

Abstract
Both multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE), its animal model, involve inflammatory attack on central nervous system (CNS) white matter, leading to demyelination and axonal damage. Changes in astrocytic morphology and function are also prominent features of MS and EAE. Resting astrocytes form a network that is interconnected through gap junctions, composed mainly of connexin43 (Cx43) protein. Although astrocytic gap junctional connectivity is known to be altered in many CNS pathologies, little is known about Cx43 expression in inflammatory demyelinating disease. Therefore, we evaluated the expression of Cx43 in spinal cords of EAE mice compared with healthy controls. Lumbar ventral white matter areas were heavily infiltrated with CD11β-immunoreactive monocytes, and within these infiltrated regions loss of Cx43 immunoreactivity was evident. These regions also showed axonal dystrophy, demonstrated by the abnormally dephosphorylated heavy-chain neurofilament proteins. Astrocytes in these Cx43-depleted lesions were strongly glial fibrillary acidic protein reactive. Significant loss (38%) of Cx43 protein in EAE mouse at the lumbar portion of spinal cords was confirmed by Western blot analysis. Decreased Cx43 transcript level was also observed on cDNA microarray analysis. In addition to changes in Cx43 expression, numerous other genes were altered, including those encoding adhesion and extracellular matrix proteins. Our data support the notion that, in addition to damage of myelinating glia, altered astrocyte connectivity is a prominent feature of inflammatory demyelination.
Keywords: white matter, inflammation, immunohistochemistry, microarray, experimental autoimmune encephalomyelitis, Cx43
Multiple sclerosis (MS) is an immune-cell-mediated inflammatory demyelinating disease of the central nervous system (CNS; Miller and Karpus, 1994; Ewing and Bernard, 1998; Hohlfeld and Wekerle, 2001). MS and experimental autoimmune encephalomyelitis (EAE), an animal model of MS (Honegger and Langemann, 1989; Glabinski et al., 1997; Deloire-Grassin et al., 2000), are characterized by white matter (WM) inflammation accompanied by disturbed central nerve conductance and motor and sensory impairments (Juhler, 1988; Whitaker and Mitchell, 1997).
Pathological findings in MS and EAE include infiltration of activated peripheral inflammatory cells into the CNS (Bauer et al., 1995; Owens et al., 1998; Imrich and Harzer, 2001), loss of myelin (Adams and Kubik, 1952; Raine et al., 1984; Whitaker and Mitchell, 1997), axonal damage (Ferguson et al., 1997; Trapp et al., 1998; Kornek et al., 2000; Bitsch et al., 2000; Deloire-Grassin et al., 2000), axonal loss (D’amelio et al., 1990; Landete et al., 2000; Bjartmar et al., 2001), edema (Claudio et al., 1990; Levine et al., 1966), and astrocytic activation (Smith et al., 1983; Eng et al., 1992; Rafalowska et al., 1992). Notably, swelling of astrocytes is an early key event that precedes macrophage infiltration to the CNS in EAE (Smith et al., 1984; Eng et al., 1989, 1992; D’amelio et al., 1990).
Astrocytes in situ and in vitro are coupled by gap junctions, mainly formed by connexin43 (Cx43; Dermietzel et al.,2000; Nagy and Rash, 2000). Gap junction channels provide cytoplasmic continuity between coupled cells, thereby allowing the transfer of ions and metabolites throughout the astrocytic network (Alexander and Goldberg, 2003).
The modulation of Cx43 expression levels in various CNS pathologies has been widely investigated (Nakase et al., 2003; for comprehensive reviews see Dermietzel and Hofstadter, 1998; Rouach et al., 2002a). However, contrasting results regarding Cx43 regulation among different pathological CNS conditions have been reported. For example, in Alzheimer’s disease, Cx43 was found to be up-regulated in amyloid plaques (Nagy et al., 1996). In contrast, Cx43 down-regulation was observed after treatment of human fetal cultured astrocytes with the proinflammatory cytokine interleukin (IL)-1β (John et al., 1999) and after brain mechanical lesions (Rouach et al., 2002a) and ischemia (Nagy and Li, 2000). In EAE spinal cord, changes occur that may be expected to either increase or decrease Cx43 expression: glutamate excitoxicity (Pitt et al., 2000) and mechanical damage from edema (Paterson, 1976), ischemia (Honegger and Langemann, 1989), and the induction of inflammatory cytokines (Bettelli and Nicholson, 2000).
In systemic inflammation, induced by administration of bacterial lipopolysacharide, Cx43 expression was reported to be down-regulated in the heart (Fernandez-Cobo et al., 1999) but up-regulated in the kidney and lungs (Fernandez-Cobo et al., 1998). Thus, not only do different pathologies elicit differential effect on Cx43 expression but also similar inflammatory insult may exert diverse effects on Cx43 in different tissues. Although cDNA microarray analyses have been performed on a variety of EAE models and in MS tissue, a report on Cx43 changes either was not included (Ibrahim et al., 2001; Whitney et al., 2001; Mix et al., 2002; Matejuk et al., 2003) or was not statistically significant because of large variance (Lock et al., 2002). Moreover, none of these studies determined protein expression levels, which add valuable information on the impact of the changes in message on the actual level of protein itself. This is an important aspect; protein levels do not necessarily follow the same trend as RNA levels (Rogojina et al., 2003). Therefore, we evaluated the effect of acute CNS inflammation on Cx43 expression in spinal cords of mice with adoptive-transfer (AT)-EAE, a model of MS (Brown and McFarlin, 1981; Raine et al., 1984), by using cDNA microarrays, immunohistochemistry, and Western blotting to determine spinal cord Cx43 RNA and protein expression levels. Building on initial results (Brand-Schieber et al., 2003), this study is the first published report on Cx43 expression and localization in EAE WM that utilizes these three techniques to describe changes in Cx43 associated with autoimmune demyelination.
MATERIALS AND METHODS
Animals and Disease Induction
Female SJL/J mice were housed in a light- and temperature-controlled environment in accordance with NIH and AAALAC guidelines. All experiments were performed under an institutionally approved animal protocol. Myelin basic protein (MBP; 1 mg/mouse), a component of myelin, dissolved in sterile phosphate-buffered saline (PBS) and emulsified with an equal volume of incomplete Freund’s adjuvant supplemented with Mycobacterium tuberculosis emulsion (200 μl/mouse) was injected into the flanks of 11-week-old mice. Lymph node cells (LNC) were obtained from draining lymph nodes 10 days later. LNC were cultured for 4 days in RPMI-1640 medium in the presence of MBP (75 μg/ml). Cultured LNC were subsequently injected into the tail vein of syngeneic 6-week-old SJL/J mice at a dose of about 4 × 107 cells/mouse. Onset of disease occurred after 8–10 days, and the animals manifested a predictable pattern of ascending paralysis that correlates with the degree of spinal cord pathology (Simmons et al., 1982). Disability was scored daily as follows: 0, no clinical signs; 1, flaccid tail; 2, abnormal gait; 3, hind limb paralysis; 4, complete paralysis; 5, moribund or dead. Spinal cord tissue was harvested at the peak of disability; 11 days post-LNC transfer.
Tissue Processing
Anesthetized animals were perfused intracardially with ice-cold PBS. Spinal cords were dissected out. Spinal cord tissue was cryoprotected in Tissue-Tek and flash frozen in supercooled acetone. Tissue blocks were stored at −80°C until cut into 10-μm sections that were laid onto glass microscope slides. For RNA and Western blotting analysis, tissues were snap frozen and stored at −80°C until use.
RNA Analysis
We compared RNA samples extracted from spinal cords of control and EAE mouse samples by analyzing hybridization to 26,809 spotted mouse cDNA sequences, produced by our Einstein Microarray Facility (for full technical information see http://129.98.70.229). Total RNA was extracted according to an established, standardized protocol (see http://www.aecom.yu.edu/home/molgen/funcgenomic.html; Iacobas et al., 2003). We used four sets of two spinal cords each from EAE mice (E1, E2, E3, E4; eight spinal cords total) and from control mice (C1, C2, C3, C4; eight spinal cords total) to ensure statistical relevance of the study. From each control or experimental set, 60 μg RNA was reverse transcribed into cDNA by using fluorescent dUTPs [Cy3-dUTP (green; g) or Cy5-dUTP (red; r)].
We hybridized four microarrays with the following combinations: C1(r)C2(g), C3(r)C4(g), E1(r)E2(g), E3(r)E4(g) (‘‘multiple yellow’’ design). The labeled cDNAs were hybridized overnight at 50°C. After hybridization, the slides were washed at room temperature, in solutions containing 0.1% sodium dodecyl sulfate (SDS) and 1% SSC (3 M NaCl + 0.3 M sodium citrate) to remove the nonhybridized cDNAs. Slides were immediately scanned, with the same accelerating voltages in the two channels for all slides: 760V(r) and 670V(g).
The images were acquired and initially analyzed with GenePix Pro 4.1 software. Raw data were further normalized and mined through in-house-developed algorithms (Iacobas et al., 2003, 2005a, 2005b). Sequences flagged because of spot imperfections, saturated pixels, or high noise/background signal were eliminated from the analysis. To maintain a uniform level of significance, analysis was performed only when spots were valid in all samples and in both channels. The net fluorescence, i.e., the background subtracted from the foreground signal, of each validated spot was transformed through intra- and interchip normalization, aiming to obtain biologically meaningful comparisons between the studied specimens. [Intrachip normalization balances the averages of net fluorescence values in the two channels within each pin domain (subset of spots printed by the same pin), corrects the intensity-dependent bias (usually referred to as lowest normalization), and forces the standard distribution (mean 0 and standard deviation 1) of log2 ratios (scale normalization) of net fluorescent values in the two channels for each array. Interchip normalization assigns a ratio between the corrected net fluorescence of each valid spot and the average net fluorescence of all valid spots in both control (C1, C2, C3, C4) and experimental (E1, E2, E3, E4) arrays.]
When multiple spots probed a single gene, we used the weighted average of the ratios of all corresponding spots. The weight of a spot increased in proportion to the number of identities between the spotted sequence and the probed gene and decreased in proportion to the length of the query.
The expression ratio of a gene in EAE spinal cords compared with that in control mice was computed as the geometric average of the normalized fluorescence ratios detected by each channel for all spots probing the same gene. As shown elsewhere (Iacobas et al., 2005), the geometric average compensates for the sequence-dependent bias toward one label or the other.
The P values of fluorescence ratio intensities in comparing EAE and control samples were computed through the classical Student’s t-test of equality of the means of two distributions. To maximize the stringency of the analysis, only P < 0.01 was considered statistically significant.
Immunohistochemistry
Frozen tissue sections were processed as previously described (Brand-Schieber and Werner, 2003). Briefly, transverse lumbar spinal cord sections were air dried, fixed with supercooled acetone, permeabilized with Triton X-100, and incubated overnight with primary antibodies at 4°C. Secondary antibodies were applied for 1 hr at room temperature. To visualize biotinylated secondary antibodies, we incubated the sections for an additional 1 hr with streptavidin conjugated AlexaFluor 488 (Molecular Probes, Eugene, OR; 1:2,000). Sections were preserved with 1% n-propylgallate in 50% glycerol in PBS, coverslipped, and sealed with nail polish.
ANTIBODIES
A cocktail of mouse monoclonal antibodies to glial fibrillary acidic protein (GFAP; SM122; Sternberger Monoclonals, Baltimore, MD; 1:8,000) was used to identify astrocytes. Dystrophic axons were identified by using mouse monoclonal SMI32 (Sternberger Monoclonals; 1:8,000), which recognizes a nonphosphorylated epitope in neurofilament H. To detect monocytes, we used rat monoclonal anti-CD11β (Pharmingen, San Diego, CA; 1:10). For detection of Cx43, we used either the anti-Cx43 18-A (a gift from Dr. E. Hertzberg, Albert Einstein College of Medicine; 1:3,000) or Sigma (st. Louis, MO) C6219 (1:2,000) antibodies both rabbit polyclonals. For detection of Cx30, we used a polyclonal antibody from Zymed (South San Francisco, CA; 1:50). In control sections, we replaced the primary antibodies with corresponding preimmune IgG (Alpha Diagnostics, San Antonio, TX) at the same concentration as the primary antibodies (see Fig. 2). For secondary antibodies (all raised in goat), we used a biotinylated anti-rabbit (Vector, Burlingame, CA; 1:1,000), an anti-mouse conjugated to AlexaFluor 594 and an anti-rat conjugated to AlexaFluor 633 (both from Molecular Probes; 1:1,000).
Fig. 2.

Control sections show specificity of immunolabeling in spinal cords from mice with EAE. Confocal microscopic images depicting transverse lumbar spinal cord sections from mice with EAE that were labeled by combining all pairs of the following proteins: Cx43 (b,c; green), SMI32 (a,c; red), and CD11β (a,b; blue). In each section, the third primary antibody was replaced with a matching species/IgG subtype antibody obtained from animals that were not exposed to an antigenic peptide. The combinations are specified above each panel. a: No labeling was detected in the green channel when Cx43 antibody was replaced with a nonspecific rabbit IgG. b: Only faint labeling was detected in the red channel when SMI32 antibody was replaced with nonspecific mouse IgG1. c: No labeling was detected in the blue channel when CD11β antibody was replaced with a non-specific rat IgG2b. Scale bar = 50 μm.
Imaging
Thin optical sections were imaged using a Bio-Rad Radiance 2000 confocal microscope (Bio-Rad, Hercules, CA), equipped with a krypton/argon laser that provided excitation at 488 nm and 568 nm. Narrow-pass filters were used for detection of green and red probes. A diode laser provided excitation at 637 nm and a longpass filter was used for detection of far-red emission. We used a Nikon Eclipse 200 microscope with ×20 and ×60 1.4 N.A. plan apo objectives.
Western Blotting
Lumbar spinal cord tissues of three control and three EAE mice were sonicated in a lysis buffer containing protease inhibitors (Sigma F2714). Each sample (20 μg total protein) was electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide minigels (Bio-Rad) and then transferred to nitrocellulose membranes, as previously described (Suadicani et al., 2003). After overnight incubation of the membranes with blocking solution (1 × PBS containing 5% dry nonfat milk), blotting was performed by using a polyclonal rabbit anti-Cx43 antibody (18-A; 1:2,000) and secondary polyclonal anti-rabbit antibodies conjugated to horseradish peroxidase (1:2,000). Immunopositive bands on membranes exposed to ECL reagents were detected with X-ray films. Membranes were reblotted with anti-mouse β-actin antibodies (Chemicon , Temecula, CA; 1:5,000) for quantification of Cx43 expression levels using Scion (NIH imaging software).
RESULTS
Passive immunization of animals with MBP-primed lymph node cells led to the development of EAE, with a typical disease progression (Brown and McFarlin, 1981; Raine et al., 1984), which corresponds to ascending spinal cord inflammation. Mice began to show signs of disease 7 days after LNC transfer and progressed to develop hind limb paralysis (disease score 3 on a scale of 0–5) 3 days later. Spinal cord tissues were harvested at the peak of the disease, i.e., 11–14 days posttransfer.
To determine whether the inflammatory demyelination also affected astrocytic Cx43 expression, Cx43 immunohistochemistry was performed on transverse sections from lumbar spinal cords of control and EAE mice (Fig. 1). For triple labeling, we used the following antibodies: Cx43 (green; Fig. 1a–c); a marker of axonal dystrophy, SMI32 (red; Fig. 1d–f); and a monocyte marker, CD11β (blue; Fig. 1g–h). Labeling was specific, insofar as there was no appreciable labeling when each primary antibody was replaced with a corresponding nonspecific IgG (Fig. 2).
Fig. 1.

Regions showing reduced Cx43 in spinal cords from mice with EAE correlate with monocytes and dystrophic axons. Confocal microscopic images depicting transverse lumbar spinal cord sections from controls and two mice with EAE that were triply labeled for Cx43 (a–c; green), SMI32 (d–f; red), and CD11β (g–i; blue). Merged images are shown in j–l. Control tissue (left row) has intense Cx43 labeling but does not display SMI32 (dystrophic axons) and CD11β (monocytes) labeling. Tissue from animals sick with EAE (middle and right rows) has pockets of reduced Cx43 immunoreactivity. These regions also show increased axonal dystrophy and monocyte infiltration, indicated by enhanced SMI32 and CD11β labeling, respectively. A region clear of CD11β within an inflammatory compartment displays preserved Cx43 labeling (arrow). Scale bar = 50 μm.
In the lumbar spinal cord, Cx43 immunoreactivity was reduced in inflamed WM of mice with EAE compared with healthy controls (Fig. 1a–c). Loss of Cx43 was clearly confined to areas highly infiltrated by monocytes (CD11β; Fig. 1g–i). In contrast, in the same sections, flanking WM Cx43 labeling was not reduced. The preserved Cx43 labeling within a monocyte-free pocket, amidst a large area with infiltrated monocytes and markedly reduced Cx43 labeling (Fig. 1i, arrow), underscores the inverse correlation between monocyte infiltration and Cx43 expression.
Lesions with inflammatory activity, delineated by monocyte infiltration, also displayed axonal dystrophy as visualized by SMI32 immunoreactivity (Fig. 1e,h,f), which is consistent with previous observations in EAE WM (Pitt et al., 2000; Werner et al., 2001). Interestingly, these areas corresponded almost exactly to areas of Cx43 loss from astrocytes (compare with Fig. 1b,c).
Because astrocytes can migrate away from damaged CNS regions (Fitch et al., 1999; Ma et al., 2001; Inman et al., 2002) and can become apoptotic in response to inflammatory cytokines (Benn et al., 2001), we investigated whether the decreased Cx43 immunolabeling in inflamed regions was due to depletion of astrocytes. For these experiments, we double-labeled spinal cord tissue with Cx43 and GFAP antibodies (Fig. 3). In control tissue WM, Cx43 overlapped with GFAP-positive astrocytes featuring slender processes and unremarkable cell bodies, characteristic of resting astrocytes (Fig. 3, top). In EAE, regions with reduced Cx43 immunolabeling were not devoid of GFAP-positive astrocytes (Fig. 3, bottom). Many of the these astrocytes appeared rounded and had long, thick processes (asterisks in Fig. 3, bottom), suggestive of reactive astrocytes (Smith et al., 1983; Eng et al., 1992). These findings suggest that the reduction in Cx43 immunoreactivity most likely is due to altered expression levels of this protein in the inflamed tissue and not due to astrocyte disappearance.
Fig. 3.
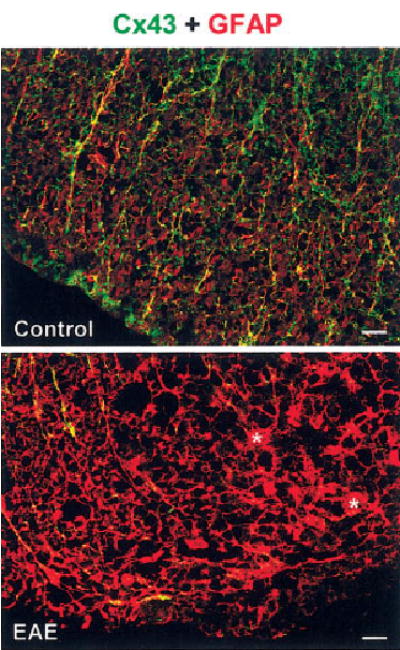
Regions showing reduced Cx43 in spinal cords from mice with EAE also display hypertrophic astrocytes. Confocal microscopic images depicting transverse lumbar spinal cord section from control (top) and EAE (bottom) that were doubly labeled for Cx43 (green) and the astrocytic marker GFAP (red). Overlap of green and red appears yellow. Control white matter shows Cx43 overlapping with typical GFAP-positive astrocytes. In EAE, regions of reduced Cx43 immunoreactivity contain intensely labeled GFAP-positive astrocytes that appear hypertrophied, with enlarged cell bodies (asterisks) and thick processes. Scale bars = 20 μm.
We used two different anti-Cx43 antibodies. The Cx43 18-A antibody (a gift from Dr. Hertzberg) was raised against an epitope (aa 346–360) that is susceptible to masking (Ochalski et al., 1995; Theriault et al., 1997). The other, a commercially available antibody (Sigma), is raised against a different epitope (aa 363–382). Both antibodies showed loss of immunoreactivity in tissue sections from mice with EAE (only the latter shown). Because the two sequences against which the antibodies are raised are adjacent regions of the carboxyl terminus of Cx43, these results do not exclude the possibility that both antibody epitopes are masked in EAE spinal cords.
To determine whether the reduction in the Cx43 immunolabeling was due to masking of antigenic sites, we measured Cx43 expression levels by Western blotting. Figure 4a shows a Western blot of spinal cord homogenates from three controls (lanes 1–3) and three mice with EAE (lanes 4–6). Densitometric analysis comparing Cx43 with β-actin revealed a 38% decrease in Cx43 protein levels (P < 0.05, t-test; n = 3 control and 3 EAE animals and 1 Western blot; Fig. 4b) in EAE spinal cord tissue homogenates.
Fig. 4.
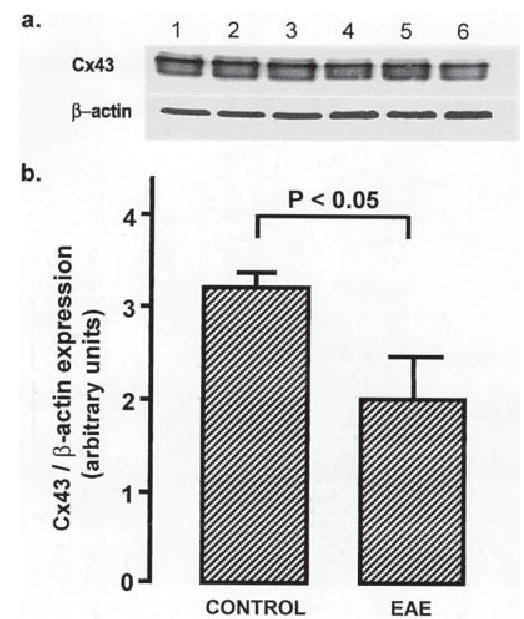
Cx43 protein is reduced in spinal cords of mice with EAE. a: A Western blot demonstrating reduced Cx43 protein in lumbar spinal cord homogenates from mice with EAE (lanes 4–6) compared with controls (lanes 1–3). b: A graph showing semiquantitative analysis based on Cx43 to β-actin ratios (mean ± SEM). There is a significant reduction of Cx43 in EAE (control 3.17 ± 0.12; EAE 1.99 + 0.41; n = 3; P < 0.05, ANOVA followed by Student’s t-test).
Because reduced levels of Cx43 could reflect increased degradation and/or reduced synthesis, we investigated whether autoimmune demyelination affects Cx43 mRNA levels, by using high-density cDNA arrays (Iacobas et al., 2003). The microarray study was performed according the standards of the Microarray Gene Expression Data Society (MGED), and data complying with the ‘‘Minimum information about microarray experiments’’ (MIAME) have been deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo, series GSE 2446) to be provided prior to publication).
We determined the differences in RNA levels of Cx43, as well as other gene products, in control vs. EAE spinal cord tissue. Among the 7,455 genes encoding known proteins that were successfully probed, 1,433 (19%) hybridized differentially (P < 0.01) in EAE vs. control, with 839 (58.5%) genes up-regulated and 594 (41.4%) down-regulated. These significantly altered genes were classified according the functions performed by their protein products, as previously described (Iacobas et al., 2003). The relative distribution of the significantly regulated genes among these functional groups is illustrated in Figure 5, which shows that the regulated genes belong to all functional classes, revealing the complexity of the transcriptomic alterations related to the EAE.
Fig. 5.
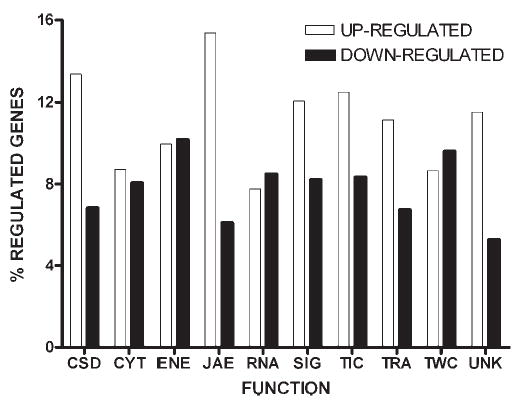
Functional categories of significantly regulated genes in spinal cords of AT-EAE mice compared to controls. A total of 1,433 out of 7,455 genes with known protein product were significantly (P < 0.05) either up- (open bars) or down-regulated (solid bars) in AT-EAE. In this histogram, numbers of genes regulated within each class are expressed as a percentage of the total number of genes within each class on the array. Genes were categorized as to the following functions: CSD: cell cycle, shape, differentiation, death, CYT: cytoskeleton, ENE: energy and metabolism, JAE: cell junction, adhesion, extracellular matrix, RNA: RNA processing, SIG: cell signaling, TIC, transport of small molecules and ions into the cell, TRA: transcription, TWC: transport of ions/molecules within the cell, UNK: function not yet assigned. Note that the JAE group has the highest percentage of up-regulated (15.4%) and the lowest percentage of down-regulated (6.3%) genes, whereas other gene categories show a more uniform distribution of up- and down-regulation.
Table I shows a selection of JAE (cell junction, adhesion, and extracellular matrix) genes that exhibited the largest change in EAE compared with control tissue. Most of the highly up-regulated genes were related to the immune response, as expected based on the inflammatory nature of EAE. Some genes showed an especially large elevation in expression, which may represent RNA originating from inflammatory infiltrates penetrating the spinal cord from the periphery. Cx43 was one of the prominently down-regulated genes in this category, with a 2.9-fold decrease in RNA level (P < 0.05) in EAE compared with controls.
TABLE I.
Most Profoundly Up- and Down-Regulated JAE (Cell Junction Adhesion and Extracellular Matrix) Genes in EAE
| Name | Symbol | Fold change |
|---|---|---|
| Up-regulated JAE transcripts | ||
| Histocompatibility 2, class II antigen A, alpha | H2-Aa | 26.37 |
| Cathepsin S | Ctss | 14.23 |
| Complement component 1, q subcomponent, alpha polypeptide | C1qa | 10.03 |
| Complement component 1, q subcomponent, beta polypeptide | C1qb | 8.45 |
| Histocompatibility 2, class II antigen A, beta 1 | H2-Ab1 | 6.67 |
| Histocompatibility 2, L region | H2-L | 6.50 |
| Histocompatibility 2, K region | H2-K | 6.33 |
| Serine (or cysteine) proteinase inhibitor, clade A, member 3G | Serpina3g | 6.17 |
| Cathepsin Z | Ctsz | 5.93 |
| Beta-2 microglobulin | B2m | 5.59 |
| Fc receptor, IgG, low affinity III | Fcgr3 | 5.52 |
| Histocompatibility 2, D region locus 1 | H2-D1 | 5.30 |
| Pinin | Pnn | 5.16 |
| Fragilis | Fgls-pend. | 4.46 |
| Complement component 3 | C3 | 4.33 |
| Histocompatibility 2, class II, locus DMa | H2-DMa | 4.22 |
| Cathepsin C | Ctsc | 4.14 |
| Down-regulated JAE transcripts | ||
| Hemoglobin, beta adult major chain | Hbb-b1 | −5.14 |
| Serine (or cysteine) proteinase inhibitor, clade B, member 1a | Serpinb1a | −4.82 |
| Ependymin 2 | Epdm2-pend. | −4.13 |
| Hemoglobin alpha, adult chain 1 | Hba-a1 | −3.97 |
| SPARC-like 1 (mast9, hevin) | Sparcl1 | −3.09 |
| Gap junction membrane channel protein alpha 1 (Cx43) | Gja1 | −2.90 |
| Sperm-associated antigen 9 | Spag9 | −2.88 |
| Secreted (semaphorin) 3C | Sema3c | −2.85 |
| Cathepsin 8 | Cts8 | −2.68 |
| Integrin beta 1 (fibronectin receptor beta) | Itgb1 | −2.53 |
| Embigin | Emb | −2.47 |
| N-ethylmaleimide sensitive fusion protein | Nsf | −2.42 |
| T-cell immunomodulatory protein | Cda08-pend. | −2.28 |
| Calsyntenin 1 | Clstn1 | −2.26 |
| Stromal Cell-derived factor receptor 1 | Sdfr1 | −2.25 |
| Myelin and lymphocyte protein, T-cell differentiation protein | Mal | −2.25 |
| Melanoma antigen, family D, 1 | Maged1 | −2.25 |
In addition to Cx43, another gap junction protein expressed in astrocytes is Cx30 (Dermietzel et al., 2000; Rash et al., 2001a,b). Although Cx30 is normally expressed in gray matter but not in WM (Nagy et al., 1999), we used immunohistochemistry to test whether the decreased Cx43 in WM astrocytes was compensated for by an up-regulation of Cx30 (Fig. 6). Our results show that, whereas Cx30 clearly labels gray matter regions (Fig. 6a), there is only little Cx30 immunolabeling in WM (Fig. 6b), mostly in regions adjacent to the gray matter. There was no increase in Cx30 immunoreactivity in regions infiltrated with monocytes in EAE (Fig. 6c). This finding is in stark contrast to findings for Cx43, which prominently labels similar WM regions in control animals (Fig. 1a) and shows a dramatic loss of reactivity in inflammation (Figs. 1b,c, 2, bottom).
Fig. 6.
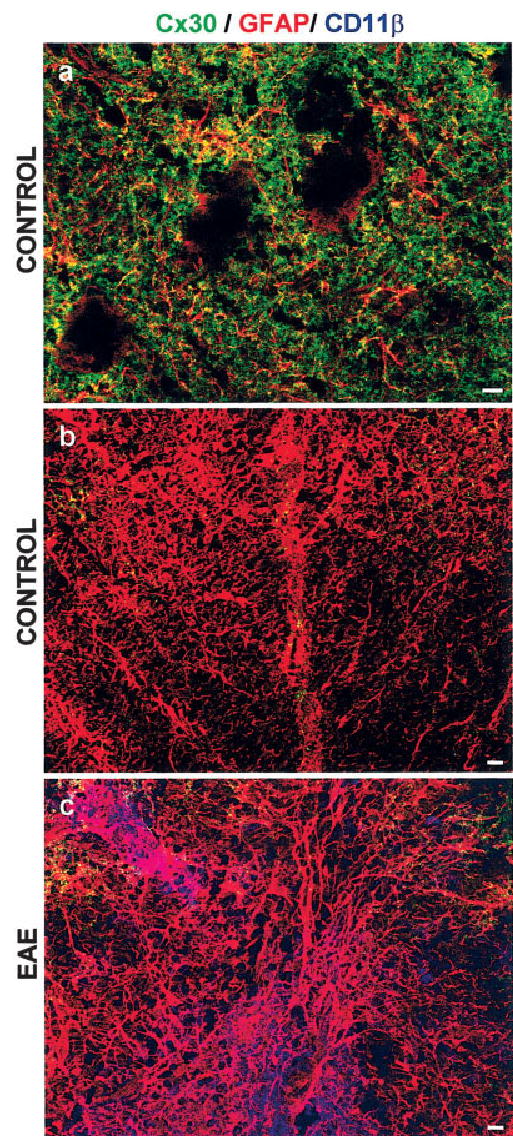
Cx30 immunoreactivity is not enhanced in inflamed white matter. Confocal microscopic images depicting transverse lumbar spinal cord sections from controls (a,b) and animals with EAE (c) labeled for Cx30 (green), GFAP (red), and CD11β (blue). Gray matter (a) is well labeled for Cx30, which overlaps (appearing yellow) with GFAP. Rounded voids most likely represent motor neurons. There is only little Cx30 immunoreactivity in white matter (b), especially adjacent to gray matter (top edges of the images). No infiltrating cells are apparent as evidenced by the absence of CD11β labeling. In EAE, Cx30 labeling is not enhanced in inflamed monocyte infiltrated white matter (c). Scale bars = 10 μm.
DISCUSSION
Our data show that, in EAE, an established model of MS (Juhler, 1988; Glabinski et al., 1997; Steinman, 2001), the expression of Cx43, the main gap junction protein of astrocytes (Dermietzel et al. 2000; Nagy and Rash, 2000, 2003), is significantly reduced. In EAE, astrocytes are activated and display major changes in shape and size, especially around inflammatory cuffs (Smith et al., 1983, 1984; Juhler, 1988; Eng et al., 1989; D’amelio et al., 1990). Given that astrocytic gap junctions have been implicated in control of the extracellular ionic concentration, in metabolite transfer, and in cell volume regulation (Spray, 1996; Rouach et al., 2002a), it is possible that changes in Cx43 expression in the inflammatory regions in EAE contribute to the reported changes in astrocytic phenotype.
Our immunohistochemical data show that the decrease in Cx43 occurs at distinct regions of the WM. In the lumbar spinal cord sections that we processed, the decrease in Cx43 immunoreactivity was located at, and closely abutting, regions heavily infiltrated with monocytes, whereas other regions appeared normal. Individual monocytes were scattered throughout the tissue but without visibly affecting Cx43 labeling.
The correlation between Cx43 disappearance and the presence of monocyte clusters suggests a cumulative localized effect. A localized effect may be mediated by a diffusible factor. In this regard, John et al. (1999) showed that the proinflammatory cytokine IL-1 down-regulated Cx43 in cultured human fetal astrocytes. Interestingly, IL-1 is a major proinflammatory cytokine found within cellular infiltrates in EAE (Bauer et al., 1993; Rajan et al., 1998). Another means for inflammatory cells to mediate localized effects is via direct cellular contact. Indeed, Rouach et al. (2002b) reported that cultured astrocytes exposed to brain macrophages reacted with a decrease in Cx43 protein only when there was a direct contact between these two cell types. The authors suggested that membrane-associated factors likely mediated the decrease in astrocytic Cx43. Whether diffusible or membrane-associated factors play a role in the EAE-induced localized Cx43 changes observed in this study is still to be determined.
Decreased Cx43 immunohistochemical labeling has been previously reported to occur as a result of epitope masking in rat brains after in vivo treatment with the excitatory amino acids kainate and N-methyl-D-aspartate (NMDA; Ochalski et al., 1995). Although both of these glutamatergic pathways are operational in EAE pathology (Bolton and Paul, 1997; Paul et al., 1997; Pitt et al., 2000), our Western blot analyses indicated that the reduction in spinal cord Cx43 immunoreactivity in EAE was not due to epitope masking.
For MS and EAE, cDNA microarray technology has been aiding research by focusing on certain pathological pathways (Steinman, 2001). Several studies of EAE and MS tissue have used microarray techniques (Ibrahim et al., 2001; Whitney et al., 2001; Mix et al., 2002; Lock et al., 2002; Matejuk et al., 2003), but we are the first to report a significant decrease in Cx43 RNA level in spinal cords from mice with EAE compared with controls. In fact, Cx43 was one of the most affected genes in its category (JAE).
We expected, based on our RNA data (almost threefold reduction in Cx43), a larger decrease in protein level than the 38% decrease found in Western blotting. However, RNA levels are not necessarily indicative of protein levels; other cellular functions such as translation and degradation are also important determinants of steady-state protein expression. These considerations were well described by Rogojina et al. (2003) when their study using cDNA microarrays comparing two retinal pigment epithelium cell lines showed discrepancies between Cx43 RNA and protein levels. In correlation, it has been shown by Eng et al. (1989) that, in EAE, morphological changes in astrocytes in inflamed regions lead to exposure of antigenic sites. Therefore, it is all the more striking that we actually see a decreased Cx43 immunoreactivity in inflamed regions and that this decrease is also accompanied by lower Cx43 levels in Western blotting as well as in the DNA microarray. This suggests that the trend toward decreased Cx43 expression is predominant in the EAE tissue. Theoretically, both increased and decreased Cx43 expression can occur at different regions of the spinal cord. Nevertheless, even if Cx43 expression is not uniform in all regions, it has no bearing on the fact that distinct regions clearly defined by cellular infiltration are showing dramatic loss of Cx43 (but not GFAP) immunolabeling compared with control. Because three different methods encompassing RNA and protein expression determination show a decrease in Cx43, it is highly likely that down-regulation of its expression in EAE, most likely in inflamed lesions, does occur.
The decreased Cx43 expression in inflamed regions is not compensated for by the other major astrocytic gap junction protein Cx30, so the consequences may be substantial. However, whether down-regulation of Cx43 in EAE is detrimental or beneficial is still to be determined. Studies in various models of brain pathology show that gap junction inhibition can be protective or detrimental in different experimental settings (Rawanduzy et al., 1997; Siushansian et al., 2001; Rami et al., 2001; Lin et al., 2002; Frantseva et al., 2002a,b). Although in EAE down-regulation and disconnection from the syncytium can minimize the spread of damage, as shown for brain infarct (Rawanduzy et al., 1997), decreased CNS tissue integrity may ease peripheral monocyte access into the CNS parenchyma. In addition, decreased Cx43 may stimulate astrocytic proliferation (Tabernero et al., 2001). Nonetheless, inhibition of reactive astrocytes was found to be detrimental in spinal cord injury (Faulkner et al., 2004). Astrocytic activation, thus, appeared to have limited the extent of tissue damage and to have promoted regeneration. Therefore, if Cx43 down-regulation is a part of reactive astrocytic phenotype, and given that the latter was proposed to be beneficial to damaged spinal cords, it is possible that reduced astrocytic Cx43 expression in heavily infiltrated regions in EAE may be important in preventing or attenuating the spread of inflammatory damage.
In summary, this study provides evidence that the expression of the major astrocytic gap junction protein Cx43 is reduced in inflamed spinal cord WM in EAE, an animal model of MS. Reduced Cx43 immunoreactivity was extensive in areas of inflammation and immune cell infiltration, marked by the presence of monocytes. This pattern of decreased Cx43 protein suggests that cell contact through membrane-bound factors or a diffusible factor with limited range (i.e., IL-1β) might participate in reducing astrocytic connectivity in EAE. A better understanding of events leading to the change in Cx43 might allow modulation of the pathological process of lesion formation in conditions of inflammatory demyelination, such as MS.
Acknowledgments
This work was supported by NIH grants NS-41023 to E.S., NS-41282 and MH-65495 to D.C.S., and NS-41056 to P.W. and National Multiple Sclerosis Society grants PP1019 to E.S. and PP0988 to P.W.
References
- Adams RD, Kubik CS. The morbid anatomy of the demyelinative disease. Am J Med. 1952;12:510–546. doi: 10.1016/0002-9343(52)90234-9. [DOI] [PubMed] [Google Scholar]
- Alexander DB, Goldberg GS. Transfer of biologically important molecules between cells through gap junction channels. Curr Med Chem. 2003;10:2045–2058. doi: 10.2174/0929867033456927. [DOI] [PubMed] [Google Scholar]
- Bauer J, Berkenbosch F, Van Dam AM, Dijkstra CD. Demonstration of interleukin-1 beta in Lewis rat brain during experimental allergic encephalomyelitis by immunocytochemistry at the light and ultra-structural level. J Neuroimmunol. 1993;48:13–21. doi: 10.1016/0165-5728(93)90053-2. [DOI] [PubMed] [Google Scholar]
- Bauer J, Huitinga I, Zhao W, Lassmann H, Hickey WF, Dijkestra CD. The role of macrophages, perivascular cells, and microglial cells in the pathogenesis of experimental autoimmune encephalomyelitis. Glia. 1995;15:437–446. doi: 10.1002/glia.440150407. [DOI] [PubMed] [Google Scholar]
- Benn T, Halfpenny C, Scolding N. Glial cells as targets for cytotoxic immune mediators. Glia. 2001;36:200–211. doi: 10.1002/glia.1109. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Nicholson L. The role of cytokines in experimental autoimmune encephalomyelitis. Arch Immunol Ther Exp. 2000;48:389–398. [PubMed] [Google Scholar]
- Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, Bruck W. Acute axonal injury in multiple sclerosis; correlation with demyelination and inflammation. Brain. 2000;123:1174–1183. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Kinkel RP, Kidd G, Rudick RA, Trapp BD. Axonal loss in normal-appearing white matter in a patient with acute MS. Neurology. 2001;57:1248–1252. doi: 10.1212/wnl.57.7.1248. [DOI] [PubMed] [Google Scholar]
- Bolton C, Paul C. MK-801 limits neurovascular dysfunction during experimental allergic encephalomyelitis. J Pharmacol Exp Ther. 1997;282:397–402. [PubMed] [Google Scholar]
- Brand-Schieber E, Werner P. AMPA/kainate receptors in mouse spinal cord cell-specific display of receptor subunits by oligodendrocytes and astrocytes and at the nodes of Ranvier. Glia. 2003;42:12–24. doi: 10.1002/glia.10136. [DOI] [PubMed] [Google Scholar]
- Brand-Schieber E, Werner P, Iacobas DA, Iacobas S, Beelitz M, Lowery SL, Spray DC, Scemes E. Cx43, the major gap junction protein of astrocytes, is down-regulated in an animal model of multiple sclerosis. J Neurochem. 2003;85(Suppl 1):7. doi: 10.1002/jnr.20474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, McFarlin DE. Relapsing experimental allergic encephalomyelitis in the SJL/J mouse. Lab Invest. 1981;45:278–284. [PubMed] [Google Scholar]
- Claudio L, Kress Y, Factor J, Brosnan CF. Mechanisms of edema formation in experimental autoimmune encephalomyelitis. The contribution of inflammatory cells. Am J Pathol. 1990;137:1033–1045. [PMC free article] [PubMed] [Google Scholar]
- D’amelio FE, Smith ME, Eng LF. Sequence of tissue responses in the early stages of experimental allergic encephalomyelitis (EAE): immunohistochemical, light microscopic, and ultrastructural observations in the spinal cord. Glia. 1990;3:229–240. doi: 10.1002/glia.440030402. [DOI] [PubMed] [Google Scholar]
- Deloire-Grassin MSA, Brochet B, Petry KP. Severe protracted and relapsing encephalomyelitis autoimmune experimental: a model for axonal pathology in multiple sclerosis. Rev Neurol. 2000;(Suppl 3):3S70. [Google Scholar]
- Dermietzel R, Hofstadter F. Gap junctions in health and disease. Virchows Arch. 1998;432:177–186. doi: 10.1007/s004280050153. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Gao Y, Scemes E, Vieira D, Urban M, Kremer M, Bennett MV, Spray DC. Connexin43 null mice reveal that astrocytes express multiple connexins. Brain Res Brain Res Rev. 2000;32:45–56. doi: 10.1016/s0165-0173(99)00067-3. [DOI] [PubMed] [Google Scholar]
- Eng LF, D’amelio FE, Smith ME. Dissociation of GFAP intermediate filaments in EAE: observations in the lumbar spinal cord. Glia. 1989;2:308–317. doi: 10.1002/glia.440020504. [DOI] [PubMed] [Google Scholar]
- Eng LF, Yu AC, Lee YL. Astrocytic response to injury. Prog Brain Res. 1992;94:353–365. doi: 10.1016/s0079-6123(08)61764-1. [DOI] [PubMed] [Google Scholar]
- Ewing C, Bernard CCA. Insight into the aetiology and pathogenesis of multiple sclerosis. Immunol Cell Biol. 1998;76:47–54. doi: 10.1046/j.1440-1711.1998.00718.x. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120:393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- Fernandez-Cobo M, Gingalewski C, De Maio A. Expression of the connexin 43 gene is increased in the kidneys and the lungs of rats injected with bacterial lipopolysaccharide. Shock. 1998;10:97–102. doi: 10.1097/00024382-199808000-00003. [DOI] [PubMed] [Google Scholar]
- Fernandez-Cobo M, Gingalewski C, Drujan D, De Maio A. Downregulation of connexin 43 gene expression in rat heart during inflammation. The role of tumour necrosis factor. Cytokine. 1999;11:216–224. doi: 10.1006/cyto.1998.0422. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci. 1999;19:8182–8198. doi: 10.1523/JNEUROSCI.19-19-08182.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantseva MV, Kokarovtseva L, Naus CG, Carlen PL, MacFabe D, Perez Velazquez JL. Specific gap junctions enhance the neuronal vulnerability to brain traumatic injury. J Neurosci. 2002a;22:644–653. doi: 10.1523/JNEUROSCI.22-03-00644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantseva MV, Kokarovtseva L, Perez Velazquez JL. Ischemia-induced brain damage depends on specific gap-junctional coupling. J Cereb Blood Flow Metab. 2002b;22:453–462. doi: 10.1097/00004647-200204000-00009. [DOI] [PubMed] [Google Scholar]
- Glabinski AR, Tani M, Tuohy VK, Ransohoff RM. Murine experimental autoimmune encephalomyelitis: a model of immune-mediated inflammation and multiple sclerosis. Methods Enzymol. 1997;288:182–190. doi: 10.1016/s0076-6879(97)88015-9. [DOI] [PubMed] [Google Scholar]
- Hohlfeld R, Wekerle H. Immunological update on multiple sclerosis. Curr Opin Neurol. 2001;14:299–304. doi: 10.1097/00019052-200106000-00006. [DOI] [PubMed] [Google Scholar]
- Honegger CG, Langemann H. 1989. Some pathophysiological aspects of experimental autoimmune encephalomyelitis. Schweiz Rundschau Med (PRAXIS) 78. [PubMed]
- Iacobas DA, Urban-Maldonado M, Iacobas S, Scemes E, Spray DC. Array analysis of gene expression in connexin-43 null astrocytes. Physiol Genomics. 2003;15:177–190. doi: 10.1152/physiolgenomics.00062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobas DA, Iacobas S, Li WEI, Zoidl G, Dermietzel R, Spray DC. Genes controlling multiple functional pathways are transcriptionally regulated in Connexin43 null mouse heart. Physiol Genomics. 2005a;20:211–223. doi: 10.1152/physiolgenomics.00229.2003. [DOI] [PubMed] [Google Scholar]
- Iacobas DA, Iacobas S, Urban-Maldonado M, Spray DC. 2005b. Sensitivity of the brain transcriptome to connexin ablation. Biochim Biophys Acta doi: 10.1016/j.bbamem.2004.12.002. [DOI] [PubMed]
- Ibrahim SM, Mix E, Bottcher T, Koczan D, Gold R, Rolfs A, Thiesen HJ. Gene expression profiling of the nervous system in murine experimental autoimmune encephalomyelitis. Brain. 2001;124:1927–1938. doi: 10.1093/brain/124.10.1927. [DOI] [PubMed] [Google Scholar]
- Imrich H, Harzer K. On the role of peripheral macrophages during active experimental allergic encephalomyelitis (EAE) J Neural Transm. 2001;108:379–395. doi: 10.1007/s007020170060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman D, Guth L, Steward O. Genetic influences on secondary degeneration and wound healing following spinal cord injury in various strains of mice. J Comp Neurol. 2002;451:225–235. doi: 10.1002/cne.10340. [DOI] [PubMed] [Google Scholar]
- John GR, Scemes E, Suadicani SO, Liu JS, Charles PC, Lee SC, Spray DC, Brosnan CF. IL-1beta differentially regulates calcium wave propagation between primary human fetal astrocytes via pathways involving P2 receptors and gap junction channels. Proc Natl Acad Sci U S A. 1999;96:11613–11618. doi: 10.1073/pnas.96.20.11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhler M. Pathophysiological aspects of acute experimental allergic encephalomyelitis. Acta Neurol Scand. 1988;78:1–21. doi: 10.1111/j.1600-0404.1988.tb08016.x. [DOI] [PubMed] [Google Scholar]
- Kornek B, Storch MK, Weissert R, Wallstroem E, Andreas S, Olsson T, Lassmann H, et al. Multiple sclerosis and chronic autoimmune encephalomyelitis. A comparative study of axonal injury in active, inactive and remyelinated lesions. Am J Pathol. 2000;157:267–276. doi: 10.1016/S0002-9440(10)64537-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landete L, Vallet M, Vilela C, Millet E, Coret F, Casanova B. Axonal loss in early multiple sclerosis. Study of the amplitude of evoked potentials and central conduction time. Rev Neurol. 2000;(Suppl 3):3S112. [Google Scholar]
- Levine S, Simon J, Wenk EJ. Edema of the spinal cord in experimental allergic encephalomyelitis. Proc Soc Exp Biol Med. 1966;123:539–541. doi: 10.3181/00379727-123-31536. [DOI] [PubMed] [Google Scholar]
- Lin JH, Takano T, Cotrina ML, Arcuino G, Kang J, Liu S, Gao Q, Jiang L, Li F, Lichtenberg-Frate H, Haubrich S, Willecke K, Goldman SA, Nedergaard M. Connexin 43 enhances the adhesivity and mediates the invasion of malignant glioma cells. J Neurosci. 2002;22:4302–4311. doi: 10.1523/JNEUROSCI.22-11-04302.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- Ma M, Basso DM, Walters P, Stokes BT, Jakeman LB. Behavioral and histological outcomes following graded spinal cord contusion injury in the C57Bl/6 mouse. Exp Neurol. 2001;169:239–254. doi: 10.1006/exnr.2001.7679. [DOI] [PubMed] [Google Scholar]
- Matejuk A, Hopke C, Dwyer J, Subramanian S, Jones RE, Bourdette DN, Vandenbark AA, Offner H. CNS gene expression pattern associated with spontaneous experimental autoimmune encephalomyelitis. J Neurosci Res. 2003;73:667–678. doi: 10.1002/jnr.10689. [DOI] [PubMed] [Google Scholar]
- Miller SD, Karpus WJ. The immunopathogenesis and regulation of T-cell-mediated demyelinating diseases. Immunol Today. 1994;15:356–361. doi: 10.1016/0167-5699(94)90173-2. [DOI] [PubMed] [Google Scholar]
- Mix E, Pahnke J, Ibrahim SM. Gene-expression profiling of experimental autoimmune encephalomyelitis. Neurochem Res. 2002;27:1157–1163. doi: 10.1023/a:1020925425780. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Patel D, Ochalski PA, Stelmack GL. Connexin30 in rodent, cat and human brain: selective expression in gray matter astrocytes, co-localization with connexin43 at gap junctions and late developmental appearance. Neuroscience. 1999;88:447–468. doi: 10.1016/s0306-4522(98)00191-2. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Li WE. A brain slice model for in vitro analyses of astrocytic gap junction and connexin43 regulation: actions of ischemia, glutamate and elevated potassium. Eur J Neurosci. 2000;12:4567–4572. [PubMed] [Google Scholar]
- Nagy JI, Rash JE. Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res Brain Res Rev. 2000;32:29–44. doi: 10.1016/s0165-0173(99)00066-1. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Rash JE. Astrocyte and oligodendrocyte connexins of the glial syncytium in relation to astrocyte anatomical domains and spatial buffering. Cell Commun Adhes. 2003;10:401–406. doi: 10.1080/15419060390263191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Li W, Hertzberg EL, Marotta CA. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res. 1996;717:173–178. doi: 10.1016/0006-8993(95)01526-4. [DOI] [PubMed] [Google Scholar]
- Nakase T, Fushiki S, Sohl G, Theis M, Willecke K, Naus CC. Neuroprotective role of astrocytic gap junctions in ischemic stroke. Cell Commun Adhes. 2003;10:413–417. doi: 10.1080/cac.10.4-6.413.417. [DOI] [PubMed] [Google Scholar]
- Ochalski PA, Sawchuk MA, Hertzberg EL, Nagy JI. Astrocytic gap junction removal, connexin43 redistribution, and epitope masking at excitatory amino acid lesion sites in rat brain. Glia. 1995;14:279–294. doi: 10.1002/glia.440140405. [DOI] [PubMed] [Google Scholar]
- Owens T, Hassan-Zahraee M, Krakowski M. Immune cell entry to the CNS-focus for immunoregulation of EAE. Res Immunol. 1998;149:781–789. doi: 10.1016/s0923-2494(99)80005-4. [DOI] [PubMed] [Google Scholar]
- Paterson PY. Experimental allergic encephalomyelitis: role of fibrin deposition in immunopathogenesis of inflammation in rats. Fed Proc. 1976;35:2428–2434. [PubMed] [Google Scholar]
- Paul C, Scott GS, Barbour MJ, Seiler N, Bolton C. N-methyl-D-aspartate receptor-mediated events contribute to neurovascular breakdown during experimental allergic encephalomyelitis. Biochem Soc Trans. 1997;25:167S. doi: 10.1042/bst025167s. [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- Rafalowska J, Krajewski S, Dolinska E, Dziewulska D. Does damage of perivascular astrocytes in multiple sclerosis plaques participate in blood–brain barrier permeability? Neuropatol Pol. 1992;30:73–80. [PubMed] [Google Scholar]
- Raine CS, Mokhtarian F, McFarlin DE. Adoptively transferred chronic relapsing experimental autoimmune encephalomyelitis in the mouse. Lab Invest. 1984;51:534–546. [PubMed] [Google Scholar]
- Rajan AJ, Klein JD, Brosnan CF. The effect of gammadelta T cell depletion on cytokine gene expression in experimental allergic encephalomyelitis. J Immunol. 1998;160:5955–5962. [PubMed] [Google Scholar]
- Rami A, Volkmann T, Winckler J. Effective reduction of neuronal death by inhibiting gap junctional intercellular communication in a rodent model of global transient cerebral ischemia. Exp Neurol. 2001;170:297–304. doi: 10.1006/exnr.2001.7712. [DOI] [PubMed] [Google Scholar]
- Rash JE, Yasumura T, Davidson KG, Furman CS, Dudek FE, Nagy JI. Identification of cells expressing Cx43, Cx30, Cx26, Cx32 and Cx36 in gap junctions of rat brain and spinal cord. Cell Commun Adhes. 2001;8:315–320. doi: 10.3109/15419060109080745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash JE, Yasumura T, Dudek FE, Nagy JI. Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J Neurosci. 2001;21:1983–2000. doi: 10.1523/JNEUROSCI.21-06-01983.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawanduzy A, Hansen A, Hansen TW, Nedergaard M. Effective reduction of infarct volume by gap junction blockade in a rodent model of stroke. J Neurosurg. 1997;87:916–920. doi: 10.3171/jns.1997.87.6.0916. [DOI] [PubMed] [Google Scholar]
- Rogojina AT, Orr WE, Song BK, Geisert EE., Jr Comparing the use of Affymetrix to spotted oligonucleotide microarrays using two retinal pigment epithelium cell lines. Mol Vis. 2003;9:482–496. [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Avignone E, Meme W, Koulakoff A, Venance L, Blomstrand F, Giaume C. Gap junctions and connexin expression in the normal and pathological central nervous system. Biol Cell. 2002a;94:457–475. doi: 10.1016/s0248-4900(02)00016-3. [DOI] [PubMed] [Google Scholar]
- Rouach N, Calvo CF, Glowinski J, Giaume C. Brain macrophages inhibit gap junctional communication and downregulate connexin 43 expression in cultured astrocytes. Eur J Neurosci. 2002b;15:403–407. doi: 10.1046/j.0953-816x.2001.01868.x. [DOI] [PubMed] [Google Scholar]
- Simmons RD, Bernard CCA, Singer G, Carnegie PR. Experimental autoimmune encephalomyelitis. An anatomically-based explanation of clinical progression in rodents. J Neuroimmunol. 1982;3:307–318. doi: 10.1016/0165-5728(82)90034-0. [DOI] [PubMed] [Google Scholar]
- Siushansian R, Bechberger JF, Cechetto DF, Hachinski VC, Naus CC. Connexin43 null mutation increases infarct size after stroke. J Comp Neurol. 2001;440:387–394. doi: 10.1002/cne.1392. [DOI] [PubMed] [Google Scholar]
- Smith ME, Somera FP, Eng LF. Immunocytochemical staining for glial fibrillary acidic protein and the metabolism of cytoskeletal proteins in experimental allergic encephalomyelitis. Brain Res. 1983;264:241–253. doi: 10.1016/0006-8993(83)90822-3. [DOI] [PubMed] [Google Scholar]
- Smith ME, Somera FP, Swanson K, Eng LF. 1984. Glial fibrillary acidic protein in acute and chronic relapsing experimental allergic encephalomyelitis (EAE). In: Experimental allergic encephalomyelitis: a useful model for multiple sclerosis. New York: Alan R. Liss, Inc. p 139–144. [PubMed]
- Spray DC. Molecular physiology of gap junction channels. Clin Exp Pharmacol Physiol. 1996;23:1038–1040. doi: 10.1111/j.1440-1681.1996.tb01165.x. [DOI] [PubMed] [Google Scholar]
- Steinman L. Gene microarrays and experimental demyelinating disease: a tool to enhance serendipity. Brain. 2001;124:1897–1899. doi: 10.1093/brain/124.10.1897. [DOI] [PubMed] [Google Scholar]
- Suadicani SO, Pina-Benabou MH, Urban-Maldonado M, Spray DC, Scemes E. Acute downregulation of Cx43 alters P2Y receptor expression levels in mouse spinal cord astrocytes. Glia. 2003;42:160–171. doi: 10.1002/glia.10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabernero A, Jimenez C, Velasco A, Giaume C, Medina JM. The enhancement of glucose uptake caused by the collapse of gap junction communication is due to an increase in astrocyte proliferation. J Neurochem. 2001;78:890–898. doi: 10.1046/j.1471-4159.2001.00476.x. [DOI] [PubMed] [Google Scholar]
- Theriault E, Frankenstein UN, Hertzberg EL, Nagy JI. Connexin43 and astrocytic gap junctions in the rat spinal cord after acute compression injury. J Comp Neurol. 1997;382:199–214. [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. 2001;50:169–180. doi: 10.1002/ana.1077. [DOI] [PubMed] [Google Scholar]
- Whitaker JN, Mitchell GW. 1997. Clinical features of multiple sclerosis. In: Raine CS, McFarland HF, Tourtellotte WW, editors. Multiple sclerosis: clinical and pathogenic basis. London: Chapman and Hall. p 3–20.
- Whitney LW, Ludwin SK, McFarland HF, Biddison WE. Microarray analysis of gene expression in multiple sclerosis and EAE identifies 5-lipoxygenase as a component of inflammatory lesions. J Neuroimmunol. 2001;121:40–48. doi: 10.1016/s0165-5728(01)00438-6. [DOI] [PubMed] [Google Scholar]