

To the Editor:
In the March issue of the Journal, Seaman and Müller-Myhsok (2005) proposed a method for rapid simulation of P values in association studies. The authors kindly discussed my article (Lin 2005), which was electronically published in September 2004. Unfortunately, their discussion is inaccurate. In particular, their assertion that the variance formula given in my article ignores the variation due to the estimation of nuisance parameters is untrue.
Both my article (Lin 2005) and that of Seaman and Müller-Myhsok (2005) are based on the simulation of the (same) joint distribution for a set of test statistics, although the actual simulation procedures are somewhat different. If a statistic is asymptotically normal, then it can be approximated by a sum of independent terms. Specifically, the statistic for testing the jth null hypothesis Hj:βj=0 can be written as
 |
up to an asymptotically negligible term, where Uji involves only the data from the ith subject, and n is the sample size. Under Hj:βj=0 (j=1,…,J), the set of statistics (U1,…,UJ) is asymptotically multivariate zero-mean normal with covariance
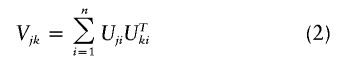 |
between Uj and Uk (j,k=1,…,J). In my article, I proposed to simulate this joint distribution by
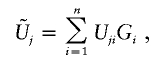 |
where G1,…,Gn are independent standard normal random variables, because 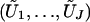 has the same joint distribution as (U1,…,UJ). Seaman and Müller-Myhsok proposed to fit a joint model that includes all βj terms and to simulate the joint distribution of (U1,…,UJ) by a multivariate normal random vector with mean 0 and with covariance matrix {Vjk;j,k=1,…,J}. The two proposals simulate from (essentially) the same joint distribution and are both very fast. In particular, my proposal involves the evaluation of the
has the same joint distribution as (U1,…,UJ). Seaman and Müller-Myhsok proposed to fit a joint model that includes all βj terms and to simulate the joint distribution of (U1,…,UJ) by a multivariate normal random vector with mean 0 and with covariance matrix {Vjk;j,k=1,…,J}. The two proposals simulate from (essentially) the same joint distribution and are both very fast. In particular, my proposal involves the evaluation of the  terms, which is of the order nJ and which can be done in seconds or minutes, even for large values of n and J. One advantage of my proposal is that missing genotype data for one statistic do not affect any other statistics. (If the ith subject has no genotype data for calculating the jth statistic, then we simply set Uji to 0. There is no need to impute missing data.) By contrast, the proposal of Seaman and Müller-Myhsok can include in the analysis only those subjects with complete genotype data on all the SNPs, unless all the missing genotypes are imputed. Imputation can adversely affect the type I error and power.
terms, which is of the order nJ and which can be done in seconds or minutes, even for large values of n and J. One advantage of my proposal is that missing genotype data for one statistic do not affect any other statistics. (If the ith subject has no genotype data for calculating the jth statistic, then we simply set Uji to 0. There is no need to impute missing data.) By contrast, the proposal of Seaman and Müller-Myhsok can include in the analysis only those subjects with complete genotype data on all the SNPs, unless all the missing genotypes are imputed. Imputation can adversely affect the type I error and power.
Seaman and Müller-Myhsok (2005) focused on the parametric statistics under generalized linear models, whereas my article (Lin 2005) covered all possible statistics, parametric or nonparametric. As described in the appendix of my article, all the commonly used association statistics can be written in the form of equation (1), in which Uji is the ith subject’s efficient score function for βj. In the special case of parametric statistics,
where Sβj,i and Sαj,i are the ith subject’s score functions for βj and αj, αj is the set of nuisance parameters, and Aβjαj and Aαjαj are the appropriate submatrices of the limiting Fisher information matrix for βj and αj. As mentioned in the appendix of my article, this expression can be found in mathematical statistics texts, such as that by Bickel et al. (1993, p. 28). It was also given as equation (A1) of Lin and Zou (2004). In this case, 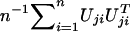 converges to Aβjβj-AβjαjA-1αjαjAαjβj, the limiting covariance matrix of n-1/2Uj, and the joint distribution of
converges to Aβjβj-AβjαjA-1αjαjAαjβj, the limiting covariance matrix of n-1/2Uj, and the joint distribution of 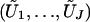 indeed provides a valid approximation to that of (U1,…,UJ). Thus, Seaman and Müller-Myhsok’s statement that my variance formula ignores the term AβjαjA-1αjαjAαjβj is simply untrue. Had I used the wrong variance formula, the numerical results presented in my article would not have been sensible.
indeed provides a valid approximation to that of (U1,…,UJ). Thus, Seaman and Müller-Myhsok’s statement that my variance formula ignores the term AβjαjA-1αjαjAαjβj is simply untrue. Had I used the wrong variance formula, the numerical results presented in my article would not have been sensible.
Seaman and Müller-Myhsok (2005) might have been confusing score functions with efficient score functions. The score function for βj involves the nuisance parameters αj, which are replaced by 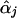 , the maximum-likelihood estimators of αj under Hj:βj=0. To account for the extra variation caused by this estimation, we use the Taylor series expansion to express the score function for βj (with αj replaced by
, the maximum-likelihood estimators of αj under Hj:βj=0. To account for the extra variation caused by this estimation, we use the Taylor series expansion to express the score function for βj (with αj replaced by 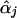 ) as a sum of independent terms, which is in the form of equation (1) with Uji as given in equation (3), so that equation (2) provides the correct variance-covariance expression (Lin and Zou 2004). The efficient score functions Uji involve the unknown parameters αj. When αj in Uji is replaced by
) as a sum of independent terms, which is in the form of equation (1) with Uji as given in equation (3), so that equation (2) provides the correct variance-covariance expression (Lin and Zou 2004). The efficient score functions Uji involve the unknown parameters αj. When αj in Uji is replaced by 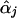 , the resulting Uj, Vjj, and Tj are (essentially) the same as the Uβ(l), Vβ(l), and Tl given by Seaman and Müller-Myhsok (2005). Again, the framework of my article (Lin 2005) extends far beyond the parametric setting.
, the resulting Uj, Vjj, and Tj are (essentially) the same as the Uβ(l), Vβ(l), and Tl given by Seaman and Müller-Myhsok (2005). Again, the framework of my article (Lin 2005) extends far beyond the parametric setting.
In fact, the parametric setting considered by Seaman and Müller-Myhsok (2005) does not demonstrate the full power of the simulation approach. In their setting, the calculation of each statistic is of the order n, so that the permutation test is very feasible, even for large values of n. There is a stronger case for the simulation approach when the calculation of each statistic is time consuming or when the null distribution cannot be properly generated by permutation, as discussed in my article (Lin 2005).
Incidentally, equation (2) in Seaman and Müller-Myhsok (2005) is confusing. The term in the middle is the score function for β, which is a function of α, whereas the term on the far right involves 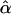 instead.
instead.
References
- Bickel PJ, Klassen CAJ, Ritov Y, Wellner JA (1993) Efficient and adaptive estimation in semiparametric models. The Johns Hopkins University Press, Baltimore [Google Scholar]
- Lin DY (2005) An efficient Monte Carlo approach to assessing statistical significance in genomic studies. Bioinformatics 21:781–787 10.1093/bioinformatics/bti053 [DOI] [PubMed] [Google Scholar]
- Lin DY, Zou F (2004) Assessing genomewide statistical significance in linkage studies. Genet Epidemiol 27:202–214 10.1002/gepi.20017 [DOI] [PubMed] [Google Scholar]
- Seaman SR, Müller-Myhsok B (2005) Rapid simulation of P values for product methods and multiple-testing adjustments in association studies. Am J Hum Genet 76:399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]