

Summary
Cellular responses to stress-like stimuli require the IKK signalsome (IKKα, IKKβ and NEMO/IKKγ) to activate NF-κB dependent genes. IKKβ and NEMO/IKKγ are required to release NF-κB p65/p50 heterodimers from IκBα, resulting in their nuclear migration and sequence specific DNA binding; but IKKα was found to be dispensable for this initial phase of canonical NF-κB activation. Nevertheless, IKKα(−/−) MEFs fail to express NF- κB targets in response to pro-inflammatory stimuli, uncovering a nuclear role for IKKα in NF-κB activation. However, it remains unknown if the global defect in NF-κB dependent gene expression of IKKα(−/−) cells is caused by the absence of IKKα kinase activity. We show by gene expression profiling that rescue of near physiological levels of Wt. IKKα in IKKα(−/−) MEFs globally restores expression of their canonical NF-κB target genes. To prove that IKKα’s kinase activity was required on a genomic scale, the same physiological rescue was performed with a kinase dead, ATP binding domain IKKα mutant [IKKα(K44M)]. Remarkably, the IKKα(K44M) protein rescued ~28% of these genes, albeit in a largely stimulus independent manner with the notable exception of several genes that also acquired TNFα responsiveness. Thus the IKKα containing signalsome unexpectedly functions in the presence and absence of extracellar signals in both kinase dependent and independent modes to differentially modulate the expression five distinct classes of IKKα/NF-κB dependent genes.
Introduction
The NF-κB pathway is important for a host of cellular processes including its central role in responses to stress-like stimuli, the anti-apoptotic cascade, the initiation and maintenance of immune responses, embryonic and adult tissue development and cell cycle progression [reviewed in (1–11)]. In mammals the NF-κB family of transcription factors is comprised of 5 subunits characterized by the presence of a conserved Rel homology DNA binding domain [reviewed in (2,12,13)]. The RelA/p65, c-Rel and RelB NF-κB subunits are fully functional transcriptional activators, whereas the p50 and p52 subunits lack a transcriptional activation domain [reviewed in (12,13). NF-κBs function as specific hetero- or homo-dimers that bind to a GGGRNWTYCC consensus DNA sequence found in the promoters or enhancers of NF-κB target genes [reviewed in (12,13)]. Transcriptional activating NF-κB subunits are normally sequestered in the cytoplasm of unstimulated cells in a complex with one of the IκB family proteins, which block their nuclear import and DNA binding activity. A large variety of extra-cellular activating stimuli induce the proteosomal dependent destruction of IκBs thereby differentially freeing NF-κBs to bind DNA and activate the transcription of their genomic targets [reviewed in (14)]. Stress-like inducers of NF-κB (including pro-inflammatory cytokines like TNFα and IL-1) function in a classical monophasic capacity to rapidly drive the canonical NF-κB activation pathway, which largely involves the activation of p65(RelA)/p50 DNA binding activity and transcriptional competence [reviewed in (1,2,8,15)]. More recently a distinct class of NF-κB stimuli (exemplified by LTβ, BAFF and CD40 ligand), which also contribute to the implementation of differentiation programs and the adaptive phase of immune responses, have been shown to function as biphasic activators initially acting via the rapid canonical pathway and subsequently feeding into a delayed non-canonical protein synthesis dependent route characterized by the activation of RelB/p52 heterodimers [reviewed in (8,9,15,16)].
With the exceptions of UV radiation and the effects of some DNA damaging agents (17,18), the release of NF-κBs from IκBs is mediated by the cytoplasmic signalsome complex, which consists of two serine-threonine kinases (IKKα, IKKβ) and NEMO/IKKγ, a regulatory/docking protein [reviewed in (1,7,8,15)]. IKKβ is essential for the phosphorylation of IκBs on a pair of amino terminal serines (residues 32 and 36 in IκBα) thereby targeting IκB for ubiquitination and subsequent proteosomal destruction [reviewed in (1,7,8)]. In contrast, IKKα is not required for the phosphorylation of IκBs via the canonical NF-κB activation pathway in vivo, with the exception of RANK ligand signaling in mammary epithelial cells (19). Rather, IKKα plays an essential role in epidermal keratinocyte differentiation independent of both its kinase activity and NF-κB activation and has also recently been found to play NF-κB dependent and independent roles in tooth development (20–22). With respect to its physiological role in NF-κB signaling pathways, IKKα is instead essential for the activation of the non-canonical NF- κB activation pathway, which requires neither IKKβ nor NEMO/IKKγ [reviewed in (8,15,16)]. In this context, via NIK (NF-κB inducing kinase) dependent signaling, IKKα phosphorylates multiple serines of the p100 precursor of the p52 subunit, thereby inducing its proteosome dependent processing into mature p52 subunits which are then freed to activate NF-κB target genes as p52/RelB heterodimers (8,9,23,24). We and other groups have also found IKKα is required to activate the transcription of canonical NF-κB target genes (25–29). The latter dependency on IKKα is independent of IκBα destruction, and instead appears to involve one or more nuclear targets perhaps including histone H3 (27,28) and the SMRT transcriptional co-repressor (29) resulting in the de-repression of NF-κB target genes
In this report we have investigated the physiological requirement of IKKα’s kinase activity for the expression of NF-κB dependent genes on a genomic scale in IKKα null MEFs. Physiological expression of Wt. IKKα in IKKα(−/−) MEFs by retroviral transduction resulted in the rescue of specific NF-κB dependent genes in the presence and absence of TNF-α stimulation. Comparative microarray screens with NF-κB compromised MEFs [p50(−/−) and Wt. + IκBα(S32A, S36A)] revealed that the large majority of these IKKα rescued genes are either dependent on basal or TNFα inducible NF-κB, thus demonstrating that: (1) IKKα plays an essential role in controlling the expression of both signal induced and basal NF-κB dependent genes and 2) IKKα does not appear to influence the expression of a large number of genes outside of the NF-κB pathway. Comparable physiological rescue with a kinase dead IKKα mutant protein [(IKKαK44M)] showed that most of these genes are dependent on IKKα kinase activity for their stimulus dependent and independent expression. However, the expression of up to 28% of these NF-κB dependent genes was also surprisingly rescued by the kinase inactive IKKα(K44M) mutant. Furthermore both wild type and mutant IKKα are also required for the basal levels of expression of specific NF-κB dependent genes. Thus, our findings collectively reveal that the levels of expression of different downstream NF-κB dependent genes are differentially co-dependent on catalytically active IKKα in the presence or absence of extracellular stimuli.
Experimental Procedures
Tissue Culture
Growth of IKKα(−/−) MEFs and their stimulation with TNF-α was performed as previously described. Wt. IKKα/CHUK-HA or IKKα(K44M)-HA (a kinase inactive ATP binding domain mutant with lysine 44 mutated to methionine) with carboxy-terminal HA epitope tags (30–32) were introduced into IKKα(−/−) MEFs by transduction with a retroviral vector co-expressing a puromycin resistance gene followed by 6–8 days of puromycin selection (26,33). IKKα(−/−) cells harboring an empty retroviral vector (EV cells) were simultaneously generated as a matched negative control.
RNA Preparation:
Total cellular RNAs were extracted from cell lysates with an RNeasy kit (Qiagen). Purified RNAs were converted to double-stranded cDNA with a Super Script Double Stranded cDNA synthesis kit (Invitrogen) and an oligo-dT primer containing a T7 RNA polymerase promoter (GENSET). Biotin-labeled cRNAs were generated from the cDNA samples by in vitro transcription with T7 RNA polymerase (Enzo kit, Enzo Diagnostics). The labeled cRNAs were fragmented to an average size of 35 to 200 bases by incubation at 94°C for 35 min. Hybridization (16 hr), washing and staining protocols have been described [Affymetrix Gene ChipR Expression Analysis Technical Manual; (34)].
DNA Microarrays and Clustering Analysis:
We employed Affymetrix MG-U74Av2 chips that include 12400 genes. Chips were stained with streptavidin-phycoerythrin (Molecular Probes) and scanned with a Hewlett-Packard Gene Array Scanner. DNA microarray chip data analysis was performed using MAS5.1 software (Affymetrix) and as previously described (26). Levels of gene expression were quantitated from the hybridization intensities of 16 pairs of perfectly matched (PM) and mismatched (MM) control probes (35) (Affymetrix Inc.). The average of the differences (PM minus MM) for each gene-specific probe family were calculated and expressed as Signal values. The software computes how each transcript’s expression level has changed between the baseline and experimental samples (Difference Call/Change Call). Change Call is a qualitative call that describes whether a transcript in an experimental array has changed compared to a baseline array. One array is designated as the experimental and another array is designated as the baseline. Wilcoxon’s Signed Rank is used to generate a Change p-value. A Change call is assigned based on analysis parameters. Change p-values between 0.00 and 0.0025 are given an Increase (I) call. Change p-values between 0.0025 and 0.003 are given a Marginal Increase (MI) call. Change p-values between 0.997 and 0.998 are given a Marginal Decrease (MD) call. Change p-values between 0.998 and 1.00 are given a Decrease (D) call (Affymetrix User manual). For a comparative chip file (such as TNFα stimulated IKKα(−/−) MEF + Wt.IKKα vs. IKKα(−/−) MEF + EV (empty vector), the experimental file [Wt.IKKα 2T] was compared to the baseline file [EV 2T]. We employed the following stringent selection criteria to identify significant changes in gene expression: (1) a change call of “increase” or “marginal increase” in both samples; and (2) average fold change values of 1.5 or greater (minimum of 1.3 fold each) in two independently stimulated samples of IKKα(−/−) MEF + Wt.IKKα 2T vs. IKKα(−/−) MEF + EV 2T. The following additional criteria were employed to identify the spectrum of these genes that were also dependent upon NF-κB for their expression: (1) A change call of either “Increase” or “Marginal Increase” in Wt. MEF 2T vs. Wt. MEF + IκBαSR-Ires-Neomycin or (2) a change call of either “Increase” or “Marginal Increase” in Wt. MEF 2T vs. p50 (−/−) MEF 2T and (3) a valid presence call of “P” in Wt. 2T screens. By this analysis we define NF-κB dependency to represent genes whose expressions either directly (true direct targets of NF- κB subunits) or indirectly (other downstream genes whose expressions are effected by the NF-κB pathway) require NF-κB. The TNFα inducibilities of genes rescued by either Wt. IKKα or IKKα(K44M) were based on a combination of the following stringent criteria: (1) Increase calls in duplicate 2T vs. US microarray screens of IKKα(−/−) MEFs rescued by Wt. IKKα or IKKα(K44M) and/or (2) TaqMan ‘real time’ PCR analysis performed at least in duplicate and an increase call in one out of two screens. By these criteria, the vast majority of IKKα rescued genes that responded to TNFα did so with fold change values of 2.0 and higher in duplicate screens with a minimum of average fold change value of 1.7.
Hierarchical clustering was performed with the Cluster program (available at http://rana.lbl.gov/) as described previously (36). Genes that have double “Increase calls” and are induced >1.5 fold (avg. fold values) in the duplicate primary Wt. IKKα vs. EV rescued IKKα(−/−) MEFs (see above description) were selected. The Signal values (equivalent to the quantities of mRNAs, see “above”) of the selected genes were median centered by subtracting the median observed value, and normalized by genes to the magnitude (sum of the squares of the values) of a row vector to 1.0. The normalized data were clustered by average linkage clustering analysis of Y axis (genes) using an uncentered correlation similarity metric, as described in the program Cluster. Signal values of 50 or less were set to 50 before centering and normalization. The clustered data were visualized with the Treeview program (available at http://rana.lbl.gov/).
TaqMan Real-Time Quantitative PCR
TaqMan Real-time quantitative PCRs were performed as previously described (26,37). Data from TaqMan PCR analyses were normalized based on GAPDH mRNA copy numbers using rodent GAPDH control reagents (Applied Biosystems). TaqMan probe sets were designed for the following genes using either Primer Express 1.5 or Bio. Rad Beacon Designer 2.0 software: ATF3, A20, ISG15, MyD118/GADD45β, SAA3 and VCAM1. Murine IL-6 and RANTES TaqMan reagents were obtained from ABI. TaqMan ‘real time PCR’ experiments were performed in an ABI PRISM 7700 sequence detector or in a Bio. Rad iCycler. DNA sequences for each of these probe sets are available upon request.
Western Blotting:
Cell lysates were prepared in an isotonic lysis buffer containing 1% NP-40 supplemented with protease inhibitors. SDS-10%PAGE transfer to PVDF membranes was performed as previously described, and membranes were probed with primary antibodies to either IKKα (Cell Signaling Technology) or NEMO (Santa Cruz) followed by an anti-rabbit-Horseradish Peroxidase conjugated secondary antibody (Amersham). Blots were developed using a Lumi-Light Plus kit (Roche).
Immunostaining.
Prior to immunostaining cells were maintained in 10 cm tissue culture treated plates in their regular growth media. Cells were trypsinized, resuspended in 12 ml of growth medium, and plated at 3 ml/well in 6-well plates containing 22 mm glass microscope coverslips (VWR) pre-coated with poly-L-lysine (Sigma). Cells were incubated overnight at 37°C with 5% CO2. The following day, cells were washed and then fixed in 50% methanol/50% acetone for 10 min. These and all subsequent washes were with PBS. The fixed cells were rehydrated in PBS, and the coverslips were washed, blocked with PBS containing 10% heat denatured fetal bovine serum for 1 hr, and washed again. In situ expression of retrovirally transduced Wt. IKKα-HA and IKKα(K44M)-HA proteins in IKKα(−/−) cells were specifically detected with a primary anti-HA 12CA5 antibody (38). 12CA5 antibody was diluted in blocking solution and applied to the cells, followed by 1 hr incubation at room temperature. Following washing, alkaline-phosphatase conjugated goat anti-mouse IgG (Jackson) secondary antibody, diluted 1:2000 in block solution, was applied for 1 hr incubation at room temperature. The coverslips were washed and NBT/BCIP developing substrate applied (Roche). Cells were visualized on a Nikon Diaphot phase contrast microscope and photographed using a Nikon D1X digital camera.
NF-κB DNA Binding:
Activation of NF-κB p65 dependent DNA binding activity was quantitated with an ELISA-based kit (Active Motif Inc)(39). Nuclear extracts were prepared from unstimulated or 2 hr TNFα stimulated wild type, IKKα null and Wt. IKKα or IKKα(K44M) rescued IKKα(−/−) MEFs and applied to 96 well plates containing an immobilized NF-κB DNA binding consensus oligonucleotide according to the manufacturer’s instructions (Active Motif Inc). DNA bound NF-κB was detected with a p65 specific primary antibody followed by addition of a secondary antibody conjugated to horseradish peroxidase (HRP) and absorbence quantitated at 450 nm with a microplate spectrophotometer. The specificity of NF-κB DNA binding was confirmed by competitions with wild type and mutant NF-κB binding sequences. Negative controls for stimulus dependent NF-κB nuclear localization and DNA binding activity also included nuclear extracts prepared from NEMO (−/−) and p65/p50 (−/−) MEFs.
Results
Physiological rescue of IKKα(−/−) MEFs with Wt. IKKα and IKKα(K44M) proteins does not interfere with the induction of NF-κB DNA binding activity
By employing DNA microarray chip technology, we previously reported that the IKKα protein was as essential as the IKKβ and NEMO/IKKγ signalsome subunits for the genomic NF-κB dependent transcriptional response induced by TNF-α or IL-1 stimulation. Our finding of a strict requirement for IKKα in the regulation of NF-κB dependent transcription in MEFs was controversial, because earlier studies had shown that cells derived from IKKα null mice exhibited no significant defect in the stimulus dependent induction of NF-κB nuclear localization and DNA binding activity. However, in agreement with our observations, two other studies had shown that IKKα was required for the TNFα and IL-1 dependent transcriptional induction of IL-6 gene expression (25,40). Subsequent to these reports, chromatin immunoprecipitation experiments showed that IKKα’s role in engendering DNA bound NF-κB with transcriptional competence was associated with its TNFα dependent binding to the IκBα gene’s promoter and with its ability to directly phosphorylate histone H3 on serine 10 in vitro (27,28) and more recently to also phosphorylate and thereby facilitate the release of the SMRT co-repressor from specific NF-κB target gene promoters (29).
To determine if the intrinsic defect of IKKα(−/−) MEFs to express NF-κB dependent genes on a genomic scale was solely due to the absence of a functional IKKα kinase, we employed retroviral transduction to rescue physiological levels of Wt. IKKα expression in a large population of IKKα (−/−) MEFs. To determine if IKKα’s kinase activity was required to rescue the expression of their NF-κB dependent genes, we also derived a similar population of IKKα(−/−) MEFs expressing near physiological levels of a kinasedead IKKα mutant [IKKα(K44M)](31,32). To rule out any effects of stable retroviral transduction, we also generated a matched negative control population of IKKα(−/−) MEFs harboring the empty retroviral vector [IKKα(−/−)-EV cells]. Western blotting revealed that the levels of Wt. IKKα and IKKα(K44M) expression in IKKα(−/−) rescued MEFs were similar to the expression of endogenous IKKα in wild type NF-κB competent MEFs (see Figure 1). Importantly as shown in Figure 2, in situ immunostaining also revealed uniform expression of either the Wt. IKKα-HA or mutant IKKα(K44M)-HA proteins in their respective stably transduced cell populations. These stable populations of IKKα(−/−) cells expressing physiologically comparable levels of either Wt. IKKα-HA or IKKα(K44M)-HA were used in all subsequent experiments.
Figure 1. Physiological levels of expression of Wt. IKKα and IKKα(K44M) in rescued populations of IKKα(−/−) MEFs.
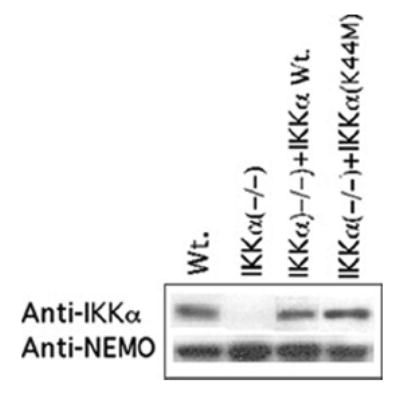
Populations of IKKα(−/−) MEFs were retrovirally transduced to stably express either murine Wt. IKKα-HA or an IKKα(K44M)-HA mutant proteins. SDS-PAGE was performed to determine the levels of IKKα expression obtained in the infected populations (top) (see Experimental Procedures). The membrane was stripped and reprobed with a NEMO specific antibody as a reference control (bottom), which showed comparable expression in each cell background.
Figure 2. Immunostaining of Wt. IKKα-HA and IKKα(K44M)-HA proteins in retrovirally transduced populations of IKKα(−/−) cells.
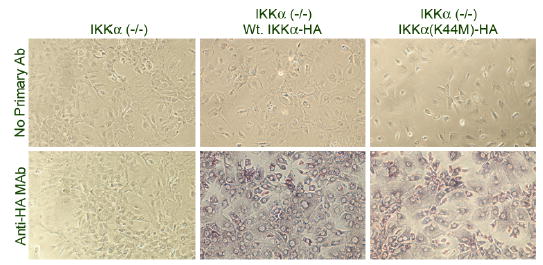
Wt. IKKα-HA and IKKα(K44M)-HA proteins, stably expressed by populations of retrovirally transduced IKKα(−/−) MEFs, were visualized by in situ immunostaining. Cells were plated on coverslips coated with poly-L-lysine, fixed and immunostained using 12CA5 anti-HA monoclonal antibody or no primary antibody as a negative control. Stained cells were viewed by a phase contrast Nikon Diaphot microscope and photographed using a Nikon D1X digital camera, as described in "Experimental Procedures". In the absence of primary antibody (top panels) or in IKKα(−/−) parental cells, no immunostaining is seen, while populations of IKKα(−/−) cells stably transduced by either Wt. IKKα-HA or IKKα(K44M)-HA expressing retroviruses show uniform cytoplasmic staining.
As discussed above, studies with IKKα null mice have proven that the loss of IKKα has no effect on the induction of NF-κB DNA binding activity by proinflammatory cytokines (40–42). Consequently, before proceeding to compare the effects of exogenous wild type and kinase dead IKKα on the TNFα induced NF-κB dependent transcriptional response of IKKα null MEFs, it was necessary for us to verify that our retrovirally derived cell populations exhibited comparable TNFα induced NF-κB DNA binding activity to their IKKα(−/−) counterparts and Wt. MEFs. To this end, we employed a quantitative ELISA-based DNA binding assay to directly compare levels of NF-κB p65 subunit DNA binding activity induced by TNFα stimulation. Nuclear extracts of NEMO/IKKγ(−/−) and p65/p50(−/−) MEFs were employed as negative controls. As shown in Figure 3, comparable levels of NF-κB p65 DNA binding activity were induced upon TNFα stimulation of wild type, IKKα(−/−) + EV, IKKα(−/−) + Wt.IKKα and IKKα(−/−) + IKKα(K44M) MEFs. To validate the specificity of the DNA binding reactions, NF-κB p65 dependent DNA binding was abolished in nuclear extracts of TNFα induced IKKα(−/−) + Wt.IKKα and IKKα(−/−) + IKKα(K44M) cells by an excess of a wild type NF-κB binding oligonucleotide but not by a mutant NF-κB binding sequence (Figure 3). Importantly, this experiment demonstrates that when expressed at near-physiological levels, the IKKα(K44M) mutant protein does not function in a general dominant negative manner with respect to the nuclear localization and activation of NF- κB DNA binding activity.
Figure 3. Physiological expression of Wt. IKKα or IKKα(K44M) in IKKα(−/−) MEFs does not interfere with stimulus dependent NF-κB DNA binding.
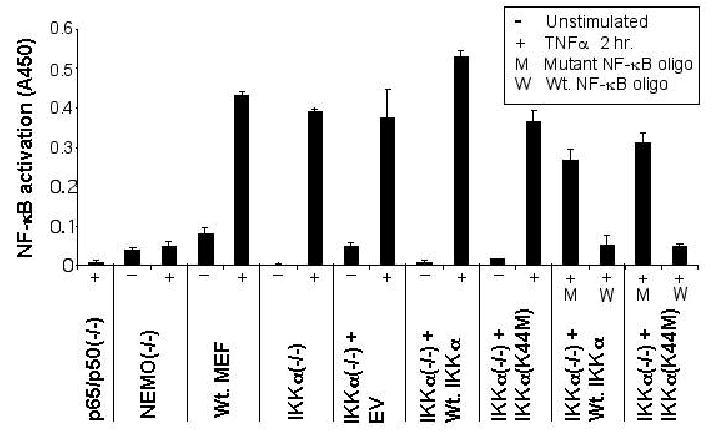
RelA/p65 DNA binding was assayed using the TransAM NF-κB p65 Transcription factor assay kit (Active Motif), following the manufacturer’s instructions for the preparation of nuclear extracts. All samples are presented as (−) unstimulated or (+) stimulated for 2 hours with 20 ng/ml TNFα prior to lysis and nuclear extract preparation. Data shown represent each data point done in quadruplicate, with standard deviations presented as error bars. Nuclear p65 was measured as the absorbance at 450 nm, with a reference wavelength of 650 nm using a fluorescent plate reader. Specificity of p65- DNA binding within the Wt. IKKα and IKKα(K44M) infected populations was determined by competition for binding using an excess of either Wt. (W) or mutant (M) NF-κB synthetic oligonucleotide.
Physiological rescue of IKKα(−/−) MEFs with Wt. IKKα is sufficient to restore the global expression of NF-κB dependent genes
To identify the cohort of TNFα responsive genes in IKKα(−/−) MEFs, which are not expressed due to their IKKα deficiency, we compared DNA microarray screens of IKKα(−/−) MEFs rescued with Wt. IKKα to IKKα(−/−) cells expressing an empty retroviral vector (EV) as a matched negative control. IKKα(−/−) + Wt. IKKα screens were performed in duplicate to rule out any gene specific variations and were compared to TNFα stimulated IKKα(−/−)EV cells. 118 genes were rescued based on the stringent criteria of double increase calls (Affymetix MAS 5.1) and average fold change values of 1.5 or greater. Hierarchical clustering of the signal values of these 118 genes shows that two independent TNFα stimulations of the Wt. IKKα rescued IKKα(−/−) cell population have very similar expression profiles (see columns 1 and 2 of Figure 4). Some variations in the degrees of expression of specific genes were observed but more importantly all of these genes are expressed at significantly higher levels in the two Wt. IKKα rescued samples compared to TNF stimulated IKKα(−/−) cells harboring the empty retroviral vector or to parental IKKα(−/−) cells.
Figure 4. Hierarchical cluster image of the NF-κB dependencies of genes rescued by Wt. IKKα in IKKα(−/−) MEFs.
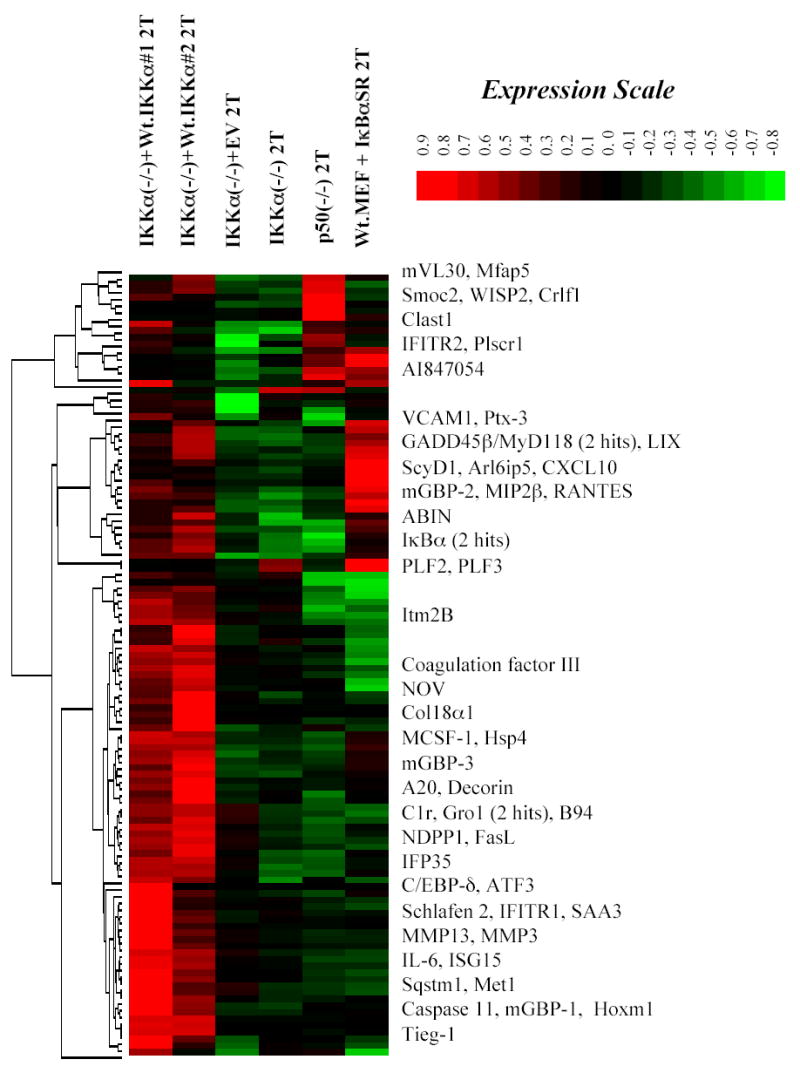
Signal values of 118 IKKα dependent genes derived from two independent microarray screens of IKKα(−/−) cells expressing Wt. IKKα (Columns 1 and 2) were subjected to hierarchical clustering in comparison to the following samples: Column 3: IKKα(−/−) MEFs+ empty vector (EV) 2T, Column 4: IKKα(−/−) MEFs 2T, Column 5: p50(−/−) 2T and Column 6: Wt. MEFs + IκBα(S32A, S36A) 2T. The 118 IKKα dependent genes employed in these comparisons were selected on the basis of average fold change values of 1.5 or greater (minimum of 1.3 fold each) and difference calls of “increase” or “marginal increase” in two independent samples of 2 hr TNFα (2T) stimulated IKKα(−/−) MEF + Wt.IKKα 2T vs. IKKα(−/−) MEF + empty vector (EV) 2T MEFs. The locations of a number of genes are indicated. Signal values were all derived from MAS5.0 calculation and normalized as described in Experimental Procedures. Gene expression values are shown in color according to the indicated expression scale bar.
To determine if these IKKα dependent genes were also dependent on NF-κB, we employed hierarchical clustering analysis to cross compare these screens to others performed with two varieties of NF-κB compromised MEFs: (1) Wt. MEFs stably expressing an IκBα(S32A, S36A) super-repressor (26) and (2) NF-κB p50(−/−) MEFs (43). Both cell lines were stimulated with TNFα for 2 hrs and compared to their wild type counterparts. Some variation in the degrees of NF-κB dependence of subsets of genes in the p50(−/−) and Wt. + IκBαSR screens were observed (see Figure 4 Treeview image). This latter effect was most likely due to a combination of the penetrance of the IκBα super represssor as well as the different thresholds of specific NF-κB subunits required for the expression of their specific downstream target genes. Known genes and ESTs with significant decreases in expression in either the IκBαSR expressing MEFs or in p50(−/−) MEFs compared to Wt. MEFs were judged to be dependent on NF-κB for their activity.
The fold change values of a representative set of 40 genes rescued by restoring Wt. IKKα expression in IKKα null MEFs are shown in Table I. The relative NF-κB dependencies of these genes are presented as positive fold change values in one of two microarray screens [Wt. MEF vs. Wt. MEF + IκBαSR or Wt. MEF vs. p50(−/−)]. Importantly many of these genes were also rescued to levels of expression observed in wild type MEFs. Comparisons of the relative mRNA expression levels of genes in duplicate TNFα stimulated and un-stimulated samples revealed that the genes rescued by Wt. IKKα protein in IKKα null cells fell into two distinct stimulus dependent and independent classes (Table I, Figure 5 and data not shown). The signal values (equivalent to mRNA quantities) of three representative examples of the stimulus dependent (Fas ligand, C/EBP-δ and CXCL10) and independent (Clast1, NOV and C1r) classes of these Wt. IKKα rescued genes are presented in bar graph format in Figure 5. Collectively, these results reveal that the expression of a large number of NF-κB dependent genes was restored in IKKα null MEFs by retroviral transduction of a Wt. IKKα protein in a TNFα responsive manner including: IL-6, GADD45β, RANTES, ScyB5/LIX, A20, IκBα, IFITR-1, C/EBP-δ, ATF3, Fas ligand, Caspase 11, M/CSF-1, Serum amyloid A3, MIP2β, VCAM, JunB, ScyD1, ISG15, Gro1, MMP3, MMP13 and Met 1 (see Table I and selected signal value comparisons in Figure 5A). Amongst these forty representative genes rescued by the Wt. IKKα protein, examples of signal independent rescues by Wt. IKKα include ClastI/LR8, C1r, IFP35, Plf2, Plf3, Itm2b, NOV/CNN3, Decorin, Snx10 and IFITR2 (see genes highlighted in Gray in Table I and also selected signal value comparisons in Figure 5B). In agreement with these results, the expression of this same class of genes without extracellular stimulation was also found to be similarly reduced in Wt. MEFs harboring a constitutively expressed IκBα super repressor or in p50 null MEFs in comparison to their wild type counterparts (data not shown). Thus these observations show that the Wt. IKKα containing signalsome is required for both the stimulus dependent and basal levels of expression of NF-κB dependent genes. Because this class of NF-κB dependent genes required IKKα without TNFα stimulation, they define a novel class of IKKα dependent genes that are downstream of basally activated NF-κB.
Table I. A Wt. kinase competent IKKα protein rescues the expression of two classes of NF-κB dependent genes in IKKα null cells.
Forty representative IKKα dependent genes, which met the stringent selection criteria outlined in Experimental Procedures are shown. The first data column shows the fold change values of each genes obtained in two independent microarray screens of IKKα(−/−) +Wt. IKKα 2T vs. IKKα(−/−) + EV 2T. Column 2 displays their relative dependencies on IKKα in the context of wild type MEFs (i.e., Wt. MEF 2T vs. IKKα(−/−) 2T as previously described)(26)(and data not shown). Columns 3–4 show NF-κB dependencies of each gene by comparing their induced expressions in Wt. MEF 2T compared to either p50 null MEF 2T (Column 3) or Wt. MEF + IκBαSR(super repressor) 2T (26)(Column 4). The criteria for assigning the TNFα responsiveness of each rescued gene was determined on the basis of duplicate S (2T) vs. US microarray screens of Wt. IKKα rescued IKKα(−/−) MEFs (see description of criteria in Experimental Procedures). Examples of signal values of three genes are shown in Figure 5 and TaqMan ‘real time’ PCR analysis of selected genes are shown in Figure 9. Genes induced by TNFα are assigned a (+) sign and genes whose expressions were not significantly stimulated by TNFα were given a (−) sign. The expressions of nine of this representative group of forty genes were rescued independent of TNFα stimulation and are highlighted in gray.
| Accession# | Genes | |||||
|---|---|---|---|---|---|---|
| Inflammation/Stress & Immune-like Responses | IKKα(−/−) + Wt. IKKα vs IKKα( −/−) + EV | Wt. MEF vs IKKα( −/−) | Wt. MEF vs p50(−/−) | Wt. MEF vs Wt. MEF + IκBαSR | TNFα (Wt. IKKα) | |
| U43084 | IFITR-1 | 26.4/20.1 | 8.6 | 4.2 | 8.0 | + |
| AJ007970 | Guanylate binding protein 2/mGBP-2 | 19.3/22.8 | 24.3 | 56.3 | 6.4 | + |
| AF065947 | ScyA5/RANTES | 15.5/5.1 | 45.0 | 52.8 | 3.6 | + |
| X03505 | Serum amyloid A3 | 10.8/4.9 | 22.7 | 498.5 | 450.7 | + |
| M55544 | Guanylate binding protein 1/mGBP-1 | 9.2/13.3 | 22.6 | 15.2 | 14.8 | + |
| X66402 | Matrix metalloproteinase 3/MMP3 | 6.3/2.1 | 3.2 | 8.8 | 5.7 | + |
| M33266 | CXCL10/ScyB10 | 5.8/6.7 | 2.8 | 6.0 | 2.0 | + |
| X53798 | Macrophage inhibitory protein 2β/MIP2β/ScyB2 | 5.3/5.0 | 2.0 | 3.2 | 2.8 | + |
| U27267 | ScyB5/LIX | 4.6/8.1 | 21.9 | 133.9 | 5.7 | + |
| AW047476 | Guanylate binding protein 3/mGBP-3 | 3.6/5.1 | 3.8 | 3.9 | 1.7 | + |
| X66473 | Matrix metalloproteinase 13/MMP13 | 2.8/1.5 | 1.1 | 1.1 | 1.0 | + |
| AB031386 | Clast1/LR8 | 2.3/1.5 | 11.3 | 7.2 | 7.8 | - |
| J04596 | GRO1 oncogene (2 hits) | 2.2/2.3 | 1.5 | 5.2 | 2.4 | + |
| X83601 | Pentaxin related gene/Ptx3 | 2.1/3.7 | 38.2 | 12.6 | 7.8 | + |
| U92565 | ScyD1 | 2.1/1.5 | 24.2 | 18.4 | 2.2 | + |
| X56602 | ISG15 (2 hits) | 2.1/1.3 | 7.0 | 12.3 | 13.9 | + |
| X54542 | Interleukin-6 | 2.0/2.2 | 6.5 | 50.4 | 65.5 | + |
| L24118 | TNF-α induced protein 2/B94 | 1.9/2.3 | 4.0 | 20.2 | 4.4 | + |
| AW121732 | Interferon-induced protein 35/IFP35 | 1.9/1.7 | 2.0 | 6.3 | 1.4 | − |
| NF-κB Regulation | ||||||
| U19463 | A20/somatostatin receptor 1 | 5.8/12.6 | 5.7 | 5.7 | 3.3 | + |
| U57524 | IκBα (2 hits) | 1.8/2.2 | 2.2 | 4.9 | ND | + |
| AJ242778 | ABIN/Tnip1 | 1.7/1.6 | 1.4 | 2.2 | 1.4 | + |
| Growth & Develoment/Differentiation & Cell Fate | ||||||
| AF099973 | Schlafen 2 | 11.9/3.2 | 8.6 | 16.6 | 22.9 | + |
| U19118 | Activating transcription factor 3/ATF3 | 2.8/2.1 | 1.7 | 3.6 | 1.5 | + |
| X61800 | CCAAT/enhancer binding protein δ | 1.6/1.5 | 6.8 | 5.2 | 8.9 | + |
| Growth Arrest & Apoptosis | ||||||
| Y13089 | Caspase 11 | 12.4/12.3 | 4.9 | 4.3 | 5.5 | + |
| M83649 | Fas antigen | 3.2/4.4 | 8.1 | 14.0 | 8.3 | + |
| X54149 | GADD45β/MyD118 (2hits) | 2.2/2.8 | 4.3 | 4.5 | 2.2 | + |
| U76253 | Integral membrane protein 2/Itm2B/BRI | 1.6/1.5 | 1.9 | 2.5 | 2.7 | − |
| Proliferation & Survival | ||||||
| X16009 | Proliferin 3/PLF3 | 3.7/4.5 | 5.4 | 264.5 | 3.8 | − |
| Y09257 | Nephroblastoma over expressed/NOV/CCN3 | 2.1/3.3 | −1.0 | 4.0 | 6.0 | − |
| K03235 | Proliferin 2/PLF2 | 2.0/2.5 | 5.3 | 397.4 | 3.4 | − |
| M21952 | M-CSF1 | 1.9/2.1 | 2.7 | 6.0 | 3.3 | + |
| Adhesion/Extracellular matrix | ||||||
| L22545 | Collagen XVIII alpha 1/Endostatin | 5.0/7.3 | 1.9 | 2.8 | 4.7 | + |
| X53929 | Decorin | 2.0/2.8 | 8.9 | 87.7 | 27.1 | − |
| Metabolic Pathways | ||||||
| D78354 | Phospholipid scramblase 1/Plscr1 | 5.2/3.9 | 8.0 | 2.0 | 1.6 | + |
| V00835 | Metallothionein 1/Met-1 | 2.3/1.9 | 6.7 | 19.3 | 7.7 | + |
| AI746846 | Sorting nexin 10/Snx10 | 1.6/1.4 | 1.7 | −1.3 | 4.6 | − |
| Miscellaneous | ||||||
| C85523 | mVL30/Murine retrotransposable element | 2.4/1.4 | 2.9 | 2.7 | 4.6 | + |
| U43085 | IFITR-2/GARG39 | 2.3/1.9 | 2.6 | 1.5 | 2.5 | − |
Figure 5. Relative mRNA expression levels of examples of stimulus dependent and independent classes of genes only rescued by a Wt. IKKα protein.
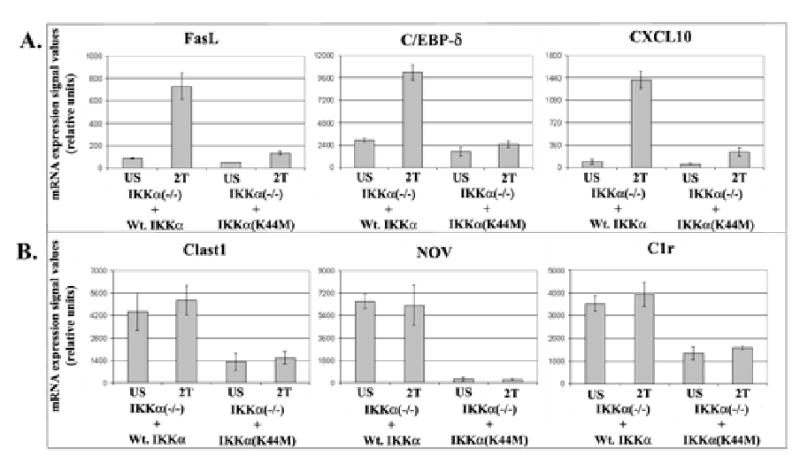
Comparisons of signal values of three NF-κB dependent genes, which were rescued only by a kinase competent Wt. IKKα protein, in a TNFα responsive manner (Panel A). Signal value comparisons of three additional genes, whose basal levels of NF-κB dependent expression were rescued only by Wt. IKKα and were unaffected by TNFα stimulation (Panel B).
In addition, the vast majority of the 118 genes rescued by physiological restoration of IKKα in IKKα null MEFs were co-dependent on NF-κB, based on their reduced or severely compromised expression in either Wt. MEFs expressing an IκBα super repressor or NF-κB p50 (−/−) MEFs (Figure 4 and Table I). Thus our global expression results also show that IKKα is not likely required for the transcription of a large number of genes outside of the NF-κB pathway. It also directly follows that in spite of IKKα’s reported ability to facilitate potentially more general aspects of chromatin activation by either phosphorylating serine 10 of histone H3 or the SMRT transcriptional co-repressor (27–29), IKKα must somehow still remain preferentially targeted to NF-κB dependent genes .
A kinase inactive IKKα mutant rescues a portion of the genes dependent on wild type IKKα in IKKα(−/−) MEFs
Because IKKα’s mechanism of action to activate NF-κB dependent transcription in the canonical pathway remains poorly understood, we next investigated whether the kinase activity of IKKα was essential for the expression of the 118 genes rescued by the Wt. IKKα protein in IKKα(−/−) MEFs. Lysine 44 in IKKα’s kinase domain is essential for its binding of ATP and its mutation to methionine prevents ATP binding thereby completely destroying IKKα kinase activity (32,44). To determine the contribution of IKKα’s kinase function for the rescue of NF-κB dependent genes, we used duplicate microarray analysis to determine the ability of a kinase dead IKKα(K44M) mutant to rescue IKKα/NF-κB dependent targets when expressed at near physiological levels in the IKKα(−/−) cells (see Hierarchical Treeview comparisons of Wt. IKKα and IKKα(K44M) expressing IKKα(−/−) cells in columns 1 and 2 and columns 3 and 4 respectively in Figure 6). These results demonstrate that ~72% of the genes rescued by Wt. IKKα in IKKα(−/−) cells were unaffected by the presence of comparable levels of an IKKα(K44M) mutant protein. Surprisingly, ~28% (33 of the 118 IKKα dependent genes) were reproducibly rescued by the IKKα(K44M) mutant. Most of these 33 IKKα(K44M) rescued genes can be visualized as two co-clustered groups in the upper and lower portions of the hierarchical treeview image presented in Figure 6.
Figure 6. Hierarchical cluster image of genes rescued by Wt. IKKα in comparison to an IKKα(K44M) mutant.
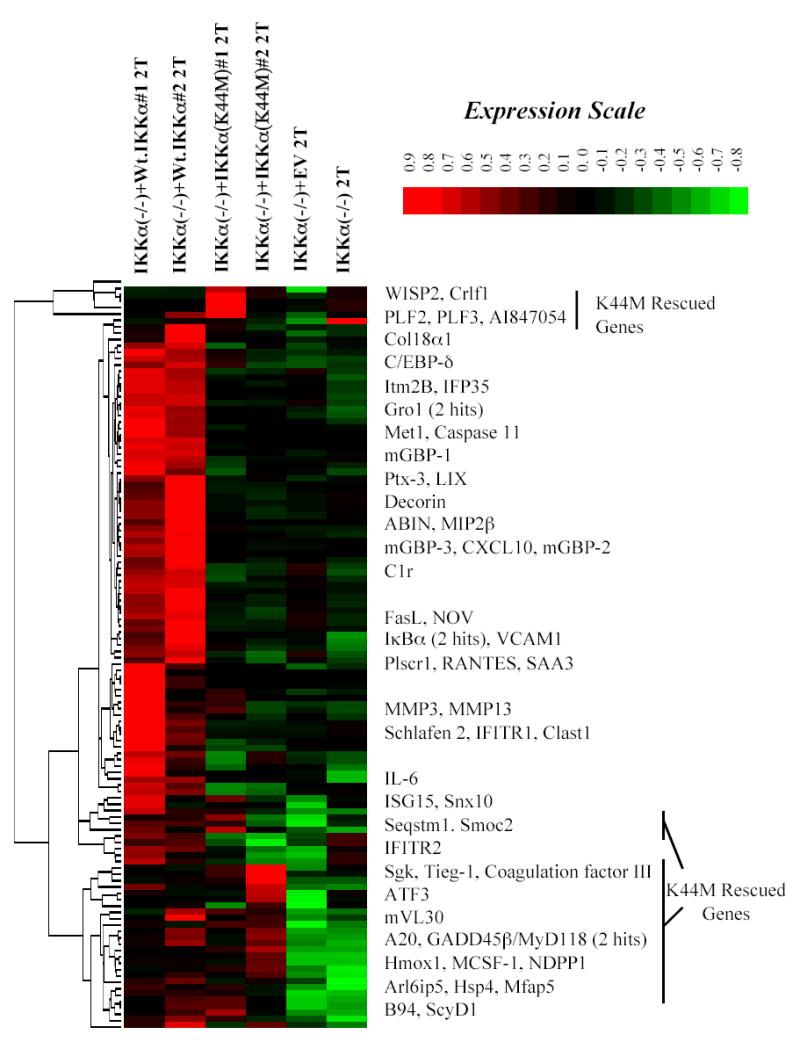
Signal values of the 118 genes rescued by Wt. IKKα in duplicate screens (columns 1 and 2) were evaluated by hierarchical clustering (as described in Figure 3) in comparison to their signal values in duplicate screens of 2 hr TNFα (2T) stimulated IKKα (−/−) MEFs expressing a kinase inactive IKKα(K44M) mutant (columns 3 and 4). As in Figure 4, the IKKα specificities of the rescues can be visualized in columns 5 and 6, which display the signal values of the 118 genes in IKKα(−/−) null MEFs or the same cells expressing an empty retroviral (EV).
The fold change values of 20 representative genes rescued by the IKKα(K44M) mutant protein are presented in Table I. Comparable degrees of rescue of each of these genes were achieved with Wt. IKKα and the kinase inactive IKKα mutant proteins. However, in contrast to the expression of genes restored by Wt. IKKα, pairs of TNFα stimulated and unstimulated microarray screens of IKKα(K44M) transduced IKKα null cells revealed that only a small fraction of the genes rescued by the IKKα(K44M) mutant responded to TNFα stimulation (see examples of A20, M/CSF-1 and VL30 highlighted in gray in Table II). Bar graphs of signal values (comparable to amounts of mRNAs) of three representative examples of the stimulus dependent (A20, mVL30 and B94) and independent (Coagulation factor III, Sgk and NDPP1) classes of IKKα(K44M) rescued genes are shown in Figure 7. In addition to these two distinct classes of genes, a third class of IKKα(K44M) rescued genes only responded to TNFα stimulation after rescue by Wt. IKKα and not if rescued by the IKKα(K44M) kinase inactive mutant (see examples GADD45β, ATF3 and JunB in Table II). Interestingly, in comparison to the NF-κB dependent genes solely rescued by the Wt. IKKα protein, a larger fraction of the IKKα/NF-κB dependent genes rescued by the kinase inactive IKKα(K44M) mutant preferentially encoded proteins associated with NF-κB autoregulation, growth arrest, apoptosis, proliferation and survival (see representative genes in Table II and discussion section). In addition, most of these NF-κB dependent genes, which are rescued by wild type and kinase inactive IKKα, depend on the IKKα for their basal levels of expression in the absence of an extracellular stimulus. Thus our global expression profiling analysis reveals the surprising result that different NF-κB dependent genes differentially require IKKα kinase activity for their basal and TNFα dependent expression, with the majority of genes encoding proteins associated with pro-inflammatory stress-like responses requiring IKKα kinase activity.
Table II. NF-κB dependent genes rescued by IKKα independent of its kinase activity.
Twenty representative genes meeting the criteria of being rescued by both the Wt . IKKα and IKKα(K44M) proteins are shown. Fold Change values of these twenty genes obtained from duplicate microarray comparisons using independent samples of IKKα(−/−) + IKKα(K44M) 2T vs. IKKα(−/−) + EV 2T are indicated in Column 1. Column 2 shows the results obtained for the same genes in duplicate microarray comparisons of IKKα(−/−) + Wt. IKKα 2T vs. IKKα(−/−) + EV 2T. Column 3 displays their relative dependencies on IKKα in the context of wild type MEFs (i.e., Wt. MEF 2T vs. IKKα(−/−) 2T as previously described)(26)(and data not shown). Columns 4–5 shows their NF-κB dependencies by comparing their expressions in Wt. MEF 2T compared to either p50 null MEF 2T (Column 4) or Wt. MEF + IκBαSR(super repressor) 2T (26)(Column 5). The TNFα dependencies for the IKKα rescued expression of each gene are shown in the context of their independent rescues by physiological levels of either IKKα(K44M) (Column 6) or Wt. IKKα (Column 7). The criteria for assigning the TNFα responsiveness of each rescued gene was determined on the basis of duplicate S (2T) vs. US microarray screens [for Wt. IKKα or IKKα(K44M) rescued IKKα(−/−) MEFs as indicated] (see description of criteria in Experimental Procedures). Examples of signal values of three genes are shown in Figure 7 and TaqMan ‘real time’ PCR analyses of selected genes are in Figure 9. Genes induced by TNFα are assigned a (+) sign and genes whose expression was not significantly stimulated by TNFα were given a (−). The IKKα(K44M) mutant in a TNFα responsive manner comparable to that achieved by Wt. IKKα rescued three genes highlighted in gray.
| Accession# | Genes | |||||||
|---|---|---|---|---|---|---|---|---|
| Inflammation/Stress & Immune-like Responses | IKKα( −/−) + IKKα (K44M) vs IKKα( −/−) | IKKα( −/−) + Wt. IKKα vs IKKα( −/−) + EV | Wt. MEF vs IKKα( −/−) | Wt. MEF vs p50(−/−) | Wt. MEF vs Wt. MEF + IκBαSR | TNFα (K44M) | TNFα (Wt. IKKα) | |
| M26071 | Coagulation factor III | 2.2/2.6 | 1.7/1.6 | 4.2 | 2.8 | 4.8 | − | − |
| X56824 | Heme oxygenase 1/Hmox1/HO-1 | 1.8/2.1 | 1.5/1.6 | 4.6 | 6.0 | 9.2 | − | − |
| AA615831 | Heat shock protein 4/Hsp4/Apg-2 | 1.5/1.4 | 1.5/1.5 | 2.0 | 2.6 | 1.6 | − | − |
| NF-κB Regulation | ||||||||
| U19463 | A20/somatostatin receptor 1 | 18.6/6.8 | 5.8/12.6 | 5.7 | 5.7 | 3.3 | + | + |
| U40930 | Sequestosome 1/Sqstm1/p62 | 1.7/1.3 | 2.3/1.8 | 1.4 | 2.3 | 2.4 | − | − |
| D10727 | NPC derived proline rich protein 1/NDPP1/CARD8 | 1.5/1.8 | 1.4/1.7 | 1.1 | 2.9 | 1.5 | − | − |
| Growth & Develoment/Differentiation & Cell Fate | ||||||||
| U19118 | Activating transcription factor 3/ATF3 | 3.8/2.0 | 2.8/2.1 | 1.7 | 3.6 | 1.5 | − | + |
| Growth Arrest & Apoptosis | ||||||||
| X54149 | GADD45β/MyD118 (2hits) | 2.8/1.9 | 2.2/2.8 | 4.3 | 4.5 | 2.2 | − | + |
| AF064088 | TGFβ inducible early growth response/Tieg1/GIF | 2.2/3.9 | 1.6/1.3 | 4.0 | 5.0 | 6.1 | − | − |
| U20735 | Jun-B oncogene | 1.7/2.2 | 1.5/1.5 | 1.5 | 21.7 | 5.6 | − | + |
| Proliferation & Survival | ||||||||
| X16009 | Proliferin 3/PLF3 | 5.0/83.3 | 3.7/4.5 | 5.4 | 264.5 | 3.8 | − | − |
| K03235 | Proliferin 2/PLF2 | 2.6/22.3 | 2.0/2.5 | 5.3 | 397.4 | 3.4 | − | − |
| AA270365 | Cytokine receptor-like factor 1/Crlf1 | 14.1/4.9 | 2.0/1.8 | 3.6 | −1.3 | 2.6 | − | − |
| M21952 | M-CSF1 | 2.2/1.7 | 1.9/2.1 | 2.7 | 6.0 | 3.3 | + | + |
| Adhesion/Extracellular matrix | ||||||||
| AW121179 | Microfibrillar associated protein 5/Mfap5/Magp2 | 5.0/3.8 | 3.6/4.5 | −1.2 | −6.8 | 4.4 | − | − |
| AA980164 | SPARC related modular calcium binding 2/Smoc2 | 3.4/2.5 | 2.3/2.2 | −1.6 | −11.8 | 4.0 | − | − |
| Metabolic Pathways | ||||||||
| AW046181 | Serum/glucocorticoid regulated kinase | 2.4/3.8 | 1.7/2.0 | 3.1 | 4.6 | 2.2 | − | − |
| AW049647 | ADP-ribosylation factor-like 6 interacting protein 5/Arl6ip5/GTRAP 3-18 | 1.5/1.4 | 1.5/1.4 | 1.9 | 2.1 | 1.2 | − | − |
| Miscellaneous | ||||||||
| C85523 | mVL30/Murine retrotransposon | 2.1/2.1 | 2.4/1.4 | 2.9 | 2.7 | 4.6 | + | + |
| AI847054 | EST | 1.9/1.7 | 2.0/2.4 | 22.6 | 19.7 | 32.1 | − | − |
Figure 7. Relative mRNA expression levels of examples of stimulus dependent and independent classes of genes rescued by the IKKα protein regardless of its kinase activity.
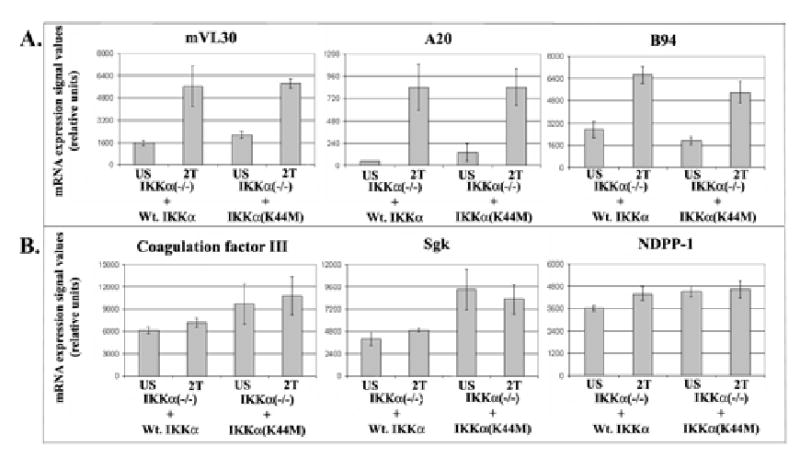
Comparisons of signal values of three NF-κB dependent genes, which were comparably rescued by both Wt. IKKα and the IKKα(K44M) mutant, in a TNFα responsive manner (Panel A). Signal value comparisons of three additional NF-κB target genes, whose basal levels of NF-κB dependent expression were rescued by either a wild type or a kinase inactive IKKα mutant but were unresponsive to TNFα stimulation (Panel B).
TaqMan ‘real time’ PCR validations of Wt. IKKα and IKKα(K44M) rescued genes
As an additional validation of the duplicate microarray screens, TaqMan ‘real time’ PCR experiments were performed on eight NF-κB target genes (IL-6, ISG15, RANTES, SAA3, VCAM1, GADD45B, A20 and ATF3). Importantly, TaqMan validations were performed at least in duplicate on a third independent set of unstimulated and 2 hr (2T) TNFα stimulated samples, which were not employed in the duplicate microarray screens. Figure 8 shows the absolute expression levels in the context of TNFα stimulation of each of these eight genes as mRNA copy numbers in IKKα (−/−) MEFs expressing Wt. IKKα, IKKα(K44M) or an empty retroviral vector (EV) compared to Wt. MEFs. Each of these genes are expressed in Wt. MEFs and Wt. IKKα rescued IKKα (−/−) MEFs above their low to negligible levels in IKKα(−/−) +EV cells. Some variations in the degrees of the IKKα dependent rescues in comparison to wild type control cells are noted with some genes being expressed at higher levels in the wild type control and others expressed at higher levels in the Wt. IKKα rescued cells. Figure 9 illustrates the TNFα dependencies of the same eight genes. In agreement with the duplicate microarray screens, A20, ATF3, and MyD118/GADD45β, were rescued by the IKKα(K44M) mutant in comparison to IKKα(−/−)EV MEFs, while IL-6, ISG15, RANTES, SAA3 and VCAM1 failed to exhibit expression above background in IKKα(K44M) expressing IKKα(−/−) cells. Of these three IKKα(K44M) rescued genes, only A20 significantly responded to TNFα stimulation. In agreement with our duplicate microarray screens, after their rescue by Wt. IKKα each of these eight genes was confirmed to be TNFα responsive.
Figure 8. TaqMan ‘real time’ PCR analysis of selected genes differentially rescued by Wt. IKKα and IKKα(K44M).
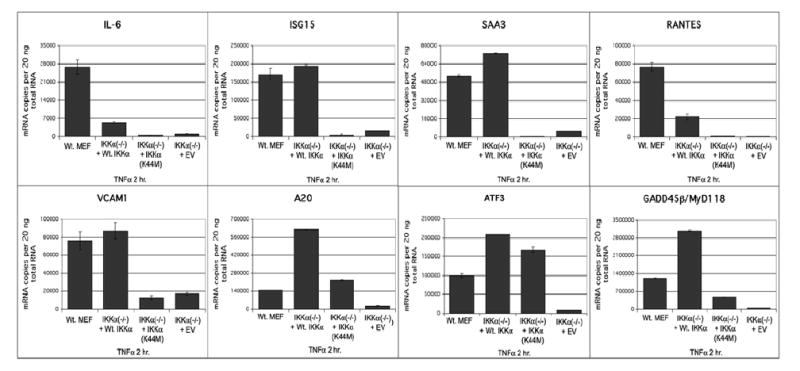
Independent samples of Wt. MEFs and IKKα(−/−) MEF expressing Wt. IKKα, EV (empty retroviral vector) or IKKα(K44M) were stimulated with 20 ng/ml TNFα for 2 hrs. and RNAs were prepared and subjected to TaqMan real time PCR analysis. The levels of expression of eight representative genes (IL-6, ISG15, RANTES, SAA3, VCAM1, GADD45β, ATF3 and A20) were quantitatively compared. Each bar represents data obtained at least in duplicate with the indicated standard deviations. All samples were normalized to a GAPDH probe set and mRNA copy numbers were determined in comparison to a genomic DNA standard for each probe set.
Figure 9. TaqMan ‘real time’ PCR analysis of the TNFα dependencies of selected genes rescued by Wt. IKKα or IKKα(K44M).
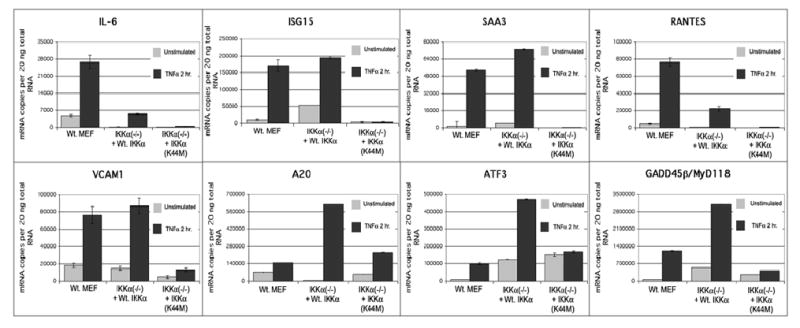
The relative levels of expression in TNFα stimulated and unstimulated cells of the eight selected genes in Figure 8 (IL-6, ISG15, RANTES, SAA3, VCAM1, GADD45β, ATF3 and A20) are shown. TaqMan PCR reactions were performed and quantitated as described in Experimental Procedures and in the legend of Figure 8.
Discussion
Studies of IKKα(−/−) and IKKβ(−/−) MEFs have definitively shown that IKKβ is the in vivo IκBα kinase, and that IKKα is not needed for IκB degradation, NF-κB nuclear localization nor for inducing NF-κB DNA binding activity in response to proinflammatory NF-κB stimuli like TNFα. However, IKKα functions in the canonical NF- κB pathway to ensure or modulate the transcriptional competence of DNA bound NF-κB. In support of this view, we have shown herein that restoration of IKKα(−/−) MEFs with near physiological levels of a wild type IKKα kinase globally and specifically activated NF-κB dependent genes in response to TNFα stimulation. In addition, these experiments also revealed a hitherto unknown requirement for IKKα to maintain the basal levels of expression of specific NF-κB dependent genes in the absence of an extracellular stress-like stimulus. Furthermore, the ability of a kinase inactive IKKα mutant to rescue a portion of these NF-κB target genes reveals that even though the IKKα protein is globally required for the expression of NF-κB dependent genes, its role as a functional kinase is also target gene specific. In summary our findings show that genes dependent on IKKα and NF-κB can be formally divided into five distinct classes of responsive genes: (1) genes that require a functional Wt. IKKα kinase for their stimulus dependent, NF-κB dependent expression; (2) genes that require a functional Wt. IKKα kinase for their stimulus independent basal NF-κB dependent expression; (3) genes that require a functional Wt. IKKα kinase for their signal dependent rescue but only require an IKKα protein for their basal, stimulus independent expression, (4) genes that require the IKKα protein regardless of its kinase activity for their stimulus dependent, NF-κB dependent expression and (5) genes that require an IKKα protein regardless of its kinase activity for their stimulus independent, basal NF-κB dependent expression.
Modifications of NF-κB subunits and other post-translational nuclear processes are also necessary for the induction of NF-κB target genes and a number of reports have implicated IKKα in these events. Phosphorylation of serines 276 (in the Rel Homology domain/RHD) and serines 529 and 536 in the transcriptional activation domain (TAD) of the NF-κB p65/RelA subunit have been suggested to play activating roles, and a number of kinases have been directly implicated in this step including the IKKs (45–52). Transactivation by p65/RelA in response to TNFα was localized to serine 529 within the p65/RelA TAD and found to mediate its transcriptional activation independent of NF- κB’s ability to bind DNA [Wang, 1998 #404]. Phosphorylation of serine 276 was also found to be involved in the activation of p65/RelA at least in part by controlling its association with the p300/CBP co-activator or the histone deacetylase HDAC-1 (53). IKKβ and IKKα were both implicated as downstream effectors of Akt- dependent signaling targeted to serines 529 and 536 in the p65/RelA TAD, which in part appeared to involve the engagement of the CBP/p300 transcriptional co-activator [Madrid, 2000 #838; Madrid, 2001 #1245]. PTEN, a negative upstream effector of Akt, was also reported to inhibit TNFα induced NF-κB activation [Koul, 2001 #1304;Gustin, 2001 #1305], which was subsequently shown to occur solely at the level of p65 transactivation [Mayo, 2002 #1746]. Additionally, IL-1 and Akt mediated NF-κB activation was found to involve p65 TAD phosphorylations with a co-dependency on IKKα and IKKβ [Sizemore, 2002 #1745]. TNFα induced phosphorylation of p65/RelA serine 536 was recently shown to be dependent on both IKKα and IKKβ and mediated by TRAF2, TRAF5 and TAK1 signaling (54) and a requirement for IKKα in p65 serine 529 phosphorylation and NF-κB dependent transcriptional activation in response to LTβ stimulation has also recently been reported (55). In addition, the activation of NF-κB by the HTLV-Tax1 protein involves the specific phosphorylation of p65 serines 529 and 536, requiring IKKα, but not IKKβ (56). IKKα’s mechanism of action in the canonical NF-κB pathway has also been proposed to be purely nuclear in nature. In this context, IKKα has been shown to migrate into the nucleus (57) and associate with the promoters of NF-κB dependent genes upon TNFα stimulation (27,28). More recently mitogenic activation of the c-fos gene by epidermal growth factor dependent signaling was found to require the constitutive and induced recruitment of p65/RelA and IKKα respectively to the c-fos promoter (58). Because IKKα was also found to phosphorylate serine 10 of histone H3 in vitro and the phosphorylation status of histone H3 in IKKα(−/−) MEFs was enhanced by introduction of wild type IKKα (27,28), these results suggested that IKKα might be functioning by enhancing the transcriptional accessibility of the chromatin of NF-κB target genes and potentially a broader range of genes that also respond to TNFα stimulation. More recently, IKKα was also shown to be critical for the signal dependent expression of the cIAP-2 and IL-8 NF-κB dependent genes in several cellular contexts by virtue of its required ability to phosphorylate the SMRT co-repressor, thereby also inducing chromatin activation by facilitating the exchange of transcriptional corepressors for co-activators (29).
Wild type IKKα kinase restores the expression of NF-κB dependent genes in IKKα null MEFs
Our comparative genomic analysis of the abilities of wild type IKKα and a kinase dead IKKα(K44M) ATP binding domain mutant indicate that IKKα’s kinase activity is required for the activation of most but not all genes whose expressions are dependent on NF-κB. Indeed, we find that IKKα kinase activity is required for the induction of numerous TNFα responsive NF-κB target genes (including RANTES, IκBα, IL-6, ISG- 15, IFITR-1, mGBP-2, mGBP-3, Gro1, Fas ligand, Jun-B, Caspase 11, ScyD1, Serum amyloid A3, MMP3, MMP13, ScyB5/LIX, Ptx3, Schaflen 2, Met1 and C/EBPδ), many of which we previously identified as targets of pro-inflammatory cytokine mediated NF- κB activation in Wt. MEFs but not in IKKα null MEFs (26).
The expression of the majority of IKKα rescued NF-κB target genes in IKKα null MEFs were rescued by Wt. IKKα to within 2 fold of their levels in wild type MEFs (see fold change values of representative set of 40 genes in Table I) with a portion of NF-κB targets being expressed at higher levels in wild type MEFs. This latter observation is not that surprising given that long term inactive genes are known to differentially acquire the attributes of transcriptionally inaccessible chromatin states, which is unlikely to be fully overcome by IKKα re-expression. In addition, some NF-κB dependent genes were also expressed at higher levels in IKKα rescued IKKα (−/−) cells compared to their wild type counterpart (see fold change values for IFITR-1, Gro1 and Caspase 11 in Wt. IKKα rescued IKKα null cells compared to their levels of expression in Wt. MEFs in Table I), which could reflect intrinsic differences in the absolute levels of NF-κB target gene expression in different cellular backgrounds. A fraction of the TNFα responsive genes rescued by Wt. IKKα in IKKα null fibroblasts did not appear to be dependent on NF-κB for their expression employing the criteria of being unaffected by the inhibitory effects of the IκBα super repressor in Wt. MEFs nor by the loss of the NF-κB p50 subunit. However, because genes such as MMP13, an NF-κB target gene in other cellular contexts (59–61), were amongst this latter gene subset (see Figures 4 and 6 and data not shown), this supports our conclusion that the large majority of TNFα responsive, IKKα dependent genes in these cells are also NF-κB dependent.
Surprisingly, a number of NF-κB dependent genes were dependent on IKKα for their basal, stimulus independent expression in IKKα compromised MEFs (21 of the 53 representative genes rescued by IKKα in Tables I and II). Furthermore, these same genes were also expressed at significantly lower levels in NF-κB compromised MEFs compared to wild type MEFs (data not shown). Thus our findings show that the IKKα containing signalsome is also required to maintain the basal, intrinsic expression levels of specific NF-κB dependent genes. Interestingly, 14 out of 21 of these stimulus independent/Wt. IKKα rescued genes were also preferentially rescued by the IKKα(K44M) mutant in the absence of stimulation (see Table II), also demonstrating that IKKα’s kinase activity is not required to restore their basal levels of NF-κB dependent gene expression in most but not all cases.
IKKα does not always function as a kinase to ensure the expression of NF-κB dependent genes
We also employed duplicate microarray screens to compare the abilities of Wt. IKKα or a kinase dead IKKα(K44M) mutant protein to rescue NF-κB dependent gene expression in IKKα null fibroblasts on a genomic scale. These findings, in combination with quantitative TaqMan real time PCR assays on selected genes, reveal that even though IKKα’s kinase activity is required to activate the majority of NF-κB dependent genes, it is not directly functioning as a kinase for the expression of ~28% of the NF-κB dependent genes in this cell background. Interestingly most of the latter subset of IKKα(K44M) rescued genes were unresponsive to TNFα, with the exception of several genes (including M/CSF-1, A20, mL30 and B94) (see Table II, Figures 7 and 9). In contrast to their TNFα independent rescues by the IKKα(K44M) mutant, GADD45β/MyD118, ATF3 and JunB were rescued by Wt. IKKα in a TNFα dependent manner (Table II and Figure 9). It is also noteworthy in this context that our quantitative TaqMan analysis revealed that Wt. IKKα in comparison to IKKα(K44M) also rescued higher levels of GADD45β, ATF3 and A20 expression (Figures 8 and 9). Taken together these observations show that the nature of the Wt. IKKα vs. IKKα(K44M) mediated rescues of GADD45β, ATF3, JunB and perhaps even A20 are intrinsically different from each other, revealing that their dependencies on the IKKα protein occurs at more than one regulatory level.
Of particular interest, a larger proportion of the IKKα rescued genes, which were also rescued by the kinase inactive IKKα(K44M) mutant, encode proteins with functional properties in either NF-κB autoregulation, cellular proliferation, growth arrest, apoptosis or cellular survival (M/CSF-1, PLF2, PLF3, GADD45β/MYD118, A20, Sequestosome/p62, NDPP1/CARD 8, JunB and ATF3). GADD45β/MyD118 was first described to function as an effector of myeloid cell differentiation and as a member of a class of cell cycle checkpoint protein arresting cellular growth in response to DNA damage or in association with terminal cellular differentiation (62,63). In contrast and more recently, GADD45β has also been shown to be an NF-κB dependent anti-apoptotic effector in response to TNFα, where it appears to act by directly blocking MKK7/JNKK2 activity thereby leading to the suppression of the c-Jun N-terminal kinase (JNK) cascade in response to TNFα stimulation (64,65)and also by suppressing Fas/CD95/APO-1 induced caspase activation in response to CD40 triggering in B lymphocytes (66). Our results show that the induction of GADD45β by TNFα requires IKKα kinase activity for its NF-κB dependent expression. However, because we also find that a kinase inactive IKKα(K44M) mutant restored unstimulated levels of GADD45β expression in IKKα null MEFs, our results also indicate that other intrinsic properties of the IKKα protein contribute to the expression of GADD45β under different physiological circumstances, perhaps leading to other functional properties ascribed to GADD45β/MyD118. Sequestosome/p62, an atypical protein kinase C (a PKC) interacting protein, interacts with RIP (receptor interacting protein) linking it to TNFα mediated NF-κB induction with its inhibition or down-modulation also interfering with IL-1 and TRAF 6 dependent NF-κB activation (67). A20 is a TNFα and IL-1 induced zinc-finger protein, which has been reported to act as a negative effector of NF-κB (68–70), mediated by RIP and TRAF2 signaling (70). A20 was also recently shown to attenuate TNFα mediated NF- κB activation via the cooperative action of its two intrinsic ubiquitin-editing domains resulting in the polyubiquitination of receptor interacting protein (RIP), an essential mediator of the TNF receptor 1 (TNFRI) signaling complex, thereby targeting RIP’s proteasomal destruction (71). Akin to GADD45β, A20 has also been shown to block TNFα dependent apoptosis at least in part by preventing TRADD and RIP recruitment to TNFRI (72,73). NDPP1 a novel member of the caspase-associated recruitment domain (CARD) family of proteins has also been reported to either promote or suppress apoptotic responses in specific cell types and to block TNFα induced NF-κB activation (74,75). ATF3, a member of the ATF/CREB family of leucine zipper transcription factors (also known as LRF-1/TI-241) (76,77) and a stress induced transcriptional repressor (78–80), has recently been shown to be dependent on both the NF-κB and JNK signaling pathways for its expression in response to TNFα and nitric oxide (81). ATF3 has also been shown to play roles in either protection against or induction of apoptotic responses, dependent on the nature of the signal and cellular context (79,82,83). Finally, Proliferin 2 and Proliferin 3/Mitogen-regulated protein 3, which were also rescued by both Wt. IKKα and IKKα(K44M) in a stimulus independent manner, are members of the prolactin growth hormone family of proliferins whose functional properties have been associated with cellular proliferation and migration, wound healing and angiogenesis (84–88). Because an IKKα(K44M) kinase inactive mutant was found to be incapable of phosphorylating serine 10 of histone H3 (28), our observations also indicate that not all genes whose expressions are co-dependent on IKKα and NF-κB require IKKα’s kinase activity for histone H3 phosphorylation.
In summary, our global expression profiling analysis shows that IKKα functions in both kinase dependent and independent modes, thereby revealing the existence of five distinct classes of genes co-dependent on IKKα and NF-κB in mouse embryonic fibroblasts. In addition to the importance of a Wt. IKKα containing signalsome for the activities of many stimulus dependent target genes of NF-κB, the Wt. IKKα protein also plays a role in the regulation of a distinct class of NF-κB dependent genes requiring only basal levels of active NF-κB for their regulated expression. We envision that IKKα’s kinase independent mode of action to ensure the expression of a subset of NF-κB dependent genes may be attributable to a novel regulatory/docking-like property of the IKKα protein, which facilitates the recruitment of other regulatory factors required for the expression of specific downstream targets of the NF-κB pathway.
Acknowledgments
We gratefully acknowledge Dr. Michael Karin for providing us with IKKα(−/−) and IKKγ/NEMO (−/−) MEFs and Dr. Amer Beg for p65/p50(−/−) and p50(−/−) MEFs. This research was supported in part by an NIH grant awarded to KBM and the Fondazione Cassa di Risparmio di Bologna. KBM is also a senior scholar of the Institute of Advanced Studies of the University of Bologna.
References
- 1.Karin M, Ben-Neriah Y. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh, S., and Karin, M. (2002) Cell109 Suppl, S81–96 [DOI] [PubMed]
- 3.Karin M, Lin A. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 4.Li Q, Verma IM. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 5.Kucharczak J, Simmons MJ, Fan Y, Gelinas C. Oncogene. 2003;22:8961–8982. doi: 10.1038/sj.onc.1207230. [DOI] [PubMed] [Google Scholar]
- 6.Weih F, Caamano J. Immunol Rev. 2003;195:91–105. doi: 10.1034/j.1600-065x.2003.00064.x. [DOI] [PubMed] [Google Scholar]
- 7.Karin M, Yamamoto Y, Wang QM. Nat Rev Drug Discov. 2004;3:17– 26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- 8.Bonizzi G, Karin M. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 9.Patke A, Mecklenbrauker I, Tarakhovsky A. Curr Opin Immunol. 2004;16:251–255. doi: 10.1016/j.coi.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Weil R, Israel A. Curr Opin Immunol. 2004;16:374–381. doi: 10.1016/j.coi.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 11.Hayden MS, Ghosh S. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 12.Baldwin A., Jr Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh S, May MJ, Kopp EB. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 14.Pahl HL. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto Y, Gaynor RB. Trends Biochem Sci. 2004;29:72–79. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 16.Pomerantz JL, Baltimore D. Mol Cell. 2002;10:693–694. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- 17.Kato T, Delhase M, Hoffmann A, Karin M. Mol Cell. 2003;12:829–839. doi: 10.1016/s1097-2765(03)00358-7. [DOI] [PubMed] [Google Scholar]
- 18.Tergaonkar V, Bottero V, kawa M, Li Q, Verma IM. Mol Cell Biol. 2003;23:8070–8083. doi: 10.1128/MCB.23.22.8070-8083.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, Karin M. Cell. 2001;107:763–775. doi: 10.1016/s0092-8674(01)00599-2. [DOI] [PubMed] [Google Scholar]
- 20.Hu Y, Baud V, Oga T, Kim KI, Yoshida K, Karin M. Nature. 2001;410:710–714. doi: 10.1038/35070605. [DOI] [PubMed] [Google Scholar]
- 21.Sil AK, Maeda S, Sano Y, Roop DR, Karin M. Nature. 2004;428:660–664. doi: 10.1038/nature02421. [DOI] [PubMed] [Google Scholar]
- 22.Ohazama A, Hu Y, Schmidt-Ullrich R, Cao Y, Scheidereit C, Karin M, Sharpe PT. Dev Cell. 2004;6:219–227. doi: 10.1016/s1534-5807(04)00024-3. [DOI] [PubMed] [Google Scholar]
- 23.Dejardin E, Droin NM, Delhase M, Haas E, Ca Y, Makris C, Karin M, Ware CF, Green DR. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- 24.Amir RE, Haecker H, Karin M, Ciechanover A. Oncogene. 2004;23:2540–2547. doi: 10.1038/sj.onc.1207366. [DOI] [PubMed] [Google Scholar]
- 25.Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. J Biol Chem. 2002;277:3863–3869. doi: 10.1074/jbc.M110572200. [DOI] [PubMed] [Google Scholar]
- 26.Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, Mische S, Li J, Marcu KB. J Biol Chem. 2002;277:45129–45140. doi: 10.1074/jbc.M205165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Nature. 2003;423:655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- 29.Hoberg JE, Yeung F, Mayo MW. Mol Cell. 2004;16:245–255. doi: 10.1016/j.molcel.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 30.Connelly MA, Marcu KB. Cell Mol Biol Res. 1995;41:537–549. [PubMed] [Google Scholar]
- 31.Geleziunas R, Ferrell S, Lin X, Mu Y, Cunningham EJ, Grant M, Connelly MA, Hambor JE, Marcu KB, Greene WC. Mol Cell Biol. 1998;18:5157–5165. doi: 10.1128/mcb.18.9.5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKenzie FR, Connelly MA, Balzarano D, Muller JR, Geleziunas R, Marcu KB. Mol Cell Biol. 2000;20:2635–2649. doi: 10.1128/mcb.20.8.2635-2649.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgenstern JP, Land H. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahadevappa M, Warrington JA. Nat Biotechnol. 1999;17:1134–1136. doi: 10.1038/15124. [DOI] [PubMed] [Google Scholar]
- 35.Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 36.Eisen MB, Spellman PT, Brown PO, Botstein D. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K. PCR Methods Appl. 1995;4:357–362. doi: 10.1101/gr.4.6.357. [DOI] [PubMed] [Google Scholar]
- 38.Field J, Nikawa J, Broek D, MacDonald B, Rodgers L, Wilson IA, Lerner RA, Wigler M. Mol Cell Biol. 1988;8:2159–2165. doi: 10.1128/mcb.8.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Renard P, Ernest I, Houbion A, Art M, Le Calvez H, Raes M, Remacle J. Nucleic Acids Res. 2001;29:E21. doi: 10.1093/nar/29.4.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Q, Lu Q, Hwang JY, Buscher D, Lee KF, zpisua-Belmonte JC, Verma IM. Genes Dev. 1999;13:1322–1328. doi: 10.1101/gad.13.10.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Science. 1999;284:316–320. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 42.Takeda K, Takeuchi O, Tsujimura T, tami S, Adachi O, Kawai T, Sanjo H, Yoshikawa K, Terada N, Akira S. Science. 1999;284:313–316. doi: 10.1126/science.284.5412.313. [DOI] [PubMed] [Google Scholar]
- 43.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Cell. 1995;80:321– 330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 44.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 45.Gerristen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. Proc Natl Acad Sci U S A. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ. Science. 1997;275:523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- 47.Zhong H, Suyang H, Erdjument-Bromage P, Tempst P, Ghosh S. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 48.Zhong H, Voll RE, Ghosh S. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 49.Bergmann M, Hart L, Lindsay M, Barnes PJ, Newton R. J Biol Chem. 1998;273:6607–6610. doi: 10.1074/jbc.273.12.6607. [DOI] [PubMed] [Google Scholar]
- 50.Sizemore N, Leung S, Stark GR. Mol Cell Biol. 1999;19:4798–4805. doi: 10.1128/mcb.19.7.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang D, Westerheide SD, Hanson JL, Baldwin AS., Jr J Biol Chem. 2000;275:32592–32597. doi: 10.1074/jbc.M001358200. [DOI] [PubMed] [Google Scholar]
- 52.Vermeulen L, De Wilde G, Damme PV, Vanden Berghe W, Haegeman G. Embo J. 2003;22:1313–1324. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhong H, May MJ, Jimi E, Ghosh S. Mol Cell. 2002;9:625–636. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- 54.Sakurai H, Suzuki S, Kawasaki N, Nakano H, Okazaki T, Chino A, Doi T, Saiki I. J Biol Chem. 2003;278:36916–36923. doi: 10.1074/jbc.M301598200. [DOI] [PubMed] [Google Scholar]
- 55.Jiang, X., Takahashi, N., Matsui, N., Tetsuka, T., and Okamoto, T. (2003) J Biol Chem278, 919–926 Epub 2002 Nov 2004 [DOI] [PubMed]
- 56.O’Mahony AM, Montano M, Van Beneden K, Chen LF, Greene WC. J Biol Chem. 2004;279:18137–18145. doi: 10.1074/jbc.M401397200. [DOI] [PubMed] [Google Scholar]
- 57.Birbach A, Gold P, Binder BR, Hofer E, de Martin R, Schmid JA. J Biol Chem. 2002;277:10842–10851. doi: 10.1074/jbc.M112475200. [DOI] [PubMed] [Google Scholar]
- 58.Anest, V., Cogswell, P. C., and Baldwin, A. S. (2004) J. Biol. Chem., M404380200 [DOI] [PubMed]
- 59.Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Arthritis Rheum. 2000;43:801–811. doi: 10.1002/1529-0131(200004)43:4<801::AID-ANR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 60.Vincenti, M. P., and Brinckerhoff, C. E. (2002) Arthritis Res4, 157–164 Epub 2001 Nov 2023 [DOI] [PMC free article] [PubMed]
- 61.Liacini A, Sylvester J, Li WQ, Huang W, Dehnade F, Ahmad M, Zafarullah M. Exp Cell Res. 2003;288:208–217. doi: 10.1016/s0014-4827(03)00180-0. [DOI] [PubMed] [Google Scholar]
- 62.Abdollahi A, Lord KA, Hoffman-Liebermann B, Liebermann DA. Oncogene. 1991;6:165–167. [PubMed] [Google Scholar]
- 63.Liebermann DA, Hoffman B. Oncogene. 1998;17:3319–3329. doi: 10.1038/sj.onc.1202574. [DOI] [PubMed] [Google Scholar]
- 64.De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G. Nature. 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 65.Papa S, Zazzeroni F, Bubici C, Jayawardena S, Alvarez K, Matsuda S, Nguyen DU, Pham CG, Nelsbach AH, Melis T, Smaele ED, Tang WJ, D’Adamio L, Franzoso G. Nat Cell Biol. 2004;6:146–153. doi: 10.1038/ncb1093. [DOI] [PubMed] [Google Scholar]
- 66.Zazzeroni F, Papa S, Algeciras-Schimnich A, Alvarez K, Melis T, Bubici C, Majewski N, Hay N, De Smaele E, Peter ME, Franzoso G. Blood. 2003;102:3270–3279. doi: 10.1182/blood-2003-03-0689. [DOI] [PubMed] [Google Scholar]
- 67.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. Embo J. 2000;19:1576– 1586. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ferran C, Stroka DM, Badrichani AZ, Cooper JT, Wrighton CJ, Soares M, Grey ST, Bach FH. Blood. 1998;91:2249–2258. [PubMed] [Google Scholar]
- 69.Grey ST, Arvelo MB, Hasenkamp W, Bach FH, Ferran C. J Exp Med. 1999;190:1135–1146. doi: 10.1084/jem.190.8.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heyninck K, De Valck D, Berghe WV, Van Criekinge W, Contreras R, Fiers W, Haegeman G, Beyaert R. J Cell Biol. 1999;145:1471–1482. doi: 10.1083/jcb.145.7.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wertz, I. E., O’Rourke, K. M., Zhou, H., Eby, M., Aravind, L., Seshagiri, S., Wu, P., Wiesmann, C., Baker, R., Boone, D. L., Ma, A., Koonin, E. V., and Dixit, V. M. (2004) Nature [DOI] [PubMed]
- 72.He KL, Ting AT. Mol Cell Biol. 2002;22:6034–6045. doi: 10.1128/MCB.22.17.6034-6045.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Malewicz M, Zeller N, Yilmaz ZB, Weih F. J Biol Chem. 2003;278:32825–32833. doi: 10.1074/jbc.M304000200. [DOI] [PubMed] [Google Scholar]
- 74.Razmara M, inivasula SM, Wang L, Poyet JL, Geddes BJ, DiStefano PS, Bertin J, Alnemri ES. J Biol Chem. 2002;277:13952–13958. doi: 10.1074/jbc.M107811200. [DOI] [PubMed] [Google Scholar]
- 75.Zhang H, Fu W. Int J Oncol. 2002;20:1035–1040. [PubMed] [Google Scholar]
- 76.Hsu JC, Laz T, Mohn KL, Taub R. Proc Natl Acad Sci U S A. 1991;88:3511–3515. doi: 10.1073/pnas.88.9.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nawa T, Nawa MT, Cai Y, Zhang C, Uchimura I, Narumi S, Numano F, Kitajima S. Biochem Biophys Res Commun. 2000;275:406–411. doi: 10.1006/bbrc.2000.3332. [DOI] [PubMed] [Google Scholar]
- 78.Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U. Gene Expr. 1999;7:321–335. [PMC free article] [PubMed] [Google Scholar]
- 79.Mashima T, Udagawa S, Tsuruo T. J Cell Physiol. 2001;188:352–358. doi: 10.1002/jcp.1130. [DOI] [PubMed] [Google Scholar]
- 80.Nawa T, Nawa MT, Adachi MT, Uchimura I, Shimokawa R, Fujisawa K, Tanaka A, Numano F, Kitajima S. Atherosclerosis. 2002;161:281– 291. doi: 10.1016/s0021-9150(01)00639-6. [DOI] [PubMed] [Google Scholar]
- 81.Hartman MG, Lu D, Kim ML, Kociba GJ, Shukri T, Buteau J, Wang X, Frankel WL, Guttridge D, Prentki M, Grey ST, Ron D, Hai T. Mol Cell Biol. 2004;24:5721–5732. doi: 10.1128/MCB.24.13.5721-5732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nobori K, to H, Tamamori-Adachi M, Adachi S, Ono Y, Kawauchi J, Kitajima S, Marumo F, Isobe M. J Mol Cell Cardiol. 2002;34:1387–1397. doi: 10.1006/jmcc.2002.2091. [DOI] [PubMed] [Google Scholar]
- 83.Nakagomi S, Suzuki Y, Namikawa K, Kiryu-Seo S, Kiyama H. J Neurosci. 2003;23:5187–5196. doi: 10.1523/JNEUROSCI.23-12-05187.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Connor AM, Waterhouse P, Khokha R, Denhardt DT. Biochim Biophys Acta. 1989;1009:75–82. doi: 10.1016/0167-4781(89)90081-x. [DOI] [PubMed] [Google Scholar]
- 85.Groskopf JC, Syu LJ, Saltiel AR, Linzer DI. Endocrinology. 1997;138:2835–2840. doi: 10.1210/endo.138.7.5276. [DOI] [PubMed] [Google Scholar]
- 86.Fassett JT, Nilsen-Hamilton M. Endocrinology. 2001;142:2129–2137. doi: 10.1210/endo.142.5.8132. [DOI] [PubMed] [Google Scholar]
- 87.Toft DJ, Rosenberg SB, Bergers G, Volpert O, Linzer DI. Proc Natl Acad Sci U S A. 2001;98:13055–13059. doi: 10.1073/pnas.231364798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Corbacho AM, Martinez De La Escalera G, Clapp C. J Endocrinol. 2002;173:219–238. doi: 10.1677/joe.0.1730219. [DOI] [PubMed] [Google Scholar]