

Abstract
Vitelliform macular dystrophy (VMD/Best disease; MIM*153700) is an early-onset autosomal dominant disorder in which accumulation of lipofuscin-like material within and beneath the retinal pigment epithelium is associated with a progressive loss of central vision. Bestrophin, the protein product of the VMD gene, has four predicted transmembrane domains. There are multiple bestrophin homologues in the human, Drosophila, and Caenorhabditis elegans genomes, but no function has previously been ascribed to these proteins, and they show no detectable homology to other proteins of known function. Using heterologous expression, we show here that human, Drosophila, and C. elegans bestrophins form oligomeric chloride channels, and that human bestrophin is sensitive to intracellular calcium. Each of 15 missense mutations asscociated with VMD greatly reduces or abolishes the membrane current. Four of these mutant bestrophins were coexpressed with the wild type and each dominantly inhibited the wild-type membrane current, consistent with the dominant nature of the disease. These experiments establish the existence of a new chloride channel family and VMD as a channelopathy.
In humans and other higher primates, high-acuity vision is subserved by the central (or macular) region of the retina. Macular degeneration caused by progressive changes in the retina and/or the underlying retinal pigment epithelium (RPE) produces a corresponding loss of high-acuity vision. At present, macular degeneration is the most common cause of vision loss in older individuals in industrialized countries. Although the exact pathogenic mechanisms responsible for age-related macular degeneration (AMD) remain poorly defined, current evidence suggests that progressive dysfunction of the RPE and an associated accumulation of debris within and beneath it are likely to be important contributing factors (1).
One fruitful approach to defining pathogenic mechanisms in complex disorders involves an analysis of phenotypically similar monogenic traits. In the field of macular degeneration, a few such monogenic disorders have been identified and targeted for intensive study both because of their intrinsic importance and because they may shed light on the more common age-related forms of this disease (2). These monogenic disorders include Stargardt disease (both recessive and dominant types), Sorsby's fundus dystrophy, dominant familial drusen, and vitelliform macular dystrophy (VMD, Best disease). VMD, the subject of the present work, is characterized by a distinctive accumulation of lipofuscin-like material within and beneath the RPE (3, 4).
The VMD gene was isolated by positional cloning several years ago (5, 6), but the function of its product, bestrophin, has been enigmatic. Bestrophin is homologous to at least 3 other proteins coded within the human genome, 4 in the Drosophila genome, and 24 in the Caenorhabditis elegans genome, but no function has yet been ascribed to any members of this family, and they show no detectable homology to any protein of known function. In considering the possible function(s) of bestrophin, three observations suggested to us that it may be an oligomeric Cl channel: (i) VMD is associated with a reduction in the slow light peak of the electrooculogram (7, 8), a component thought to reflect an increase in Cl conductance across the basolateral membrane of the RPE (9, 10); (ii) the amino acid sequence of bestrophin suggests a multispan transmembrane protein; and (iii) among the 48 disease-associated bestrophin mutations reported to date, all are dominant and almost all are missense mutations that cluster in or near the predicted transmembrane domains (5, 6, 11–14). Recently, Marmorstein et al. (15) reported that bestrophin is localized to the basolateral membrane of the RPE, strengthening our speculation that bestrophin may be involved in the conductance relevant to the aberrant electrooculogram. Taken together, these observations suggest a model in which mutant and wild-type bestrophins coassemble into a multisubunit Cl channel with altered properties, resulting in a decrement in Cl conductance across the basolateral membrane of the RPE.
Materials and Methods
Cloning of Bestrophins and Construction of Expression Plasmids.
hBest1 was cloned from human ocular cDNA by PCR. dmBest1 was obtained from expressed sequence tag clone LD22528. ceBest1 was obtained by reverse transcription–PCR using total RNA from an unsynchronized C. elegans population. A partial-length cDNA clone coding for hBest2 was provided by H. Hata of the University of Tokyo, and the full-length hBest2 coding region was assembled by adding the first coding exon, obtained from human genomic DNA by PCR. For reference purposes, the first 12 residues in each bestrophin are as follows: hBest1, MTITYTSQVANA; hBest2, MTVTYTARVANA; dmBest1, MTITYTGEVATC; and ceBest1, MTVNYNLDVSSA. A NotI site was engineered immediately after the last residue of each bestrophin clone, and a Rim-tag (NETYDLPLHPRTAGA, recognized by mAb Rim3F4; ref. 16) for all bestrophins, a 6× myc-tag for hBest1, and a 3× myc-tag for hBest2, dmBest1, and ceBest1 were inserted at the NotI site. Mutagenesis of hBest1 was performed by PCR. For expression in 293 cells, bestrophin cDNAs were inserted into the pRK5 vector.
Whole-Cell Recordings.
HEK 293 cells were transfected with a bestrophin plasmid (myc- or Rim-tagged) mixed with an EGFP plasmid at a 4:1 ratio (see Figs. 1, 2, and 6) or a 10:1 ratio (see Fig. 7) by using Fugene-6 (Roche Molecular Biochemicals) at 2 μg of DNA per 3.5-cm plate. The EGFP plasmid alone (2 μg) was used as a transfection control. At ≈24 h after transfection, the cells were detached from the plates with 5 mM EDTA and 1× PBS, and were replated at lower density. At ≈60 h after transfection, whole-cell recordings were performed at room temperature (22–25°C) on single, isolated green cells identified under an inverted fluorescence microscope. Isolated cells were chosen because electrical coupling between adjacent cells would otherwise lead to space-clamp problems and also incomplete dialysis of the coupled cell interior with the pipette solution. Standard extracellular solution contained (in millimolar): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 Na-Hepes (pH 7.4). Standard pipette solution contained (in millimolar): 148 CsCl, 2 MgCl2, 0.5 CaCl2, 2 EGTA, and 10 Na-Hepes (pH 7.3), giving free [Ca2+] ≈40 nM. CsCl was chosen to block endogenous K+ currents. Equimolar N-methyl-D-glucamine was used for replacing Na+, and equimolar sodium gluconate was used for replacing Cl−. For the selectivity experiments with inorganic anions, 140 mM NaCl in the extracellular solution was replaced with 140 mM NaNO3, NaI, or NaBr (Fig. 2a Center); equimolar substitutions with lower NaNO3 were also used (Fig. 2a Right). Liquid junction potentials of typically several millivolts were measured with a 3 M KCl electrode. These were corrected for in the ion-substitution experiments only.
Figure 1.

Whole-cell currents from 293 cells transiently transfected with different bestrophin cDNAs. (a) Averaged and normalized current–voltage (I–V) relations for hBest1 (n = 16 cells), ceBest1 (n = 7), dmBest1 (n = 7), and hBest2 (n = 7); error bars indicate standard deviation (SD). For each bestrophin, the I–V curves for each cell were normalized to a value of −1.0 at −150 mV before averaging was done. Recordings were performed with standard extracellular and pipette solutions. Insets show sample current traces produced by 320-ms voltage steps from a holding potential of −50 mV to voltages between −150 and +80 mV in 10-mV increments. Current amplitudes at the end of the voltage steps were used for I–V plots. Open symbols in a Left show the averaged I–V relation from 11 control EGFP-transfected cells, after scaling it at −150 mV in proportion to the average hBest1 current; the SDs for the control cells are too small to be discernible. (b) Current amplitudes ± SD at −150 mV for the cells in a. The wide variation in the currents recorded from individual cells presumably reflects variation in plasmid uptake and level of bestrophin production in transiently transfected cells.
Figure 2.

Ion permeability and DIDS sensitivity of bestrophin currents. (a Left and Center) Dependence of the whole-cell current reversal potential recorded from hBest1-transfected cells on extracellular Na+ and Cl− concentrations. Na+ was replaced by equimolar N-methyl-d-glucamine (2 cells, each represented by a different symbol), and Cl− was replaced by gluconate (mean ± SD from 5 cells). (Right) Permeability ratio of NO to Cl− for 293 cells transfected with hBest1 (4 cells) and hBest2 (3 cells) determined by replacing Cl− with equimolar NO
to Cl− for 293 cells transfected with hBest1 (4 cells) and hBest2 (3 cells) determined by replacing Cl− with equimolar NO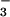 . The curves are derived from the Goldman–Hodgkin–Katz equation with PNO3/PCl ratios of 2.7 and 5.8, respectively. Initial absolute currents in standard extracellular solution and at +80 mV were 293, 307, 366, 1562, and 2320 pA for the cells in Center, and 155, 251, 545, 904 pA (hBest1), and 218, 238, 519 pA (hBest2) for the cells in Right. (b) Effect of the Cl channel blocker DIDS on the whole-cell current of a hBest1-transfected cell. DIDS (0.5 mM) was bath applied. In four experiments, 89% ± 1% (mean ± SD) of the current at +80 mV was reversibly suppressed.
. The curves are derived from the Goldman–Hodgkin–Katz equation with PNO3/PCl ratios of 2.7 and 5.8, respectively. Initial absolute currents in standard extracellular solution and at +80 mV were 293, 307, 366, 1562, and 2320 pA for the cells in Center, and 155, 251, 545, 904 pA (hBest1), and 218, 238, 519 pA (hBest2) for the cells in Right. (b) Effect of the Cl channel blocker DIDS on the whole-cell current of a hBest1-transfected cell. DIDS (0.5 mM) was bath applied. In four experiments, 89% ± 1% (mean ± SD) of the current at +80 mV was reversibly suppressed.
Figure 6.

Calcium regulation of hBest1 current. (a) Under whole-cell recording, a hBest1-transfected cell was dialyzed with a caged-Ca2+ solution containing 4 mM NPEGTA/0.4 mM Ca2+ (free [Ca2+] ≈10 nM). Two light flashes, each of 2-s duration, were applied at the times indicated. From a holding potential of −50 mV, voltage-ramp pulses from −100 to +80 mV and then back to −100 mV were applied before and after each flash. (b) Conductance increase induced by the photorelease of Ca2+. The I–V relations marked “1” and “2” were replotted from the ramp pulses 1 and 2 in a. (c--e) Control experiments showing that the photo-induced current was caused by a rise in intracellular Ca2+ concentration. (c) A pipette solution containing BAPTA in addition to NPEGTA was used instead, to suppress the photo-released [Ca2+] in a hBest1-transfected cell. (d) Instead of NPEGTA, a photo-insensitive chelator, EGTA, was loaded into a hBest1-transfected cell. (e) NPEGTA was loaded into a control EGFP-transfected cell; otherwise as in a. In these control experiments, current amplitudes were almost identical before and after the flash marked “F”. (f) Collected data from the experiments in a–e. In each case, the difference in current amplitude at +80 mV before and after the first flash is plotted as mean and SD. The number below each bar indicates the number of cells tested.
Figure 7.

Currents in 293 cells transfected with hBest1 missense mutants responsible for VMD. (a) Locations of 15 disease-associated mutations in the hBest1 model of Bakall et al. (11). (b) Immunoblot of Rim3F4-tagged wild-type and 15 mutant hBest1 proteins from transiently transfected cells. “Control” indicates untransfected cells. (c) Whole-cell currents at +80 mV from 293 cells transfected with wild-type (“WT”) or with 11 mutant hBest1 plasmids, or with EGFP expression plasmid only, measured as in Fig. 1a. Dots indicate data from individual cells, and bars represent the mean ± SD. (d–f) Four hBest1 missense mutants responsible for vitelliform macular dystrophy coassemble with wild-type hBest1 and decrease whole-cell current. (d) Coimmunoprecipitation of wild-type hBest1-M together with either wild-type or mutant hBest1-R from cotransfected cells as described for Fig. 3. IP, immunoprecipitate; IB, immunoblot. (e) Whole-cell recordings from transfected 293 cells showing that the four indicated mutant proteins gave smaller currents when transfected alone, and inhibited the wild-type current when cotransfected with wild-type hBest1. For transfection in a 3.5-cm-diameter dish, a total volume of 4 μl containing 0.2 μg of EGFP plasmid and either 2 μg of wild-type or mutant hBest1 plasmid (for single transfections) or 1 μg of each plasmid (for cotransfections) was mixed with 5 μl of Fugene 6 reagent. Indicated current amplitude was at +80 mV, measured as in Fig. 1a. (f) Experiment showing that inhibition of whole-cell current in the cotransfection experiments of e did not simply arise from competition between wild-type and mutant at the level of protein or RNA synthesis. Each 3.5-cm-diameter dish received 2 μg of wild-type hBest1 (Left), 1 μg of wild-type + 1 μg R92C mutant (Center), or 1 μg of wild-type + 1 μg Fz8-IgG (Right) plasmid, together with 0.2 μg of EGFP plasmid, in a total volume of 2 μl with 5 μl of Fugene 6 reagent. All data in an individual panel were obtained from transfections performed side by side. Each transfection of mutant alone or mutant plus wild-type produced a set of whole-cell currents that were significantly smaller than the wild-type controls measured in parallel (P < 0.05, two-tailed t test).
For caged-Ca2+ experiments, the cells were gently detached from the plastic dish with a transfer pipette and replated onto a recording chamber, the bottom of which was made of a glass coverslip coated with Con A. In these experiments, the pipette solution contained (in millimolar): 2 MgCl2 and 40 Hepes (adjusted to pH 7.2 with NaOH), plus (a) 121 CsCl, 0.4 CaCl2, and 4 o-nitrophenyl EGTA-K4 (NPEGTA, Molecular Probes); (b) 113 CsCl, 0.58 CaCl2, 4 NPEGTA-K4, and 4 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate (BAPTA)-Na4; or (c) 121 CsCl, 0.21 CaCl2, and 4 EGTA. In all cases, the calculated free [Ca2+] was ≈10 nM (based on WINMAXC software E-posted by C. Patton of Stanford University). For Kd values of unphotolyzed NPEGTA for Ca2+ and Mg2+, 8.0 × 10−8 and 9.0 × 10−3 M, respectively, were used (17). For photo-uncaging, UV light from a Hg arc lamp (RatioArc HBO 100-W, Zeiss) was guided by the epi-illumination system of the inverted microscope (Axiovert 135, Zeiss), passed through a heat filter and a dichroic mirror (cutoff at 395 nm), and focused on the recorded cell by the objective lens (F FLUAR 40×). The duration of the UV flash was controlled by the Attofluor RatioVision system (Atto Instruments, Rockville, MD). The light spot was ca. 25 μm and slightly off-center on the cell to avoid irradiating the pipette.
Sulfhydryl Modification.
After establishment of stable whole-cell recording, 2.5 mM 2-aminoethylmethanethiosulfonate (MTSEA) or 1 mM 2-trimethylammonioethylmethanethiosulfonate (MTSET) as the bromide salt (Toronto Research Chemicals, Toronto, Canada) in standard extracellular solution was added to the recording chamber in place of the standard solution (18). For these experiments, individual preweighed aliquots of reagent were dissolved only immediately before use. All other aspects of the recording arrangement were as described in the legend to Fig. 1.
Purification of Bestrophins.
Twenty-four hours after transfection of a mixture of plasmids coding for myc- and Rim-tagged bestrophins, the culture medium was removed and the cells were incubated in cold 1% Triton X-100/10% glycerol/1× PBS with protease inhibitors (lysis buffer) for 15 min and swirled with a Vortex mixer. Nuclei and insoluble material were removed by centrifugation at 16,000 × g at 4°C for 30 min. The clear supernatant was added to Sepharose beads conjugated to anti-myc mAb, rotated at 4°C for 3 h, and washed five times in lysis buffer at 4°C for 1 h. Proteins bound to the beads were eluted by mixing them with 10 vol of 1× SDS sample buffer supplemented with 7 M urea at room temperature for 10 min, and immediately loaded onto an SDS gel. Immunoblots were probed with biotinylated anti-Rim [prepared by reacting purified anti-Rim IgG with EZ-link Sulfo-NHS-LC-biotin (Pierce)] followed by Extravidin-HRP (Sigma).
For analysis of radiolabeled bestrophin, transfected 293 cells were cultured in [35S]methionine-containing medium for 24 h before harvesting, and then processed with either of two protocols for immunopurification. In one protocol, detergent-solubilized proteins were prepared in 1% dodecyl maltoside/10% glycerol/150 mM NaCl/50 mM Hepes, pH 7.5/1 mM EDTA/5 mM 2-mercaptoethanol/protease inhibitors and preincubated with Sepharose beads conjugated to an irrelevant monoclonal antibody; then myc- or Rim-tagged hBest1 was immunoaffinity-purified as described above with Sepharose beads conjugated to anti-myc or anti-Rim mAb, respectively. In a second protocol, further purification was obtained by first diluting Triton X-100-solubilized proteins with an equal volume of 200 mM imidazole/1% Triton X-100/50 mM sodium phosphate, pH 8.0/20% glycerol/1 M NaCl, incubating with Ni-NTA resin in a spin column (Qiagen, Chatsworth, CA), to which native hBest1 binds with high affinity, and then washing in the same buffer with 3 M NaCl. hBest1 was eluted with 1 M imidazole, diluted with 4 vol of lysis buffer, and then immunoaffinity purified as described above. Radioactivity in 35S-labeled bands was quantitated by using a PhosphorImager.
Results
Chloride Currents Associated with Expressed Bestrophins.
Whole-cell recordings were made from HEK 293 cells transiently cotransfected with cDNA for enhanced green fluorescent protein (EGFP) and cDNA for human bestrophin (hBest1), a second human homologue (hBest2), a Drosophila homologue (dmBest1), or a C. elegans homologue (ceBest1). With each bestrophin homologue, we observed a conductance not detected in control EGFP-transfected cells (Fig. 1). The conductance associated with each expressed protein had a distinctive I–V relation, suggesting that the expressed protein constitutes part or all of the conductance rather than simply inducing or unmasking an endogenous conductance. With a whole-cell pipette solution containing predominantly CsCl (see Materials and Methods), the reversal potential of the current in each case was near zero. For the hBest1 current, this reversal potential showed practically no dependence on extracellular Na+ concentration (replaced by N-methyl-D-glucamine), suggesting that the conductance is unlikely to be permeable to cations (Fig. 2a Left). In contrast, replacing extracellular Cl− with gluconate shifted the reversal potential to more positive values, suggesting a substantial Cl permeability (Fig. 2a Center). The deviation of the data from that predicted for a pure Cl conductance (straight line, representing the Nernst equation) most probably arises from a finite permeability of the conductance to gluconate. Similar Cl−-replacement experiments on dmBest1 with gluconate gave essentially the same conclusion. For hBest1, measurements of reversal-potential shifts in substitution experiments with other inorganic anions gave the following order of permeability: NO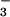 > I− > Br− > Cl−. Between hBest1 and hBest2, the relative permeability ratio PNO3/PCl is different, being 5.8 and 2.7, respectively (Fig. 2a Right). Finally, the idea that the current is carried by Cl− is further supported by its reversible blockage by 0.5 mM 4,4′-diisothiocyanostilbene-2,2′-disulfonate (DIDS), a blocker of many Cl channels (Fig. 2b). We also observed with expressed hBest1 and ceBest1 that the whole-cell current tends to run down over time, decreasing typically by ≈50% in 10–15 min. The reason for this current run-down remains unknown, although, in preliminary experiments, the run-down was slowed when nystatin-perforated-patch recording was used instead.
> I− > Br− > Cl−. Between hBest1 and hBest2, the relative permeability ratio PNO3/PCl is different, being 5.8 and 2.7, respectively (Fig. 2a Right). Finally, the idea that the current is carried by Cl− is further supported by its reversible blockage by 0.5 mM 4,4′-diisothiocyanostilbene-2,2′-disulfonate (DIDS), a blocker of many Cl channels (Fig. 2b). We also observed with expressed hBest1 and ceBest1 that the whole-cell current tends to run down over time, decreasing typically by ≈50% in 10–15 min. The reason for this current run-down remains unknown, although, in preliminary experiments, the run-down was slowed when nystatin-perforated-patch recording was used instead.
As a further test of whether expressed bestrophin constitutes an integral part of the Cl conductance, we examined the sensitivity of whole-cell currents to the sulfhydryl-reactive agents MTSEA and MTSET after transfection with either wild-type hBest1 or a mutant in which all five cysteines were replaced by alanines, a construct referred to as “cysteine-less hBest1′” (Fig. 3a). In the absence of the sulfhydryl-reactive agents, transfection of cysteine-less hBest1 produced a Cl current closely resembling that produced by wild-type hBest1. However, the current generated by cysteine-less hBest1 was resistant to inactivation by 2.5 mM MTSEA, whereas the wild-type hBest1 current was inactivated with a half-time of ≈50 sec (Fig. 3b). Similar results were obtained with the membrane-impermeant reagent MTSET when applied at 1 mM (data not shown). These data, together with those described above, indicate that the bestrophins form an integral part of the Cl conductance. In the remainder of this report, we refer to this conductance as a channel, although measurements of unitary conductance are required to definitively rule out the possibility that it functions as a carrier.
Figure 3.

Cysteine-less hBest1 is resistant to inactivation by sulfhydryl-specific reagents. (a) Locations of the 5 cysteines in hBest1 in the transmembrane topography of bestrophin proposed by Bakall et al. (11) in which the N and C termini face the cytosol. (b) Time course of whole-cell current inactivation recorded at +80 mV after addition of MTSEA to 2.5 mM at time 0 for cysteine-less hBest1 (○; n = 4 cells) or wild-type hBest1 (●; n = 4 cells). Mean current amplitudes at +80 mV recorded immediately before MTSEA addition were 1,355 ± 928 pA for cysteine-less hBest1 and 795 ± 674 pA for wild-type hBest1. At each time point, currents were recorded from −120 to +80 mV in 40-mV steps, with an interstep holding potential of 0 mV. In situations where the time points did not coincide for individual cells, linear interpolations of the measurements were made before averaging.
Oligomeric Assembly of Bestrophins.
In many channels, identical or homologous subunits associate around a pore. To test whether bestrophin assembles as oligomers, we tagged wild-type hBest1 with six tandem copies of the myc-epitope (hBest1-M) or one copy of the epitope for mAb Rim3F4 (hBest1-R). Membranes from cells cotransfected with these constructs were detergent-solubilized, subjected to immunoprecipitation with anti-myc mAb (anti-M), and then immunoblotted with mAb Rim3F4 (anti-R). This experiment showed efficient coimmunoprecipitation when hBest1-M and hBest1-R were cotransfected, versus no coimmunoprecipitation in a control experiment where detergent extracts from separately transfected cells were mixed (Fig. 4b), indicating that oligomerization is coupled to biosynthesis and is therefore specific. A splice variant of hBest1, with an in-frame deletion of 215 of the 298 amino acids in the predicted C-terminal cytosolic tail (Fig. 4a), coimmunoprecipitated with hBest1-M with the same efficiency as the full-length hBest1-R (Fig. 4c). This result implicates the N-terminal half of the protein as the principal mediator of subunit interactions. To test for subunit oligomerization among different bestrophins, we examined the relative efficiency of coimmunoprecipitation of M- or R-tagged human, Drosophila, and C. elegans bestrophins by using cells that had been triply transfected with one M-tagged bestrophin and two different R-tagged bestrophins. Fig. 4d shows that the efficiency of association with hBest1 is hBest1 ≈ hBest2 ≫ ceBest1 ≈ dmBest1. Similarly, ceBest1 associates more efficiently with itself than with hBest1, and dmBest1 associates more efficiently with itself than with hBest1. The efficient association between hBest1 and hBest2 suggests that, like the subunits of potassium channels (19) and cyclic nucleotide-gated cation channels (20), different bestrophin subunits have the potential to heterooligomerize to produce combinatorial channel diversity.
Figure 4.

Specificity of bestrophin oligomerization. (a) Proposed transmembrane topography of bestrophin (11) with dark regions indicating high levels of homology among family members. Epitope tags, Rim3F4 (R; ref. 16) and 6× myc (M) were appended at the extreme C terminus. Arrowheads demarcate the region coded by exon 10 that is missing in the “short” splice variant of hBest1. (b–d) Immunoprecipitation of different epitope-tagged bestrophins with anti-M antibodies followed by immunoblotting with anti-R antibodies. In each immunoblot, 2.5% of the total membrane protein input was loaded in lane T, and 10% of the immunopurified sample was loaded in lane IP. In the diagram to the left of each immunoblot, the circles that are touching indicate the cotranfected bestrophins and the inverted “Y” indicates the immunoprecipitating monoclonal antibody. These schematic diagrams should not be taken to imply any particular stoichiometry. (b) Cotransfection of hBest1-M and hBest1-R leads to efficient association of the two proteins (Upper), whereas mixing of detergent extracts from independently transfected cells does not (Lower). (c) Coprecipitation from cells triply transfected with full-length hBest1-M and full-length hBest1-R (represented by “L”), and the short splice variant of hBest1-R (represented by “S”) shows that association does not depend on an intact C-terminal domain. (d) Top two rows, coprecipitation from cells triply transfected with one M-tagged and two different R-tagged bestrophins. The relative efficiencies of association can be estimated by comparing the “T” and “IP” lanes. Bottom row, control experiments in which detergent extracts from cells cotransfected with two bestrophins were mixed with detergent extracts from cells transfected with a third bestrophin (shown in the schematic as the separated ball). In each case, mixing of proteins from independently transfected cells fails to produce a complex. Bars to the left of the immunoblots indicate molecular-mass markers, from top to bottom: 173, 111, 80, 61, 49, 36, and 25 kDa.
To estimate the number of subunits in a channel complex, we cotransfected 293 cells with hBest1-R and hBest1-M plasmids in different ratios and then labeled the cells with [35S]methionine, followed by immunoprecipitation of the channels with the anti-M antibody (Fig. 5 Left). As the hBest1-R/hBest1-M plasmid ratio increases, an ever-larger fraction of the channel complexes should contain only one hBest1-M subunit, the rest being hBest1-R. In this manner, the mole ratio of the two proteins in the immunoprecipitate, as measured from the ratio of radioactive methionine in the corresponding bands in an SDS gel, should asymptotically approach n − 1, where n is the number of subunits composing a channel complex. In these experiments, we observed an asymptotic ratio between 3:1 and 4:1 (Fig. 5 Lower Left), suggesting a stoichiometry of 4 or 5 subunits per complex. By contrast, with anti-R for immunoprecipitation from the same preparation, the hBest1-R/hBest1-M protein ratio increases nonasymptotically (Fig. 5 Right), as one would expect. The absence of any other predominant protein bands in the immunoprecipitates in Fig. 5 suggests that the oligomeric bestrophin complex is probably composed exclusively of bestrophin, although proteins with few or no methionines, or with low rates of synthesis, would be overlooked. In interpreting these experiments, it should be noted that the hBest1 in transfected cells is mostly localized to intracellular membranes as determined by immunofluorescent staining of fixed and permeabilized cells (data not shown). Thus, we cannot rule out the possibility that the minority residing in the plasma membrane adopts a different subunit composition or interacts with cellular proteins of comparably low abundance.
Figure 5.

Stoichiometry of hBest1 oligomeric complexes. The 293 cells were transfected with a mixture of hBest1-R and hBest1-M expression plasmids at the indicated ratios and labeled for 24 h with [35S]methionine. Bestrophin complexes were purified with either anti-M (Left) or anti-R (Right) antibodies. hBest1-R and hBest1-M monomers were resolved by SDS/PAGE, visualized by autoradiography (Upper), and the radioactivity in each bestrophin band was measured with a phosphorimager (Lower; shown as the hBest1-R/hBest1-M protein ratio). hBest1-M and hBest1-R contain the same number of methionines; the slower electrophoretic mobility of hBest1-M is caused by the presence of six tandem copies of the myc-tag. The autoradiograms and accompanying plots show the results from one experiment in which solubilized hBest1 was preenriched by using Ni-NTA resin (see Materials and Methods). (Inset) Mean and SD for four similar experiments without preenrichment. Molecular mass markers are as described for Fig. 3.
Calcium Sensitivity of hBest1 Current.
Others have suggested that the increase in the basal RPE Cl conductance associated with the electrooculogram light peak is triggered by an intracellular second messenger, with Ca2+ being one of the possibilities (9, 10). To examine whether the hBest1 channels are sensitive to intracellular Ca2+, hBest1-transfected 293 cells were loaded with the photolyzable caged-Ca2+ compound, Ca2+-NPEGTA (17), from a whole-cell pipette, and the effect of a flash on the voltage-clamp current was measured. In the experiment of Fig. 6, a voltage ramp before photolytic release of Ca2+ (marked “1” in Fig. 6a) produced a current of ≈40 pA at +80 mV (I–V relation marked “1” in Fig. 6b). After photorelease of Ca2+ by a flash, the same voltage ramp (marked “2” in Fig. 6a) gave a considerably larger current, reaching ≈250 pA at +80 mV (I–V relation marked “2” in Fig. 6b). The I–V relation was characteristic of hBest1 (compare with Fig. 1a). After this photo-induced current declined, presumably because of a subsequent decline of intracellular Ca2+, a second flash elicited another increase in the current. The Ca2+ dependence of the current was supported by control experiments showing that the flash-induced current was eliminated if the cells were preloaded with the Ca2+-chelator BAPTA in addition to Ca2+-NPEGTA (Fig. 6c), if they were preloaded with the inert Ca2+-EGTA instead of Ca2+-NPEGTA (Fig. 6d), or if Ca2+-NPEGTA was preloaded into control EGFP-transfected cells (Fig. 6e). In the collected results of Fig. 6f, the difference in current before and after flash photolysis is shown. In the experiments above, the buffered free Ca2+ concentration was adjusted to ≈10 nM before photolysis (see Materials and Methods), but the free Ca2+ concentration after photorelease of Ca2+ was not measured. Further studies with RPE cells and heterologous expression will be required to understand the Ca2+ action in more detail.
Disease-Associated hBest1 Mutants.
To investigate the mechanism by which hBest1 mutants cause VMD, we studied 15 disease-associated point mutations (Fig. 7a). Although the mutant proteins were generally similar to wild type in expression level in 293 cells (Fig. 7b), each mutant gave a considerably smaller whole-cell current (Fig. 7 c and e), and in many cases the current amplitude was indistinguishable from that of control EGFP-transfected cells. Four of these mutants (Y85H, R92C, R218S, and G299E) were studied in greater detail. On cotransfection with wild-type hBest1, each of the four assembled efficiently with the wild-type protein (Fig. 7d). When cotransfected at a 1:1 plasmid ratio with wild type (holding the total quantity of plasmid DNA constant), each hBest1 mutant also gave a reduced current relative to the wild-type control (Fig. 7e). It might be argued that the smaller current in the mutant plus wild-type cotransfection experiments merely reflects a reduced level of wild-type protein because of a 2-fold lower amount of wild-type hBest1 plasmid and/or to competition by the mutant for RNA or protein synthesis. To address this question, we performed a side-by-side comparison of wild-type hBest1 cotransfected into 293 cells with either the disease-causing R92C mutant or a secreted IgG fusion protein (Fz8-IgG), which is expressed at high levels in 293 cells (ref. 21; Fig. 7f). This experiment revealed a ≈2-fold reduction in the hBest1 current in the presence of Fz8-IgG and a ≈10-fold reduction in the hBest1 current in the presence of the R92C mutant, indicating that most of the current reductions in Fig. 7e are probably caused by active interference with wild-type function by the mutant protein. Thus, the dominance of the mutant alleles in causing disease is likely to result from the production of defective channels composed of both mutant and wild-type subunits.
Discussion
In this report, we demonstrate that (i) when expressed in a heterologous system, each of four different bestrophin family members produces a Cl conductance with a distinct I–V relationship and/or ion selectivity; (ii) the Cl current conferred by wild-type hBest1 is rapidly inactivated by sulfhydryl-reactive agents, whereas the current conferred by a cysteine-less hBest1 is resistant to such inactivation; (iii) the bestrophins oligomerize to form tetramers or pentamers; (iv) the Cl conductance observed upon expression of hBest1 is calcium-sensitive; and (v) multiple hBest1 mutants responsible for VMD dominantly inhibit the Cl conductance associated with wild-type hBest1. Moreover, earlier work showed that VMD is associated with a reduction or loss of the slow light peak in the electrooculogram in presymptomatic patients, that this light peak likely reflects the opening of Cl channels on the basolateral plasma membrane of the RPE, and that bestrophin localizes specifically to that membrane. The simplest conclusion from these diverse observations is that the bestrophins function as Cl channels and thereby define a family of these channels. Finally, we note that the high nitrate permeability observed in bestrophin-expressing cells suggests that the bestrophins are likely to conduct other physiologically significant anions such as bicarbonate.
Experiments during the past two decades have established the existence of at least three distinct families of Cl channels: the ClCs, CFTR (cystic fibrosis transmembrane conductance regulator), and the γ-aminobutyric acid and glycine receptors (22). Physiological observations imply the existence of additional classes of Cl channels, but at present their molecular identities remain undefined or controversial (23). Each of the three established Cl channel families has a distinctive architecture. The bestrophins have no detectable primary sequence homology with any of the known families of Cl channels. If the secondary-structure model of four transmembrane domains with the N and C termini facing the cytosol is correct (ref. 11; Fig. 4a), then the bestrophins would define a completely different Cl channel architecture.
The finding that hBest1 is a Cl channel (or, more generally, an anion channel) provides a mechanistic framework for understanding the pathogenesis of VMD. Most obviously, it provides a direct explanation for the electrooculographic abnormalities in patients with VMD. Less obvious is the connection between defective hBest1 function and the accumulation of lipofuscin-like material in and under the RPE. The RPE is highly active in the transport of protons, sodium, potassium, lactate, taurine, γ-aminobutyric acid, and various other molecules, and these are coupled directly or indirectly to chloride or bicarbonate movement (24). Thus, defects in chloride or bicarbonate homeostasis might create an imbalance in intracellular or intravesicular pH, or in ionic composition. Given the enormous flux of material handled by the RPE, including phagocytosed photoreceptor outer segments, nutrients from the choroidal circulation, and metabolites from the retina, such an imbalance could result in impaired transport and accumulation of debris. In this regard, Dent's disease, a disorder caused by mutations in the ClC5 chloride channel, may be instructive. In Dent's disease, excess renal excretion of calcium and proteins is likely caused by defective acidification of endosomal compartments and, perhaps, defective membrane recycling (25, 26).
In addition to the study of hBest1 and its role in RPE physiology, it will be of interest to explore the biological roles of other bestrophin family members. The presence of these proteins throughout the animal kingdom and their large number in C. elegans suggest that bestrophin channels are likely to be involved in a wide variety of cellular processes.
Acknowledgments
We thank John Williams and Luisa Cochella for assistance with cDNA cloning and construction of the cysteine-less hBest1; Dr. Robert Molday for hybridoma Rim3F4; and Mr. Haining Zhong and Drs. Jonathan Bradley, Michael Caterina, Tsung-Yu Chen, and Min Li for helpful comments on the manuscript. This work was supported by the Howard Hughes Medical Institute (J.N. and K.-W.Y.), the Foundation Fighting Blindness (J.N.), the National Institutes of Health (K.-W.Y.), and a Long-Term Fellowship from the Human Frontier Science Program (T.T.).
Abbreviations
- RPE
retinal pigment epithelium
- AMD
age-related macular degeneration
- VMD
vitelliform macular dystrophy
- EGFP
enhanced green fluorescent protein
- BAPTA
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate
- NPEGTA
o-nitrophenyl EGTA
- MTSEA
2-aminoethylmethanethiosulfonate
- MTSET
2-trimethylammonioethylmethanethiosulfonate
- DIDS
4,4′-diisothiocyanostilbene-2,2′-disulfonate
References
- 1.Berger J W, Fine S L, Maguire M G. Age-Related Macular Degeneration. St. Louis: Mosby; 1999. [Google Scholar]
- 2.Stone E M, Sheffield V C, Hageman G S. Hum Mol Genet. 2001;10:2285–2292. doi: 10.1093/hmg/10.20.2285. [DOI] [PubMed] [Google Scholar]
- 3.Weingeist T A, Kobrin J L, Watzke R C. Arch Ophthalmol. 1982;100:1108–1114. doi: 10.1001/archopht.1982.01030040086016. [DOI] [PubMed] [Google Scholar]
- 4.Frangieh G T, Green W R, Fine S L. Arch Ophthalmol. 1982;100:1115–1121. doi: 10.1001/archopht.1982.01030040093017. [DOI] [PubMed] [Google Scholar]
- 5.Marquardt A, Stohr H, Passmore L A, Kramer F, Rivera A, Weber B H. Hum Mol Genet. 1998;7:1517–1525. doi: 10.1093/hmg/7.9.1517. [DOI] [PubMed] [Google Scholar]
- 6.Petrukhin K, Koisti M J, Bakall B, Li W, Xie G, Marknell T, Sandgren O, Forsman K, Holmgren G, Andreasson S, et al. Nat Genet. 1998;19:241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- 7.Francois J, De Rouck A, Fernandez-Sasso D. Arch Ophthalmol. 1967;77:727–733. doi: 10.1001/archopht.1967.00980020728003. [DOI] [PubMed] [Google Scholar]
- 8.Deutman A F. Arch Ophthalmol. 1969;81:305–316. doi: 10.1001/archopht.1969.00990010307001. [DOI] [PubMed] [Google Scholar]
- 9.Steinberg R H, Linsenmeier R A, Griff E R. Prog Retinal Res. 1985;4:33–66. [Google Scholar]
- 10.Gallemore R P, Hughes B A, Miller S S. In: The Retinal Pigment Epithelium. Marmor M F, Wolfensberger T J, editors. Oxford: Oxford Univ. Press; 1998. pp. 175–198. [Google Scholar]
- 11.Bakall B, Marknell T, Ingvast S, Koisti M J, Sandgren O, Li W, Bergen A A, Andreasson S, Rosenberg T, Petrukhin K, Wadelius C. Hum Genet. 1999;104:383–389. doi: 10.1007/s004390050972. [DOI] [PubMed] [Google Scholar]
- 12.Krämer F, White K, Pauleikhoff D, Gehrig A, Passmore L, Rivera A, Rudolph G, Kellner U, Andrassi M, Lorenz B, Rohrschneider K, et al. Eur J Hum Genet. 2000;8:286–292. doi: 10.1038/sj.ejhg.5200447. [DOI] [PubMed] [Google Scholar]
- 13.Lotery A J, Munier F L, Fishman G A, Weleber R G, Jacobson S G, Affatigato L M, Nichols B E, Schorderet D F, Sheffield V C, Stone E M. Invest Ophthalmol Visual Sci. 2000;41:1291–1296. [PubMed] [Google Scholar]
- 14.White K, Marquardt A, Weber B H F. Hum Mutat. 2000;15:301–308. doi: 10.1002/(SICI)1098-1004(200004)15:4<301::AID-HUMU1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 15.Marmorstein A D, Marmorstein L Y, Rayborn M, Wang X, Hollyfield J G, Petrukhin K. Proc Natl Acad Sci USA. 2000;97:12758–12763. doi: 10.1073/pnas.220402097. . 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Illing M, Molday L L, Molday R S. J Biol Chem. 1997;272:10303–10310. doi: 10.1074/jbc.272.15.10303. [DOI] [PubMed] [Google Scholar]
- 17.Ellis-Davies G C, Kaplan J H. Proc Natl Acad Sci USA. 1994;91:187–191. doi: 10.1073/pnas.91.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlin A, Akabas M H. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 19.Jan L, Jan Y N. Annu Rev Neurosci. 1997;20:91–123. doi: 10.1146/annurev.neuro.20.1.91. [DOI] [PubMed] [Google Scholar]
- 20.Finn J T, Krautwurst D, Schroeder J E, Chen T-Y, Reed R R, Yau K-W. Biophys J. 1998;74:1333–1345. doi: 10.1016/S0006-3495(98)77846-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsieh J-C, Rattner A, Smallwood P M, Nathans J. Proc Natl Acad Sci USA. 1999;96:3546–3551. doi: 10.1073/pnas.96.7.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jentsch T J, Gunther W. BioEssays. 1997;19:117–126. doi: 10.1002/bies.950190206. [DOI] [PubMed] [Google Scholar]
- 23.Kidd J F, Thorn P. Annu Rev Physiol. 2000;62:493–513. doi: 10.1146/annurev.physiol.62.1.493. [DOI] [PubMed] [Google Scholar]
- 24.Hughes B A, Gallemore R P, Miller S S. In: The Retinal Pigment Epithelium. Marmor M F, Wolfensberger T J, editors. Oxford: Oxford Univ. Press; 1998. pp. 103–134. [Google Scholar]
- 25.Piwon N, Gunther W, Schwake M, Bosl M R, Jentsch T J. Nature (London) 2000;408:369–373. doi: 10.1038/35042597. [DOI] [PubMed] [Google Scholar]
- 26.Wang S S, Devuyst O, Courtoy P J, Wang X T, Wang H, Wang Y, Thakker R V, Guggino S, Guggino W B. Hum Mol Genet. 2000;9:2937–2945. doi: 10.1093/hmg/9.20.2937. [DOI] [PubMed] [Google Scholar]