individuals with B-cell CLL, with or without the segregation of additional B-cell LPD cases (B-cell NHL or HL), were ascertained. The diagnoses of B-cell CLL and other B-cell LPDs in affected family members were established, in all cases, using accepted standard clinicopathological and immunological criteria that are in accordance with current World Health Organization classification guidelines (Müller-Hermelink et al. 2001). Samples were obtained with informed consent and local ethical review board approval, in accordance with the tenets of the Declaration of Helsinki. DNA was salt extracted from patient samples by use of a standard sucrose lysis method and was quantified using PicoGreen reagents (Invitrogen).
Genotyping
A genomewide linkage search of the 115 families was undertaken using the GeneChip Mapping 10Kv1 Xba Array containing 11,560 SNP markers (Affymetrix). SNP genotypes were obtained by following the Affymetrix protocol for the GeneChip Mapping 10K Xba Array (Matsuzaki et al. 2004). In brief, 250 ng of genomic DNA isolated from peripheral blood was digested per sample, with the restriction endonuclease XbaI, for 2.5 h. Digested DNA was mixed with Xba adapters and was ligated, using T4 DNA ligase, for 2.5 h. Ligated DNA was added to four separate PCR reactions and was cycled, pooled, and purified to remove unincorporated ddNTPs. The purified PCR products were then fragmented and labeled with biotin-ddATP. Biotin-labeled DNA fragments were hybridized to the arrays for 18 h in an Affymetrix 640 hybridization oven. After hybridization, arrays were washed, stained, and scanned using an Affymetrix Fluidics Station FS450 with images obtained using an Affymetrix GeneChip 3000 scanner. Affymetrix GCOS software (v1.2) was used to obtain raw microarray feature intensities (RAS scores). RAS scores were processed using Affymetrix GDAS (v3.0.2) software to derive SNP genotypes.
Data Manipulation and Error Checking
Non-Mendelian error checking of genotypes and the generation of linkage format files from raw Affymetrix array files (with the suffix “.chp”) were performed using the program ProgenyLab (Progeny). The map order and distances between SNP markers were based on the University of California–Santa Cruz Human Genome browser (May 2004 release). The program MERLIN was employed to search for additional unlikely genotypes that are consistent with potential genotyping errors (Abecasis et al. 2002).
Investigation of Linkage Disequilibrium
Currently available linkage software for multipoint analyses assumes that all markers are in linkage equilibrium. However, for closely spaced SNP markers, this assumption may not always be correct. To identify markers in high linkage disequilibrium (LD), we calculated the pairwise LD measures r2 and D′ between consecutive pairs of SNP markers. We simplified our approach by ignoring relationships among family members and by using the expectation-maximization algorithm to estimate two-locus haplotype frequencies, from which r2 and D′ were computed. Although ignoring relationships may result in a loss of efficiency, it should not significantly bias estimates. A pair of SNPs was defined as being in high LD if they had a pairwise LD measure of r2>0.4. LD was then removed by considering each set of markers in LD (defined as sets in which each consecutive marker pair in the set had r2>0.4) and retaining one SNP from each set (the centrally positioned SNP). The impact of LD was investigated by considering linkage results calculated before and after the removal of the high-LD SNPs.
Linkage Analysis
Multipoint linkage analysis was undertaken by implementation of the Perl script SNPLINK, which performs fully automated nonparametric (mode-of-inheritance free) and parametric analyses before and after LD removal, by incorporation of the MERLIN (v0.10.1) (Abecasis et al. 2002) and ALLEGRO (v1.1) (Gudbjartsson et al. 2000) programs, respectively (Webb et al. 2005). Parametric linkage in the presence of heterogeneity was assessed using heterogeneity LOD (HLOD) scores. HLOD scores and the accompanying estimates of the proportion of linked families (α) were calculated using the statistical software package ALLEGRO. These analyses require the specification of a disease-transmission model. We derived LOD scores under both dominant and recessive models of inheritance, with reduced penetrance and three age categories dependent on age at diagnosis (<50, 50–70, or >70 years). In the absence of a genetic model and parameters from segregation analysis of familial data, we chose values that were consistent with the population age-specific risks of CLL and compatible with the observed familial risks (Goldin et al. 2004). The lifetime risk (defined at age 80 years) of being diagnosed with CLL in the U.S. population, by use of the Surveillance Epidemiology and End Results (SEER) data, is estimated to be 0.37% (Ries et al. 2003). We assumed an allele frequency of 0.0005 under the dominant model and 0.05 under the recessive model. To satisfy the constraints of the lifetime risk and familial relative risks, for the dominant model, penetrance was assumed to be 2% for individuals aged <50 years, 17% for individuals aged 50–70 years, and 36% for those aged >70 years. For the recessive model, the penetrances were assumed to be 2%, 14%, and 30% for the three liability classes. To allow for phenocopies, the penetrances for the normal genotypes under both models were set to 0.006%, 0.2%, and 0.3%.
All unaffected individuals were considered uninformative (i.e., of unknown phenotype) for the analysis. HLOD scores follow a complex statistical distribution, which can be approximated by the maximum of two independently distributed χ21 variables. To obtain significance estimates for HLODs, these variables were first converted to a χ2, where 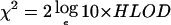 , and significance values (P1) were then derived, using the χ2 distribution with 1 df. The nominal P value for the HLOD score is then given by 0.5×[1-(1-P1)(1-P1)] (Faraway 1993).
, and significance values (P1) were then derived, using the χ2 distribution with 1 df. The nominal P value for the HLOD score is then given by 0.5×[1-(1-P1)(1-P1)] (Faraway 1993).
Multipoint nonparametric linkage (NPL) analyses were performed using the SALL statistic (Whittemore and Halpern 1994) generated by the program MERLIN. Results are reported in terms of an NPL statistic and its associated one-sided P value. Under the null hypothesis of no linkage, the NPL statistic is distributed asymptotically as a standard normal random variable. We also calculated empirical genomewide significance levels for the NPL statistics after markers in high LD were removed, using 10,000 gene-dropping simulations. In each simulation, we used MERLIN to simulate genotype data, using the original phenotypes, allele frequencies, marker spacing, and missing-data patterns. Empirical limits for genomewide significance were established at 3.59, with the suggestive linkage threshold at 2.80. The genomewide significance thresholds represent the NPL score that could be achieved with our data by chance under the null hypothesis of no linkage, at a frequency of 1 in 20 simulations, whereas the genomewide suggestive thresholds represent the values that could be obtained by chance once in every genomewide scan. MERLIN was also used to estimate IC for each chromosome provided by the marker set, by use of the entropy information described by Kruglyak et al. (1996).
Results
Description of Families Analyzed
Descriptions of the 115 families (98 with CLL; 17 with CLL/B-cell LPD) are summarized in table 1. In total, there were 261 individuals affected with CLL (International Classification of Diseases, Ninth Edition [ICD-9] of 204.1); 18 individuals affected with NHL (ICD-9 of 202), and two individuals affected with HL (ICD-9 of 201.5-9). The number of affected persons per family had a range of 2–12, and the number of affected persons per family with DNA available had a range of 2–4. There were 25 families with affected persons in two or more generations; one pedigree contained affected family members in three generations, and one pedigree contained affected individuals in four generations. All other families contained affected family members in only one generation.
Table 1.
Characteristics of the 115 Pedigrees with CLL or CLL/B-Cell LPDs[Note]
| No. of Affected Individualsper Pedigree | No. ofPedigreesa | Total No.Affectedb | Total No.DNA Typedb |
| 2 | 80 (11) | 160 (11) | 147 (10) |
| 3 | 28 (3) | 84 (5) | 57 (3) |
| 4 | 5 (2) | 20 (3) | 16 (2) |
| 5 | 1 (0) | 5 (0) | 3 (0) |
| 12 | 1 (1) |
12 (1) |
5 (1) |
| Total | 115 (17) | 281 (20) | 228 (16) |
Note.— All affected individuals had CLL, B-cell NHL, or HL.
The number of pedigrees with individuals affected with non-CLL LPDs is shown in parentheses.
The number of individuals affected with non-CLL LPDs is shown in parentheses.
In the families, the median age at diagnosis of CLL was 61 years, significantly less than the median value of 72 years for age at diagnosis observed in the general white population (Ries et al. 2003). Minimum age at diagnosis within a family is likely to be a superior indicator of the potential for existence of a susceptibility gene, since it is not influenced by older sporadic cases. In our families, the minimum age at diagnosis within the families had a range of 31–81 years, with a median value of 58 years.
Of the 115 pedigrees analyzed, 105 had successfully completed analyses (genotypes available for ⩾2 affected individuals informative for linkage). Ten pedigrees could not be analyzed completely because of poor DNA quality, which gave genotyping call rates of <85%, or because of insufficient amounts of starting material from a deceased individual. Of the 105 families used in the linkage analyses, 89 were documented as segregating CLL only, 15 as segregating both CLL and B-cell NHL, and 1 as segregating both CLL and HL. Twenty-four of the families had affected individuals in two or more generations.
Data Quality
A total of 228 Affymetrix Mapping 10Kv1 arrays were processed, which resulted in the generation of >2.45 million genotypes. We monitored a number of parameters throughout the study, to determine data quality, and all genotypes were housed within the pedigree-data-storage program ProgenyLab. The average SNP call rate per array was 92.2%. For DNA extracted from males, it was possible to examine the 309 markers on the X chromosome for errors due to miscalls or PCR contamination. No SNPs were heterozygous in male samples. Of the SNPs, 106 were excluded because their chromosomal position was unknown (Affymetrix NetAffx 10K_XBA131 [January 2005 release]), and 210 SNPs were not polymorphic in our pedigree set, which left 11,244 polymorphic markers with known map locations. After error checking with the programs ProgenyLab and MERLIN, a total of 517 and 2,826 genotypes, respectively, were deemed to be erroneous and were removed from further analysis.
Linkage Analysis
The maximum NPL statistic obtained using all 11,244 polymorphic SNPs was 3.73 (P=.0001) on chromosome 11p11. Under parametric analyses, an HLOD score of 2.66 for the dominant model and an HLOD score of 4.03 for the recessive model (which corresponds to genomewide significance at the 5% level) were achieved at the same chromosomal position. Chromosomes 5, 6, 10, 11, and 14 also attained NPL scores >2.0 and LOD scores >1.5, but these did not achieve the levels recommended to indicate genomewide suggestive linkage (Lander and Kruglyak 1995).
The presence of LD between markers has the potential to inflate multipoint linkage statistics if the vectors of inheritance have to be inferred on the basis of allele frequencies (Evans and Cardon 2004; Huang et al. 2004). This was of particular concern, because founders of many of the pedigrees were not available to genotype and a large proportion of the pedigrees were sib pairs. The two most commonly used measures of LD are D′ and r2, and the properties of both statistics have been discussed extensively elsewhere (Hedrick 1987; Devlin and Risch 1995). The behavior of these statistics is affected by a number of factors, which can bias the accuracy of LD estimation; in particular, D′ is more robust to small minor-allele frequencies, and r2 is more robust to small sample sizes. Thresholds of 0.4 and 0.7 for r2 and D′, respectively, have been advocated to define high-LD SNPs (John et al. 2004; Schaid et al. 2004). To identify high-LD SNPs, we computed pairwise LD measured by D′ and r2 between consecutive SNPs. Figure 1 shows the distributions of r2 and D′ according to the pairwise distance of SNPs within 2 Mb of each other, and figure 2 shows the distributions of r2 and D′ for all SNPs in our data set. On the basis of the distributions of these statistics, we chose to use the r2 statistic, with a threshold of 0.4, to identify high-LD SNPs. The majority of high-LD SNPs had r2 values of ∼1.0 and were within 0.3 Mb of each other (median distance 0.12 Mb; 25th–75th percentiles 0.02–0.32 Mb). Among the clusters of SNPs that had r2>0.4, only one SNP (the centrally positioned SNP) from each cluster was used in the linkage analyses. With use of these criteria, a total of 1,968 SNPs were excluded, which left a total of 9,276 SNPs.
Figure 1.

Distribution of pairwise D′ and r2 according to pairwise distance between SNP markers. The horizontal line shows the criterion used to define high-LD SNPs: r2>0.4.
Figure 2.

Distribution of pairwise D′ and r2 in the data set
NPL and LOD scores for analyses with and without the high-LD SNPs are shown in figure 3. The panels within figure 3 show that inclusion of high-LD SNPs in the analysis can lead to inflated linkage statistics; however, in most cases, the overall profile of the linkage statistics remains the same. The ICs for the analyses were virtually identical, with a genomewide IC mean of 0.64 when high-LD SNPs were included, compared with a genomewide IC mean of 0.63 when high-LD SNPs were excluded. Plots of the IC for each chromosome showed that exclusion of the high-LD SNPs had little impact on overall chromosomal IC (data not shown). Therefore, it seems reasonable to presume that any observable impact on the linkage statistics after the removal of the high-LD SNPs from our analyses is due to the effect of LD between markers rather than to decreased IC.
Figure 3.

NPL scores across each chromosome. In each plot, the dashed line shows NPL statistics obtained using all SNPs (n=11,244), whereas the solid line shows NPL statistics obtained after exclusion of high-LD SNPs (n=9,276).
The remaining 9,276 SNPs used in the linkage analyses after removal of the high-LD SNPs had a calculated median intermarker distance of 0.17 Mb (25th–75th percentile 0.06–0.38 Mb). Results of both parametric and NPL analyses are summarized in table 2. Multipoint NPL analysis of all 105 families resulted in a maximum NPL score of 3.14, with a corresponding nominal one-sided P value of .0008, at map position 11p11 (fig. 4 and table 2). This does not reach the empirical threshold for genomewide significance of 3.59, but it does attain the suggestive linkage threshold of 2.80. At the same position, an HLOD score of 1.85 (maximized with 41% of families linked) was achieved under the assumption of a dominant mode of inheritance, and an HLOD score of 2.78 (maximized with 32% of families linked) was achieved under the assumption of a recessive mode of inheritance. These results are therefore evidence of suggestive linkage at chromosome 11p11.
Table 2.
Results of Parametric and Nonparametric Analyses
|
Analysis Results by Method |
||||||
| Nonparametric |
Dominant Model |
Recessive Model |
||||
| ChromosomalRegiona | Maximum NPL | P | Maximum HLOD | αb | Maximum HLOD | αb |
| 5q22-23 | 2.01 | .022 | 1.02 | .29 | 1.35 | .20 |
| 6p22 | 2.25 | .012 | 1.05 | .29 | 1.49 | .22 |
| 10q25 | 2.12 | .017 | .94 | .28 | 1.28 | .22 |
| 11p11 | 3.14 | .0008 | 1.85 | .41 | 2.78 | .32 |
| 14q32 | 2.03 | .021 | 1.18 | .34 | .91 | .19 |
Location of maximum NPL scores >2.0 and maximum HLOD scores >1.15 (corresponding to a nominal P value <.01) after the removal of high-LD SNP markers from analyses.
The estimate of the proportion of families linked at a given genomic position.
Figure 4.

Plots of linkage statistics after the removal of high-LD SNPs for chromosomes 5, 6, 10, 11, and 14. HLOD scores under the dominant model are shown in black, HLOD scores under the recessive model are shown in red, and NPL P values transformed by 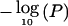 are shown in blue.
are shown in blue.
In addition, four other chromosomal positions (5q22-23, 6p22, 10q25, and 14q32) displayed HLOD scores >1.15 (corresponding to a nominal P value <.01). However, none of these regions achieved the levels required for genomewide suggestive evidence of linkage. Figure 4 shows multipoint HLOD scores generated using the dominant and recessive models and 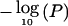 for multipoint NPL scores for all five chromosomes of interest. Restriction of the analysis to the 89 pedigrees that contained solely cases of B-cell CLL made no significant difference to the linkage statistics. Specifically, the maximum NPL score obtained at chromosome 11p11 was 3.05 after LD removal.
for multipoint NPL scores for all five chromosomes of interest. Restriction of the analysis to the 89 pedigrees that contained solely cases of B-cell CLL made no significant difference to the linkage statistics. Specifically, the maximum NPL score obtained at chromosome 11p11 was 3.05 after LD removal.
Discussion
We have shown elsewhere that the use of high-density SNP arrays provides an efficient method of conducting genomewide linkage searches to identify Mendelian disease genes (Sellick et al. 2004b). Here, we have applied the same technology to the mapping of a complex trait. When evaluating the whole genome for linkage, a stringent threshold is required. For a complex trait, LOD scores >3.6 (equating to a genomewide significance level of 5%) are generally accepted to provide significant evidence of linkage, whereas LOD scores >2.2 are said to provide suggestive evidence of linkage (Lander and Kruglyak 1995). Using a simulation approach, we established corresponding empirical limits for the NPL statistic of 3.59 and 2.80 for significant and suggestive evidence of linkage, respectively. In our genome scan, although we did not identify any regions of significant linkage, we found one region of suggestive linkage. Our results provide evidence of a major susceptibility locus on the pericentric region of chromosome 11 that influences the risk of CLL—particularly in the more densely affected families—with characteristics consistent with an autosomal recessive mode of inheritance. As expected, the multipoint analyses that test for linkage in the presence of heterogeneity (multipoint HLODs) gave the most power to detect linkage in our sample set.
Apart from the evidence of suggestive linkage to chromosome 11, we did not find any significant evidence of linkage to any of the regions of the genome commonly associated with cytogenetically detectable chromosomal losses (6q, 13q14, or 17p) or gains (trisomy 12) in CLL. There was, however, some support for linkage to chromosome 14q32, a region of the genome that has been shown to be involved in a small subset of B-cell CLL cases (Karnolsky 2000; Gozzetti et al. 2004). It is possible that there is epistatic interaction between these putative loci, but data from the analysis of additional families are required to investigate this possibility.
To date, only one genomewide linkage search of familial CLL had been conducted (Goldin et al. 2003). That study was based on 18 nuclear families containing 38 affected individuals, which inevitably had limited power to detect linkage. The authors failed to find any genomic regions that achieved statistical significance; however, six regions—on chromosomes 1q, 3q, 6q, 12q, 13q, and 17p—were reported as supporting linkage with the stipulation of a significance level of 0.02. None of these regions coincide with our observations.
Our analyses concur with the findings of Schaid et al. (2004) that the presence of LD between SNPs can lead to inflated linkage statistics, as a consequence of the fact that existing linkage software requires equilibrium between markers. The use of highly dense maps of SNPs as markers is a relatively recent advance for linkage analyses; as a result, there is as yet no consensus on the methods to be used to manage the issue of LD between SNPs. In our analysis, we excluded SNPs with high LD, defined as those with pairwise LD measure r2>0.4. The qualitative nature of our results was unaffected when an alternative definition of high LD (D′>0.7) was used, despite the differences in the distributions of the two statistics. Although the approach implemented herein is likely to address the most extreme impact of LD on the linkage results, it is still possible that any remaining LD, either pairwise values with r2<0.4 or higher-order disequilibria, influenced our results. Accepting these caveats, we did find support for linkage of CLL to the pericentric region of chromosome 11 in our genomewide scan. Previous studies by John et al. (2004) and Schaid et al. (2004) investigated the impact of LD on NPL and model-free (Kong and Cox) LOD scores, respectively. They observed differing effects with removal of LD: John et al. (2004) reported that a proportion of SNPs in high LD slightly increased NPL scores, whereas others slightly decreased NPL scores; Schaid et al. (2004) reported that the presence of SNPs in high LD inflate LOD scores. Our results show that high LD between SNPs can inflate both NPL and parametric LOD scores. We did not observe that the presence of high LD led to decreased linkage statistics. Our results also agree with those of Schaid et al. (2004) in that it is the existence of LD between the SNP markers—and not the loss of IC—that impacts the linkage statistics.
To date, no gene has been unambiguously implicated in predisposition to B-cell CLL. A role for HLA alleles has been documented for HL (Klitz et al. 1994), and some association studies have also implicated variants within or close to the major histocompatibility complex class II region in susceptibility to CLL (Machulla et al. 2001). The high frequency of somatic mutation in CLL, coupled with a high prevalence of B-cell LPDs in ataxia telangiectasia (AT [MIM 607585]) heterozygotes, suggests carriers of ATM germline mutations have an increased risk of B-cell tumors (Swift et al. 1987). Three studies have previously assessed the contribution of germline ATM mutations to CLL by screening the gene in either familial or unselected cases (Bevan et al. 1999; Stankovic et al. 1999; Yuille et al. 2002). Collectively, the data provide some support, albeit at a nonsignificant threshold, for the overrepresentation of ATM mutations in CLL. However, on the basis of the linkage data presented herein, it is unlikely that germline mutations within the ATM gene, which maps to chromosome 11q22.3, make a major contribution to the overall familial risk of CLL.
Although speculative at this juncture, there are several interesting candidate genes involved in aspects of the regulation of cellular proliferation and differentiation (PTPRJ [protein tyrosine phosphatase, receptor type J]), apoptosis (MADD [map kinase-activating death domain]) or the prevention of DNA damage (DDB2 [damage-specific DNA binding protein 2]) in the linked region on chromosome 11, some of which have already been shown to be differentially expressed in normal and neoplastic cells.
It clearly would be highly advantageous to evaluate a second series of families for linkage to the regions detected in our analyses. As a consequence of limited biospecimen availability, replication of the linkage findings presented herein in an independent set of families with CLL will be problematic. One strategy we are actively pursuing to increase the power of our existing series of families is to screen unaffected family members for monoclonal B-cell lymphocytosis. Subclinical levels of monoclonal cells with an identical phenotype to indolent CLL (termed “MCLUS” or “MBL”) have recently been shown to be detectable in ∼3% of healthy individuals from the general population, by use of flow cytometric analysis of CD5/CD20/CD79b expression on CD19-gated B cells (Rawstron et al. 2002a). We have also shown that MBL is significantly overrepresented in relatives of patients with familial CLL (detectable in ∼14% of relatives) (Rawstron et al. 2002b), which suggests that the phenotype is a surrogate marker of carrier status. Screening unaffected family members from families with CLL for MBL status therefore has the potential for facilitating gene identification, through the increased number of affected individuals within a given pedigree who would be available for future linkage analyses.
Acknowledgments
The authors thank Drs. Aitchison, Antunovic, Auger, Bell, Ben-Bassat, Berrebi, Bond, Capalbo, Chapman, Chipping, Clark, Dearden, Douglas, Esteban, Gaminara, Garcia de Coca, Garcia Diaz, Ginaldi, Jackson, Johnson, Knechtli, Lakhani, Manoharan, Mehes, Mepham, Milne, Mineur, Morgenstern, Nandi, Parker, Quabeck, Rassam, Reid, Ribeiro, Rist, Rowlands, Stark, Stewart, Stockley, Sykes, Tham, Thompson, Tiplady, Treacy, Tringham, Van Den Neste, Westerman, and Wickham, for their contribution of families with CLL; and the patients, for their participation in this study. G.S.S. was a recipient of a postdoctoral research fellowship from Leukemia Research, and E.L.W., a postdoctoral research fellowship from Cancer Research United Kingdom. A proportion of the genotyping was undertaken by Jo McBride at MRC Geneservices, U.K. The work was supported by grants from Leukemia Research and the Arbib Foundation (to R.S.H. and D.C.) and from Novo Nordic (to V.J.).
Web Resources
The URLs for data presented herein are as follows:
- Affymetrix NetAffx, http://www.affymetrix.com/analysis/index.affx
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CLL, NHL, HL, and AT)
- SNPLINK, http://www.icr.ac.uk/cancgen/molgen/MolPopGen_Bioinformatics.htm
- University of California–Santa Cruz Human Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101 [DOI] [PubMed] [Google Scholar]
- Bevan S, Catovsky D, Marossy A, Matutes A, Popat S, Antonovic P, Bell A, Berrebi A, Gaminara E, Quabeck K, Ribeiro I, Mauro FR, Stark P, Sykes H, van Dongen J, Wimperis J, Wright S, Yuille MR, Houlston RS (1999) Linkage analysis for ATM in familial B cell chronic lymphocytic leukaemia. Leukemia 13:1497–1500 [DOI] [PubMed] [Google Scholar]
- Cartwright RA, Bernard SM, Bird CC, Darwin CM, O’Brien C, Richards ID, Roberts B, McKinney PA (1987) Chronic lymphocytic leukaemia: case-control epidemiological study in Yorkshire. Brit J Cancer 56:79–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Risch N (1995) A comparison of linkage disequilibrium measure for fine-scale mapping. Genomics 29:311–322 [DOI] [PubMed] [Google Scholar]
- Evans DM, Cardon LR (2004) Guidelines for genotyping in genomewide linkage studies: single-nucleotide–polymorphism maps versus microsatellite maps. Am J Hum Genet 75:687–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraway JJ (1993) Distribution of the admixture test for the detection of linkage under heterogeneity. Genet Epidemiol 10:75–83 [DOI] [PubMed] [Google Scholar]
- Goldin LR, Ishibe N, Sgambati M, Marti GE, Fontaine L, Lee MP, Kelley JM, Scherpbier T, Buetow KH, Caporaso NE (2003) A genome scan of 18 families with chronic lymphocytic leukaemia. Br J Haematol 121:866–873 [DOI] [PubMed] [Google Scholar]
- Goldin LR, Pfeiffer RM, Li X, Hemminki K (2004) Familial risk of lymphoproliferative tumors in families of patients with chronic lymphocytic leukemia: results from the Swedish family-cancer database. Blood 104:1850–1854 [DOI] [PubMed] [Google Scholar]
- Gozzetti A, Crupi R, Tozzuoli D, Raspadori D, Forconi F, Lauria F (2004) Molecular cytogenetic analysis of B-CLL patients with aggressive disease. Hematology 9:383–385 [DOI] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13 [DOI] [PubMed] [Google Scholar]
- Gunz FW, Gunz JP, Veale AM, Chapman CJ, Houston IB (1975) Familial leukaemia: a study of 909 families. Scand J Haematol 15:117–131 [DOI] [PubMed] [Google Scholar]
- Hedrick PW (1987) Gametic disequilibrium measures: proceed with caution. Genetics 117:331–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlston RS, Sellick G, Yuille M, Matutes E, Catovsky D (2003) Causation of chronic lymphocytic leukemia—insights from familial disease. Leuk Res 27:871–876 [DOI] [PubMed] [Google Scholar]
- Huang Q, Shete S, Amos CI (2004) Ignoring linkage disequilibrium among tightly linked markers induces false-positive evidence of linkage for affected sib pair analysis. Am J Hum Genet 75:1106–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Shephard N, Liu G, Zeggini E, Cao M, Chen W, Vasavda N, Mills T, Barton A, Hinks A, Eyre S, Jones KW, Ollier W, Silman A, Gibson N, Worthington J, Kennedy GC (2004) Whole-genome scan, in a complex disease, using 11,245 single-nucleotide polymorphisms: comparison with microsatellites. Am J Hum Genet 75:54–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnolsky IN (2000) Cytogenetic abnormalities in chronic lymphocytic leukemia. Folia Med (Plovdiv) 42:5–10 [PubMed] [Google Scholar]
- Klitz W, Aldrich CL, Fildes N, Horning SH, Begovich AB (1994) Localization of predisposition to Hodgkin disease in the HLA class II region. Am J Hum Genet 54:497–505 [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed] [Google Scholar]
- Linet MS, Van Natta ML, Brookmeyer R, Khoury MJ, McCaffrey LD, Humphrey RL, Szklo M (1989) Familial cancer history and chronic lymphocytic leukaemia: a case-control study. Am J Epidemiol 130:655–664 [DOI] [PubMed] [Google Scholar]
- Machulla HK, Muller LP, Schaaf A, Kujat G, Schonermarck U, Langner J (2001) Association of chronic lymphocytic leukaemia with specific alleles of the HLA-DR4:DR53:DQ8 haplotype in German patients. Int J Cancer 92:203–207 [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Loi H, Dong S, Tsai YY, Fang J, Law J, Di X, Liu WM, Yang G, Liu G, Huang J, Kennedy GC, Ryder TB, Marcus GA, Walsh PS, Shriver MD, Puck JM, Jones KW, Mei R (2004) Parallel genotyping of over 10,000 SNPs using a one-primer assay on a high-density oligonucleotide array. Genome Res 14:414–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Hermelink HK, Montserrat E, Catovsky D, Harris NL (2001) Chronic lymphocytic leukaemia/small lymphocytic lymphoma. In: Jaffe ES, Harris NL, Stein H, Vardiman JW (eds) World Health Organization classification of tumours: pathology and genetics of tumours of haematopoietic and lymphoid tissues. IARC Press, Lyon, pp 127–130 [Google Scholar]
- Pottern LM, Linet M, Blair A, Dick F, Burmeister LF, Gibson R, Schuman LM, Fraumeni JF Jr (1991) Familial cancers associated with subtypes of leukaemia and non-Hodgkin’s lymphoma. Leuk Res 15:305–314 [DOI] [PubMed] [Google Scholar]
- Radovanovic Z, Markovic-Denic L, Jakovic S (1994) Cancer mortality of family members of patients with chronic lymphocytic leukaemia. Eur J Epidemiol 10:211–213 [DOI] [PubMed] [Google Scholar]
- Rawstron AC, Green MJ, Kuzmicki A, Kennedy B, Fenton JA, Evans PA, O’Connor SJ, Richards SJ, Morgan GJ, Jack AS, Hillmen P (2002a) Monoclonal B lymphocytes with the characteristics of “indolent” chronic lymphocytic leukemia are present in 3.5% of adults with normal blood counts. Blood 100:635–639 [DOI] [PubMed] [Google Scholar]
- Rawstron AC, Yuille MR, Fuller J, Cullen M, Kennedy B, Richards SJ, Jack AS, Matutes E, Catovsky D, Hillmen P, Houlston RS (2002b) Inherited predisposition to CLL is detectable as subclinical monoclonal B-lymphocyte expansion. Blood 100:2289–2290 [DOI] [PubMed] [Google Scholar]
- Ries LAG, Eisner MP, Kosary CL, Hankey BF, Miller BA, Clegg L, Mariotto A, Fay MP, Feuer EJ, Edwards BK (eds) (2003) SEER Cancer Statistics Review 1975–2000. National Cancer Institute, Bethesda. http://seer.cancer.gov/csr/1975_2000/ (accessed July 25, 2005)
- Schaid DJ, Guenther JC, Christensen GB, Hebbring S, Rosenow C, Hilker CA, McDonnell SK, Cunningham JM, Slager SL, Blute ML, Thibodeau SN (2004) Comparison of microsatellites versus single-nucleotide polymorphisms in a genome linkage screen for prostate cancer–susceptibility loci. Am J Hum Genet 75:948–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer M, Melies M, Ploen JE (1973) Chronic lymphocytic leukaemia in 5 siblings. Scand J Haemat 11:97–105 [DOI] [PubMed] [Google Scholar]
- Sellick G, Catovsky D, Houlston RS (2004a) Familial chronic lymphocytic leukaemia. In: Hamblin T, Johnson S, Miles A (eds) The effective management of chronic lymphocytic leukaemia. Aesculapius Medical Press, London, pp 3–15 [Google Scholar]
- Sellick GS, Longman C, Tolmie J, Newbury-Ecob R, Geenhalgh L, Hughes S, Whiteford M, Garrett C, Houlston RS (2004b) Genomewide linkage searches for Mendelian disease loci can be efficiently conducted using high-density SNP genotyping arrays. Nucleic Acids Res 32:e164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankovic T, Weber P, Stewart G, Bedenham T, Byrd OJ, Murray J, Moss PAH, Taylor AMR (1999) Inactivation of ataxia telangectasia mutated gene in B-cell chronic lymphocytic leukaemia. Lancet 353:26–29 [DOI] [PubMed] [Google Scholar]
- Swift M, Reitnauer PJ, Morrell D, Chase CL (1987) Breast and other cancers in families with ataxia-telangiectasia. N Engl J Med 316:1289–1294 [DOI] [PubMed] [Google Scholar]
- Webb EL, Sellick GS, Houlston RS (2005) SNPLINK: multipoint linkage analysis of densely distributed SNP data incorporating automated linkage disequilibrium removal. Bioinformatics 21:3060–3061 [DOI] [PubMed] [Google Scholar]
- Whittemore AS, Halpern J (1994) A class of tests for linkage using affected pedigree members. Biometrics 50:118–127 [PubMed] [Google Scholar]
- Yuille MR, Condie A, Hudson CD, Bradshaw PS, Stone EM, Matutes E, Catovsky D, Houlston RS (2002) ATM mutations are rare in familial chronic lympohcytic leukaemia. Blood 100:603–609 [DOI] [PubMed] [Google Scholar]