

Abstract
The peripheral accessory chlorophylls (Chls) of the photosystem II (PSII) reaction center (RC) are coordinated by a pair of symmetry-related histidine residues (D1-H118 and D2-H117). These Chls participate in energy transfer from the proximal antennae complexes (CP43 and CP47) to the RC core chromophores. In addition, one or both of the peripheral Chls are redox-active and participate in a low-quantum-yield electron transfer cycle around PSII. We demonstrate that conservative mutations of the D2-H117 residue result in decreased Chl fluorescence quenching efficiency attributed to reduced accumulation of the peripheral accessory Chl cation, Chl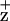 . In contrast, identical symmetry-related mutations at residue D1-H118 had no effect on Chl fluorescence yield or quenching kinetics. Mutagenesis of the D2-H117 residue also altered the line width of the Chl
. In contrast, identical symmetry-related mutations at residue D1-H118 had no effect on Chl fluorescence yield or quenching kinetics. Mutagenesis of the D2-H117 residue also altered the line width of the Chl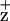 EPR signal, but the line shape of the D1-H118Q mutant remained unchanged. The D1-H118 and D2-H117 mutations also altered energy transfer properties in PSII RCs. Unlike wild type or the D1-H118Q mutant, D2-H117N RCs exhibited a reduced CD doublet in the red region of Chl absorbance band, indicative of reduced energetic coupling between P680 and the peripheral accessory Chl. In addition, transient absorption measurements of D2-H117N RCs, excited on the blue side of the Chl absorbance band, exhibited a (≈400 fs) pheophytin QX band bleach lifetime component not seen in wild-type or D1-H118Q RCs. The origin of this component may be related to delayed fast-energy equilibration of the excited state between the core pigments of this mutant.
EPR signal, but the line shape of the D1-H118Q mutant remained unchanged. The D1-H118 and D2-H117 mutations also altered energy transfer properties in PSII RCs. Unlike wild type or the D1-H118Q mutant, D2-H117N RCs exhibited a reduced CD doublet in the red region of Chl absorbance band, indicative of reduced energetic coupling between P680 and the peripheral accessory Chl. In addition, transient absorption measurements of D2-H117N RCs, excited on the blue side of the Chl absorbance band, exhibited a (≈400 fs) pheophytin QX band bleach lifetime component not seen in wild-type or D1-H118Q RCs. The origin of this component may be related to delayed fast-energy equilibration of the excited state between the core pigments of this mutant.
Among the photosynthetic organisms, there are two different, quinone-type photosynthetic reaction centers (RCs): the oxygenic type present in chloroplasts and cyanobacteria (photosystem II, PSII) and the nonoxygenic type (bacterial reaction center, BRC) as typified by that in purple-sulfur photosynthetic bacteria (for reviews, see refs. 1 and 2). In 1984, the crystal structure of the Rhodopseudomonas viridis BRC was determined at 3-Å resolution by Deisenhofer et al. (3). It soon was recognized that the L and M polypeptides of the BRC had substantial amino acid sequence similarity with the PSII D1 and D2 proteins, respectively. Early models of the D1 and D2 protein-folding topologies indicated that the PSII D1 and D2 polypeptides were structurally analogous to the L and M subunits (3–8). These models of the PSII RC polypeptides and their associated cofactors were verified by electron and x-ray diffraction of two- and three-dimensional PSII crystals (9–12). Recently, a 3.8-Å resolution structure of the oxygen-evolving PSII complex from Synechecoccus elongatus was determined (13).
The PSII RC has six chlorophylls (Chls), two pheophytins (Pheos), two quinones, and one cytochrome b559 (Cyt b559) heme (8, 13). Significantly, the PSII RC has two additional Chls, relative to the BRC. Site-directed mutagenesis and spectroscopic studies demonstrated that the additional pair of Chls present in PSII is coordinated by a pair of conserved and C2 symmetry-related histidine residues (D1-H118 and D2-H117, Chlamydomonas reinhardtii nomenclature) (14–20). Unlike the four Chls and two Pheos involved in primary charge separation, the additional pair of Chls present in the PSII RC is located on the periphery of D1 and D2 polypeptides.
Biophysical evidence for the involvement of the peripheral Chls in energy transfer first was obtained from analysis of P680 oxidation kinetics by Schelvis et al. (14). They observed a ≈30-ps P680 oxidation lifetime component, which they attributed to energy transfer from a Chl monomer to P680. Based on a Förster mechanism (21) for energy transfer, they calculated that the distance between the peripheral Chl and P680 was 30 Å, a distance confirmed by the recent PSII crystal structure. A similar distance relationship also was predicted on the basis of Chl fluorescence decay kinetics by Roelofs et al. (22). Recently, we demonstrated that the 30-ps Chl fluorescence lifetime component is altered (shifted to 10 ps) in a D2-H117N mutant (20), consistent with the assignment of the 30-ps lifetime component to energy transfer from the peripheral accessory Chl to P680.
In addition to participating in energy transfer processes, the peripheral Chls may be involved in a low-quantum-yield electron transfer pathway, leading to the reduction of P680+. Brudvig and coworkers (23–25) have proposed that a Chl monomer known as ChlZ reduces P680+ with low quantum yield. The ChlZ cation is re-reduced by Cyt b559, which, in turn, is reduced by reduced plastoquinone. Recent evidence also suggests that a β-carotene (β-Car) may participate in this PSII electron transfer cycle between ChlZ and P680 or directly reduce P680+ (26–33). The ChlZ cycle has been proposed to reduce photoinhibitory damage by facilitating the oxidation of overreduced quinone electron acceptors and reduction of long-lived P680+ states (25, 34).
The most controversial issues regarding ChlZ are whether it is a single Chl or two and, if it is a single Chl, whether it is coordinated by the D1-H118 or the D2-H117 residues. Measurements of the quantum yield of Chl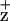 accumulation indicate, however, that only one radical equivalent is accumulated per RC at cryogenic temperatures. Stewart et al. (16) have proposed that this Chl radical is coordinated by the D1-H118. This interpretation is based on alterations in the Chl
accumulation indicate, however, that only one radical equivalent is accumulated per RC at cryogenic temperatures. Stewart et al. (16) have proposed that this Chl radical is coordinated by the D1-H118. This interpretation is based on alterations in the Chl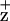 vibrational spectrum in the Synechocystis mutant, D1-H118Q. In contrast, a Synechocystis D2-H117Q mutant did not exhibit changes in the Chl
vibrational spectrum in the Synechocystis mutant, D1-H118Q. In contrast, a Synechocystis D2-H117Q mutant did not exhibit changes in the Chl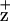 vibrational spectrum. The PSII model structures (6–8) indicate, however, that the Chl coordinated by the D2-H117 residue is closer to Cyt b559 than the Chl coordinated by the D1-H118 residue. Consistent with the interpretation that ChlZ is coordinated by the D2-H117 residue, pulsed electron spin echo measurements have indicated that the distance between the redox active tyrosine of the D2 protein (Y
vibrational spectrum. The PSII model structures (6–8) indicate, however, that the Chl coordinated by the D2-H117 residue is closer to Cyt b559 than the Chl coordinated by the D1-H118 residue. Consistent with the interpretation that ChlZ is coordinated by the D2-H117 residue, pulsed electron spin echo measurements have indicated that the distance between the redox active tyrosine of the D2 protein (Y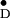 ) and Chl
) and Chl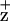 is 29 Å (35, 36). This distance is equivalent to the distance between the Chl coordinated by the D2-H117 residue and YD (redox active Tyr-160 of the D2 protein) predicted from the PSII structural models and verified by the PSII crystal structure (8, 13). In contrast, the distance between Y
is 29 Å (35, 36). This distance is equivalent to the distance between the Chl coordinated by the D2-H117 residue and YD (redox active Tyr-160 of the D2 protein) predicted from the PSII structural models and verified by the PSII crystal structure (8, 13). In contrast, the distance between Y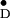 and the Chl coordinated by the D1-H118 residue is approximately 50 Å.
and the Chl coordinated by the D1-H118 residue is approximately 50 Å.
To date, a clear picture of energy transfer and charge separation within the PSII RC complex has yet to be reached. This is caused in part by the substantial spectral overlap of the six Chl and two Pheo pigments in the composite QY band of the pigments (37). As for the spectral identification of the peripheral Chls, it has been suggested that both molecules absorb near 670 nm (38). However, one of the two Chls is proposed to be red-shifted (684 nm) in its absorbance maximum (39, 40).
Additional evidence also suggests that the two peripheral accessory Chls are functionally different (41). Biochemical analyses indicate that D1-H118Q mutants have nearly wild-type (WT) rates of oxygen evolution at saturating light intensities whereas D2-H117Q mutants are light-saturated at relatively low light intensities. Significantly, both the D1-H118Q mutant and the D2-H117 mutants are more resistant to photoinhibitory light treatments than WT.
In this article, we compare the effects of D1-H118 and D2-H117 mutations on the formation of Chl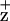 and energy coupling between the Chls coordinated by these residues and P680. Overall, the D2-H117 mutants have slower Chl fluorescence-quenching kinetics than WT or the D1-H118Q mutant. In addition, we demonstrate that the D2-H117N mutation induces changes in the electronic structure of Chl
and energy coupling between the Chls coordinated by these residues and P680. Overall, the D2-H117 mutants have slower Chl fluorescence-quenching kinetics than WT or the D1-H118Q mutant. In addition, we demonstrate that the D2-H117N mutation induces changes in the electronic structure of Chl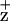 as detected by EPR whereas the D1-H118Q mutation does not. These results are consistent with ChlZ being coordinated by the D2-H117 residue in chloroplasts. It also is apparent that energy coupling between the Chl coordinated by the D2-H117 residue and nearby pigments of the RC complex is altered dramatically. In contrast, energy coupling in the D1-H118Q mutant was similar to WT. These results further confirm the functional asymmetry of these Chls.
as detected by EPR whereas the D1-H118Q mutation does not. These results are consistent with ChlZ being coordinated by the D2-H117 residue in chloroplasts. It also is apparent that energy coupling between the Chl coordinated by the D2-H117 residue and nearby pigments of the RC complex is altered dramatically. In contrast, energy coupling in the D1-H118Q mutant was similar to WT. These results further confirm the functional asymmetry of these Chls.
Materials and Methods
Generation of Mutants.
A site-directed mutation at the D1–118 residue from histidine to glutamine was introduced by changing the codon from CAC to CAG in the psbA gene. The modified psbA gene was delivered into a psbA-deficient C. reinhardtii strain, CC-741, as described by Minagawa and Crofts (42). D2-H117N and D2-H117Q mutants were generated as described (41). All of the mutations were confirmed by sequencing DNA from transgenic algae.
Thylakoid and PSII Preparations.
Thylakoids were prepared as described (41). PSII membrane fragments were prepared from C. reinhardtii similar to the methods of Berthold et al. (43). PSII membranes were depleted of Mn for Chl fluorescence-quenching experiments at 200 K (26). PSII RC complexes were purified according to the method described in ref. 20. The PSII RC preparations were desalted on prepacked Sephadex G-25 columns (Amersham Pharmacia) with a buffer of 50 mM Tris⋅HCl (pH 7.2) and 0.03% (wt/vol) n-dodecyl β-d-maltoside.
Low-Temperature Chl Fluorescence Emission Spectroscopy.
Mn-depleted, BBY-type PSII membranes were frozen in 20 mM Mes-NaOH (pH 6.0)/50% glycerol (wt/vol) at 5 μg Chl/ml, and the Chl fluorescence-quenching kinetics at 695 nm were measured at 200 K while the samples were exposed to the excitation beam (468 nm) of a Fluoro-Max spectrometer. The excitation and emission slit widths were 5 and 2 nm, respectively. At 200 K, Chl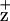 is the predominant cation that photoaccumulates (26, 44). Steady-state Chl fluorescence emission spectra of the PSII membranes before and after the chlorophyll fluorescence-quenching treatment at 200 K (as described above) were measured at 77 K by using an excitation wavelength of 468 nm with excitation and emission slit widths of 2 nm.
is the predominant cation that photoaccumulates (26, 44). Steady-state Chl fluorescence emission spectra of the PSII membranes before and after the chlorophyll fluorescence-quenching treatment at 200 K (as described above) were measured at 77 K by using an excitation wavelength of 468 nm with excitation and emission slit widths of 2 nm.
EPR Spectra of Chl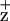 .
.
X-Band EPR spectra were recorded with a Bruker ER 300 spectrometer equipped with an Oxford liquid helium cryostat. Cyt b559 was preoxidized by 2 mM K3Fe(CN)6 in the PSII RC (≈35 μg Chl/ml). Illumination (1,000 μmol photons m−2⋅s−1) was applied to the sample at 200 K for 5 min, and the illuminated minus dark difference spectra are shown for Chl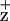 .
.
CD Spectra.
An Aviv CD Spectrometer (model 40DS/UV-VIS-IR) was used to obtain CD spectra with a bandwidth of 2 nm. The concentrations of the RC preparations from WT, D1-H118Q, and D2-H117N were adjusted to 1.3 OD cm−1 at 676 nm, and the samples were kept at 4°C with a circulated refrigerated water bath.
Time-Resolved Absorption Changes of the Pheo Qx Band.
Femtosecond transient absorption kinetics of purified RCs from WT, D1-H118Q, and D2-H117N were obtained at 4°C as described (45). Narrow-bandwidth [≈6 nm full-width half-maximum (FWHM)] sub-200-fs pulses at excitation wavelengths of 655, 665, and 687 nm were used to selectively excite the “blue” and “red” sides of the composite QY band, respectively.
Pigment concentrations of thylakoid and PSII membranes were determined by the method of Arnon (46) whereas those of purified RCs were done according to ref. 47.
Results
Measurement of the relative Chl fluorescence quantum yield induced by continuous illumination is an effective means to monitor energy migration and trapping in PSII (48). At 77 K, the fluorescence emission spectrum of PSII particles is characterized by two major Chl fluorescence emission bands centered at 685 and 695 nm (Fig. 1). The 685-nm fluorescence emission band has been attributed to multiple sources, including charge recombination in PSII (49) and emission from the outer (light-harvesting complex) and inner (CP43 and CP47) antenna protein complexes (50). Mutagenesis studies suggest that the low-energy Chl ligated by His-114 of the CP47 protein gives rise to the 695-nm Chl fluorescence emission band (51).
Figure 1.

Chl fluorescence emission spectra (77 K) of dark-adapted (solid line) and illuminated (15 min at 200 K, dotted line) from WT (A), D1-H118Q (B), D2-H117N (C), and D2-H117Q (D) PSII membranes.
Comparison of the Chl fluorescence emission spectra with and without 15-min illumination treatment revealed less quenching of the F695 emission peak relative to the F685 emission in the two D2-H117 mutants. In contrast, the F695 emission was reduced to a greater extent than the F685 emission in WT and D1-H118Q mutant PSII membranes (Fig. 1). Because the D1-H118 and D2-H117 mutations primarily affect the peripheral accessory Chls, we attribute the increased F695 emission intensity in D2-H117N/Q mutants (after 15-min illumination) to less efficient Chl fluorescence quenching by Chl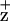 . To test this hypothesis, we monitored the time course of Chl fluorescence quenching at 695 nm (200 K). As shown in Fig. 2, Chl fluorescence (695 nm) quenching kinetics for the D2-H117N and D2-H117Q mutants were substantially and reproducibly slower than for WT or the D1-H118 mutant. These results are consistent with the relative differences in the magnitude of the 695-nm Chl fluorescence emission bands in the D1-H118Q and D2-H117N/Q mutants obtained at 77 K from dark minus light samples. Significantly, the kinetics of Chl fluorescence quenching at 685 nm were identical within experimental errors for all mutants and WT (data not shown), indicating that the effects observed at 695 nm are a result of the Chl
. To test this hypothesis, we monitored the time course of Chl fluorescence quenching at 695 nm (200 K). As shown in Fig. 2, Chl fluorescence (695 nm) quenching kinetics for the D2-H117N and D2-H117Q mutants were substantially and reproducibly slower than for WT or the D1-H118 mutant. These results are consistent with the relative differences in the magnitude of the 695-nm Chl fluorescence emission bands in the D1-H118Q and D2-H117N/Q mutants obtained at 77 K from dark minus light samples. Significantly, the kinetics of Chl fluorescence quenching at 685 nm were identical within experimental errors for all mutants and WT (data not shown), indicating that the effects observed at 695 nm are a result of the Chl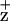 -associated quenching process (see Discussion).
-associated quenching process (see Discussion).
Figure 2.

Time course of Chl fluorescence (695 nm) quenching of WT (solid line), D1-H118Q (dashed line), D2-H117N (dotted line), and D2-H117Q (dashed, dotted line) PSII membranes during illumination at 200 K.
The Chl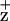 EPR signal is generated in ferricyanide-oxidized PSII RC preparations when illuminated at 200 K. Subtraction of the dark signal gives the Gaussian spectra of Chl
EPR signal is generated in ferricyanide-oxidized PSII RC preparations when illuminated at 200 K. Subtraction of the dark signal gives the Gaussian spectra of Chl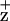 . As shown in Fig. 3, the Chl
. As shown in Fig. 3, the Chl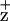 line shape of WT and the D1-H118Q mutant was identical (within experimental error), with each having a characteristic line width of around 10 G. The Chl
line shape of WT and the D1-H118Q mutant was identical (within experimental error), with each having a characteristic line width of around 10 G. The Chl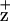 spectrum of the D2-H117N mutant, however, was broadened by 1 G. These results are consistent with the Chl fluorescence-quenching results that indicate that the Chl species that gives rise to the EPR-detectable ChlZ cation is the Chl coordinated by the D2-H117N residue. The molecular basis for the line-broadening is unexpected at the microwave frequencies used, however, because the hyperfine couplings at this frequency are dominated by the S = ½ hydrogens of the tetrapyrrole ring and not the Mg involved in ligation. A change in orientation of the Chl, however, may alter potential protein–Chl interactions with other nearby residues (see CD spectra discussion below), accounting for the line-width broadening.
spectrum of the D2-H117N mutant, however, was broadened by 1 G. These results are consistent with the Chl fluorescence-quenching results that indicate that the Chl species that gives rise to the EPR-detectable ChlZ cation is the Chl coordinated by the D2-H117N residue. The molecular basis for the line-broadening is unexpected at the microwave frequencies used, however, because the hyperfine couplings at this frequency are dominated by the S = ½ hydrogens of the tetrapyrrole ring and not the Mg involved in ligation. A change in orientation of the Chl, however, may alter potential protein–Chl interactions with other nearby residues (see CD spectra discussion below), accounting for the line-width broadening.
Figure 3.

EPR spectra of Chl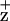 generated in PSII RC preparations from WT (solid line), D1-H118Q (dashed line), and D2-H117N (dotted line). Instrument conditions: measuring temperature, 4.7 K; microwave frequency, 9.48 GHz; modulation amplitude, 0.2 mT; power, 10 μW; and modulation frequency, 100 kHz.
generated in PSII RC preparations from WT (solid line), D1-H118Q (dashed line), and D2-H117N (dotted line). Instrument conditions: measuring temperature, 4.7 K; microwave frequency, 9.48 GHz; modulation amplitude, 0.2 mT; power, 10 μW; and modulation frequency, 100 kHz.
In addition to their role in electron transfer processes, the peripheral accessory Chls have been shown to participate in energy transfer between the proximal antennae Chl protein complexes and P680 (14, 19, 20, 41). To characterize the energetic couplings in PSII RCs, we measured the Chl (visible) CD spectra of WT and mutant RCs. Overall, the Chl CD of WT Chlamydomonas RCs (Fig. 4) resembles those from other sources (49, 52–57), with positive chirality peaking at 681 nm and a negative lobe, around 669 nm. The red edge (above 681 nm) of the positive lobe is believed to be an excitonic band associated with P680 (58–60). As shown in Fig. 4, D2-H117N RCs lost much of their CD intensity in the 681-nm region attributed to P680 whereas the D1-H118Q mutant did not. These effects plus those observed in the Soret region imply an alteration in the geometry of the pigments in both the D1 and D2 mutants. Although it was not possible to make direct comparisons between equivalent amino acid substitutions in D1 and D2 mutant RC particles (D2-H117Q RCs were not stable), it was evident that the energetic coupling between P680 and the peripheral Chl in the D1-H118Q mutant was more similar to WT, consistent with results obtained from Chl fluorescence steady-state and lifetime measurements (ref. 41; Figs. 1–3).
Figure 4.

Chl CD spectra of PSII RC preparations from WT (solid line), D1-H118Q (dashed line), and D2-H117N (dotted line) mutants at 4°C.
To examine the influence of the peripheral Chl mutations on primary energy and charge transfer kinetics, we measured the time-resolved absorption changes of the Pheo Qx band (542 nm) associated with the reduction of Pheo (Fig. 5). Lifetime fits were made 300–400 fs after t = 0 because of an interaction between the pump and probe pulses and the buffer/cell system (37, 45, 61). When pumped at 687 nm (Fig. 5A and Table 1), the Pheo QX band bleach kinetics for WT and mutant RCs were adequately fit by using three exponential decay lifetime components and a small component that does not decay (shelf) on the time scale used in this study. The lifetimes of the decay components were 2–5, 40–50, and 200–400 ps. We assign the 2- to 5-ps Pheo bleach component to the intrinsic time constant for charge separation. There are, however, numerous plausible explanations for the two slower components, including slow energy transfer to the pigment core multimer and relaxation of the charge-separated state (37). At the present time, we are unable to unambiguously distinguish which is correct. Therefore, we will restrict our analysis to the fast component, which we attribute to charge separation based on past work (37, 45). Finally, the nondecaying component is believed to reflect the fully charge-separated state of the RC (P680+–Pheo−), which has a lifetime of 36 ns at ambient temperature (45, 62, 63).
Figure 5.

Transient Pheo absorption kinetics at 542 nm in WT and peripheral Chl mutant RCs pumped at either 687 nm (A) or 655 nm (B). (Insets) Early time behavior.
Table 1.
Averages and estimated errors for the Pheo Qx band (542 nm) bleach growth kinetic of WT and peripheral Chl mutant PS II RCs
| λpump, nm | Sample | τultrafast, ps | Aultrafast | τfast, ps | Afast, ps | τint, ps | Aint | τslow, ps | Aslow | Ashelf × 104 |
|---|---|---|---|---|---|---|---|---|---|---|
| 655 | WT | 2.1 ± 0.1 | 19.0% | 39 ± 1 | 39.5% | 363 ± 12 | 33.1% | 7.9 ± 0.2 | ||
| D1-H118Q | 3.5 ± 0.2 | 20.8% | 39 ± 2 | 37.9% | 230 ± 9 | 34.4% | 2.9 ± 0.1 | |||
| D2-H117N | 0.4 ± 0.1 | 15.9% | 5.9 ± 0.8 | 14.6% | 44 ± 3 | 32.2% | 348 ± 17 | 28.5% | 8.6 ± 0.2 | |
| 665 | WT | 3.9 ± 0.1 | 23.1% | 42 ± 1 | 40.3% | 427 ± 13 | 32.0% | 4.4 ± 0.2 | ||
| D1-H118Q | 3.1 ± 0.1 | 29.8% | 39 ± 2 | 38.8% | 287 ± 13 | 30.3% | −1.1 ± 0.2 | |||
| D2-H117N | 0.4 ± 0.1 | 16.3% | 5.3 ± 0.5 | 19.5% | 49 ± 3 | 33.8% | 430 ± 22 | 25.6% | 4.4 ± 0.2 | |
| 687 | WT | 2.8 ± 0.2 | 16.3% | 46 ± 1 | 40.1% | 399 ± 12 | 42.1% | −0.7 ± 0.1 | ||
| D1-H118Q | 4.9 ± 0.4 | 19.0% | 51 ± 3 | 40.6% | 268 ± 14 | 35.4% | 2.4 ± 0.1 | |||
| D2-H117N | 4.4 ± 0.4 | 20.3% | 52 ± 3 | 40.6% | 418 ± 22 | 38.1% | −0.4 ± 0.1 |
In contrast to the red side excitation of the chlorin pigments, pumping the D2-H117N RCs at 665 and 655 nm led to the identification of an additional ≈400-fs lifetime component that was not observed in WT or D1-H118Q PSII RCs (Fig. 5B and Table 1). The 400-fs lifetime component could be associated with a delay in fast-energy equilibration between the core pigments of this mutant or formation of Pheo* (see Discussion). Overall, it is apparent that energy and electron transfer are altered in dissimilar ways in conservative D1-H118 and D2-H117 mutants.
Discussion
At cryogenic temperatures, ChlZ (200 K) and/or Car (77 K or lower) are the primary electron donors to P680+ in PSII RC complexes (26, 32, 33). Photoaccumulation of Chl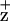 effectively quenches Chl fluorescence (695 nm) (34). The D2-H117N and D2H117Q mutation induced changes in F695 Chl fluorescence yield and quenching kinetics (200 K) thus are attributed to perturbations in the yield of Chl
effectively quenches Chl fluorescence (695 nm) (34). The D2-H117N and D2H117Q mutation induced changes in F695 Chl fluorescence yield and quenching kinetics (200 K) thus are attributed to perturbations in the yield of Chl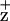 . Consistent with this interpretation, the D2-H117 mutations specifically affected quenching of only the F695 emission band (attributed to a Chl bound to the CP47 proximal antennae complex) and not that of the F685 nm Chl fluorescence emission band attributed to multiple sources including the light-harvesting complex (17, 34).
. Consistent with this interpretation, the D2-H117 mutations specifically affected quenching of only the F695 emission band (attributed to a Chl bound to the CP47 proximal antennae complex) and not that of the F685 nm Chl fluorescence emission band attributed to multiple sources including the light-harvesting complex (17, 34).
The localization of Chl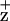 on the Chl coordinated by the D2-H117 residue was further supported by the observed mutation-induced line width broadening of the EPR-detectable Chl
on the Chl coordinated by the D2-H117 residue was further supported by the observed mutation-induced line width broadening of the EPR-detectable Chl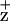 radical in the D2-H117N mutant (Fig. 3). This mutation presumably indirectly alters the electron spin density and distribution on Chl
radical in the D2-H117N mutant (Fig. 3). This mutation presumably indirectly alters the electron spin density and distribution on Chl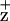 (64). Alternatively, the D2-H117 mutation may change the spatial position of ChlZ, potentially affecting magnetic dipole–dipole interactions with a nearby residue or radical, resulting in the broadening of the line width. In either case, the change of the Chl
(64). Alternatively, the D2-H117 mutation may change the spatial position of ChlZ, potentially affecting magnetic dipole–dipole interactions with a nearby residue or radical, resulting in the broadening of the line width. In either case, the change of the Chl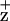 electronic structure in the D2-H117N mutant (but not its counterpart on the D1 side), together with the altered Chl fluorescence-quenching kinetics in D2-H117 mutants, allows us to assign Chl
electronic structure in the D2-H117N mutant (but not its counterpart on the D1 side), together with the altered Chl fluorescence-quenching kinetics in D2-H117 mutants, allows us to assign Chl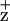 to coordination by the D2-H117 residue.
to coordination by the D2-H117 residue.
This interpretation for the location of ChlZ also is supported by recent crystallographic data from PSII core complexes (13). These studies indicate that the Cyt b559 heme is located near the D2-H117 rather than D1-H118 residue (13). Because the Cyt b559 heme is the apparent reductant of Chl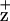 , the location of the Cyt b559 heme near the D2 polypeptide supports the interpretation that ChlZ is coordinated by the D2-H117 residue. We have not excluded the possibility, however, that both the D1-H118 and the D2-H117 Chls may be redox-active under certain conditions. It is interesting to speculate on the species-specific differences between the apparent location of ChlZ in chloroplastic and cyanobacterial PSIIs. It is possible that different strategies may have been adopted to prevent photodamage to PSII in these two types of organisms that possibly are associated with the different types of light-harvesting complexes and their physical coupling to PSII.
, the location of the Cyt b559 heme near the D2 polypeptide supports the interpretation that ChlZ is coordinated by the D2-H117 residue. We have not excluded the possibility, however, that both the D1-H118 and the D2-H117 Chls may be redox-active under certain conditions. It is interesting to speculate on the species-specific differences between the apparent location of ChlZ in chloroplastic and cyanobacterial PSIIs. It is possible that different strategies may have been adopted to prevent photodamage to PSII in these two types of organisms that possibly are associated with the different types of light-harvesting complexes and their physical coupling to PSII.
We also have investigated the effects of D1-H118 and D2-H117 mutations on energy coupling to P680. Recent studies suggest that the Chls bound to the proximal antennae complexes (associated with CP43 and CP47) are not well coupled to the RC complex (65). We have demonstrated that a slow (30-ps) Chl fluorescence decay lifetime component is associated with energy transfer from the Chl coordinated by the D2-H117 residue to P680 in PSII RC complexes (20). To characterize further the energy coupling between peripheral accessory Chls and the RC pigments, we investigated the ground-state absorption spectrum and Chl CD spectrum of isolated RC complexes. One may expect there to be some spectral shifting and redistribution of oscillator strength associated with the mutagenesis of the peripheral accessory Chl ligands. However, we did not observe any significant changes in the ground-state, room temperature absorption spectra in mutant RCs relative to WT (not shown). In contrast, we observed substantial alterations in the Chl CD spectrum of mutant RC complexes, strongly indicating that the peripheral Chls interact energetically with P680. One possible explanation for the mutation-induced change in the RC Chl CD band, without corresponding change in the ground-state absorption spectrum, is that the transition dipoles move “in plane” without significantly changing the dipolar angles relative to the membranes. However, the EPR-detectable line-width broadening of the Chl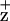 signal and the alterations in the Soret region of the CD spectrum suggest that the protein–ChlZ macrocycle interactions are altered in the D2-H117N mutant.
signal and the alterations in the Soret region of the CD spectrum suggest that the protein–ChlZ macrocycle interactions are altered in the D2-H117N mutant.
To characterize further the energy transfer properties of the two peripheral accessory Chls, we preferentially pumped either of the two peripheral accessory Chls by using red-side (687 nm) or blue-side (665 and 655 nm) excitation followed by analysis of the Pheo-bleaching kinetics. Previously, we observed no changes in Chl fluorescence decay kinetics in D1-H118Q RCs when the sample was selectively excited at 650 nm (41). This is in sharp contrast to observations made for the D2-H117N mutant (20). These results suggested that the D1-H118 Chl preferentially absorbs on the red side of the QY band whereas the D2-H117 Chl preferentially absorbs on the blue side of the composite QY band in PSII RCs. This interpretation is consistent with recent work by Small and coworkers (40), who have proposed that D1-H118 and D2-H117 Chls absorb predominantly at 684 nm and 670 nm, respectively.
No substantial differences were observed in the Pheo QX bleach growth kinetics between the two peripheral Chl mutants and WT when the samples were excited at 687 nm, however, suggesting that the D1-H118Q mutation had little effect on the ultra-fast energy transfer processes (Fig. 5 and Table 1). When 665- or 655-nm light was chosen to excite the higher-energy-absorbing Chl species, a kinetic fit of Pheo QX bleaching for D2-H117N mutant RCs required an additional lifetime component (≈400 fs) at the margin of the instrument's sensitivity. Bleaching of the Pheo Qx band can be attributed either to the formation of Pheo* or to Pheo− (37, 45). Previously, a 100-fs Pheo bleaching lifetime component has been ascribed either to rapid equilibration between red (P680 and Pheoactive) and blue (Pheoinactive) excited states of the P680 multimer core pigments or to ultrafast energy transfer from Pheoinactive to Pheoactive (37, 66–68). However, in the case of D2-H117N mutant, P680 is poorly coupled to the peripheral accessory Chl as implied by the CD spectrum (Fig. 4). Upon preferential excitation of the D2-H117 Chl (but not the D1-H118 Chl) by pumping at 665 or 655 nm, energy equilibration among the core pigments (including Pheoinactive) may be delayed from ≈100 fs (in WT) to ≈400 fs. Alternatively, formation of Pheo*, which has a bleaching spectrum similar to Pheo−, may be accelerated in the D2-H117N mutant. Recently, it has been demonstrated that β-Car transfers energy to Pheoinactive in PSII RCs (69, 70). If the position of β-Car in the PSII RC is similar to that in the BRC, then β-Car may facilitate energy transfer from the peripheral accessory Chl bound to the D2-H117 residue and Pheoinactive, thus accounting for the altered Pheo-bleaching kinetics in the D2-H117N mutant.
Acknowledgments
We thank Dr. Claudia Turro for use of the spectrofluorimeter. M.R.W. was supported by the Division of Chemical Sciences, Office of Basic Energy Sciences, Department of Energy, under Grant DE-FG02-99ER14999. R.T.S. acknowledges the Department of Energy for financial support.
Abbreviations
- (B)RC
(bacterial) reaction center
- Car
carotene
- Chl
chlorophyll
- Cyt b559
cytochrome b559
- Pheo
pheophytin
- PSII
photosystem II
- WT
wild type
References
- 1.Ruffle S V, Sayre R T. In: Molecular Biology of Chlamydomonas: Chloroplasts and Mitochondria. Cleremont-Goldshmidt M, Merchant S, Rochaix J-D, editors. Dordrecht, The Netherlands: Kluwer; 1998. pp. 287–322. [Google Scholar]
- 2.Seibert M. In: The Photosynthetic Reaction Center. Deisenhofer J, Norris J R, editors. I. San Diego: Academic; 1993. pp. 319–356. [Google Scholar]
- 3.Deisenhofer J, Epp O, Miki K, Huber R, Michel H. J Mol Biol. 1984;180:385–398. doi: 10.1016/s0022-2836(84)80011-x. [DOI] [PubMed] [Google Scholar]
- 4.Trebst A. Z Naturforsch. 1985;41:240–245. [Google Scholar]
- 5.Sayre R T, Andersson B, Bogorad L. Cell. 1986;47:601–608. doi: 10.1016/0092-8674(86)90624-0. [DOI] [PubMed] [Google Scholar]
- 6.Ruffle S V, Donnelly D, Blundell T L, Nugent J H A. Photosynth Res. 1992;34:287–300. doi: 10.1007/BF00033446. [DOI] [PubMed] [Google Scholar]
- 7.Svennson B, Etchebest C, Tuffery P, van Kan P, Smith J, Styring S. Biochemistry. 1996;35:14486–14502. doi: 10.1021/bi960764k. [DOI] [PubMed] [Google Scholar]
- 8.Xiong J, Subramanian S, Gonvindjee Photosynth Res. 1998;56:229–254. [Google Scholar]
- 9.Buchel C, Morris E, Barber J. J Struct Biol. 2000;131:181–186. doi: 10.1006/jsbi.2000.4294. [DOI] [PubMed] [Google Scholar]
- 10.Kuhl H, Kruip J, Seidler A, Krieger-Liszkay A, Bunker A, Bald D, Scheidig A J, Rogner M. J Biol Chem. 2000;275:20652–20659. doi: 10.1074/jbc.M001321200. [DOI] [PubMed] [Google Scholar]
- 11.Nield J, Kruse O, Ruprecht J, de Fonseca P, Buchel C, Barber J. J Biol Chem. 2000;275:27940–27946. doi: 10.1074/jbc.M003069200. [DOI] [PubMed] [Google Scholar]
- 12.Shen J R, Kamiya N. Biochemistry. 2000;39:14739–14744. doi: 10.1021/bi001402m. [DOI] [PubMed] [Google Scholar]
- 13.Zouni A, Witt H T, Kern J, Fromme P, Krauss N, Saenger W, Orth P. Nature (London) 2001;409:739–743. doi: 10.1038/35055589. [DOI] [PubMed] [Google Scholar]
- 14.Schelvis J P M, van Noort P I, Aartsma T J, van Gorkom H J. Biochim Biophys Acta. 1994;1184:242–250. [Google Scholar]
- 15.Hutchinson R S, Sayre R T. In: Photosynthesis: From Light to Biosphere. Mathis P, editor. Dordrecht, The Netherlands: Kluwer; 1995. pp. 471–474. [Google Scholar]
- 16.Stewart D H, Cua A, Chisholm D A, Diner B A, Bocian D F, Brudvig G W. Biochemistry. 1998;37:10040–10046. doi: 10.1021/bi980668e. [DOI] [PubMed] [Google Scholar]
- 17.Lince M T, Vermaas W. Eur J Biochem. 1998;256:595–602. doi: 10.1046/j.1432-1327.1998.2560595.x. [DOI] [PubMed] [Google Scholar]
- 18.Cua A, Stewart D H, Brudvig G W, Bocian D F. J Am Chem Soc. 1998;120:4532–4533. [Google Scholar]
- 19.Ruffle S V, Hutchison R S, Sayre R T. In: Photosynthesis: Mechanism and Effects. Garab G, editor. Dordrecht, The Netherlands: Kluwer; 1998. pp. 1013–1016. [Google Scholar]
- 20.Johnston H G, Wang J, Ruffle S V, Sayre R T, Gustafson T L. J Phys Chem. 2000;104:4777–4781. [Google Scholar]
- 21.Förster T. Ann Phys. 1947;2:55–57. [Google Scholar]
- 22.Roelofs T A, Gilbert M, Shuvalov V A, Holzwarth A R. Biochim Biophys Acta. 1991;1060:237–244. [Google Scholar]
- 23.Thompson L K, Brudvig G W. Biochemistry. 1988;27:6653–6658. doi: 10.1021/bi00418a002. [DOI] [PubMed] [Google Scholar]
- 24.Koulougliotis D, Innes J B, Brudvig G W. Biochemistry. 1994;33:11814–11822. doi: 10.1021/bi00205a018. [DOI] [PubMed] [Google Scholar]
- 25.Schweitzer R H, Brudvig G W. Biochemistry. 1997;36:11551–11559. doi: 10.1021/bi9709203. [DOI] [PubMed] [Google Scholar]
- 26.Hanley J, Deligiannakis Y, Pascal A, Faller P, Rutherford A W. Biochemistry. 1999;38:8189–8195. doi: 10.1021/bi990633u. [DOI] [PubMed] [Google Scholar]
- 27.Vrettos J S, Stewart D H, de Paula J C, Brudvig G W. J Phys Chem. 1999;103:6403–6406. [Google Scholar]
- 28.Deligiannakis Y, Hanley J, Rutherford A W. J Am Chem Soc. 2000;122:400–401. [Google Scholar]
- 29.Lakshmi K V, Reifler M J, Brudvig G W, Poluektov O G, Wagner A M, Thurnauer M C. J Phys Chem. 2000;104:10445–10448. [Google Scholar]
- 30.Faller P, Rutherford A W, Un S. J Phys Chem. 2000;104:10960–10963. [Google Scholar]
- 31.Tracewell C A, Vrettos J S, Bautista J A, Frank H A, Brudvig G W. Arch Biochem Biophys. 2001;385:61–69. doi: 10.1006/abbi.2000.2150. [DOI] [PubMed] [Google Scholar]
- 32.Tracewell C A, Cua A, Stewart D H, Bocian D F, Brudvig G W. Biochemistry. 2001;40:193–203. doi: 10.1021/bi001992o. [DOI] [PubMed] [Google Scholar]
- 33.Faller P, Pascal A, Rutherford A W. Biochemistry. 2001;40:6431–6440. doi: 10.1021/bi0026021. [DOI] [PubMed] [Google Scholar]
- 34.Schweitzer R H, Melkozernov A N, Blankenship R E, Brudvig G W. J Phys Chem. 1998;102:8320–8326. [Google Scholar]
- 35.Shigemori K, Hara H, Kawamori A, Akabori K. Biochim Biophys Acta. 1998;1363:187–198. doi: 10.1016/s0005-2728(97)00100-x. [DOI] [PubMed] [Google Scholar]
- 36.Tonaka M, Kawamori A, Hara H, Astashkin A V. Appl Magn Reson. 2000;19:141–150. [Google Scholar]
- 37.Greenfield S R, Seibert M, Wasielewski M R. J Phys Chem. 1999;103:8364–8374. [Google Scholar]
- 38.Vacha F, Joseph D M, Durrant J R, Telfer A, Klug D R, Porter G, Barber J. Proc Natl Acad Sci USA. 1995;92:2929–2933. doi: 10.1073/pnas.92.7.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang H-C, Jankowiak R, Reddy N R S, Yocum C F, Picorel R, Seibert M, Small G J. J Phys Chem. 1994;98:7725–7735. [Google Scholar]
- 40.Jankowiak R, Rätsep M, Picorel R, Seibert M, Small G J. J Phys Chem. 1999;103:9759–9769. [Google Scholar]
- 41.Ruffle S V, Wang J, Johnston H G, Gustafson T L, Hutchison R S, Minagawa J, Crofts A, Sayre R T. Plant Physiol. 2001;127:633–644. [PMC free article] [PubMed] [Google Scholar]
- 42.Minagawa J, Crofts A R. Photosynth Res. 1994;42:121–131. doi: 10.1007/BF02187123. [DOI] [PubMed] [Google Scholar]
- 43.Berthold D A, Babcock G T, Yocum C F. FEBS Lett. 1981;134:231–234. [Google Scholar]
- 44.Noguchi T, Mitsuoka Y, Inoue Y. FEBS Lett. 1994;356:179–183. doi: 10.1016/0014-5793(94)01263-6. [DOI] [PubMed] [Google Scholar]
- 45.Greenfield S R, Seibert M, Gonvindjee, Wasielewski M R. Chem Phys. 1996;210:279–295. [Google Scholar]
- 46.Arnon D I. Plant Physiol. 1949;24:1–15. doi: 10.1104/pp.24.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eijckelhoff C, Dekker J P. Photosynth Res. 1997;52:69–73. [Google Scholar]
- 48.Kurreck J, Schödel R, Renger G. Photosynth Res. 2000;63:171–182. doi: 10.1023/A:1006303510458. [DOI] [PubMed] [Google Scholar]
- 49.Braun P, Greenberg B M, Scherz A. Biochemistry. 1990;29:10376–10387. doi: 10.1021/bi00497a012. [DOI] [PubMed] [Google Scholar]
- 50.Funk C, Schröder W P, Salih G, Wiklund R, Jansson C. FEBS Lett. 1998;436:434–438. doi: 10.1016/s0014-5793(98)01179-x. [DOI] [PubMed] [Google Scholar]
- 51.Shen G, Vermaas W F J. Biochemistry. 1994;33:7379–7388. doi: 10.1021/bi00189a044. [DOI] [PubMed] [Google Scholar]
- 52.Tetenkin V L, Gulyaev B A, Seibert M, Bubin A B. FEBS Lett. 1989;250:459–463. [Google Scholar]
- 53.Newell W R, van Amerongen H, van Grondelle R, Aalberts J W, Drake A F, Udvarhelyi P, Barber J. FEBS Lett. 1988;228:162–166. [Google Scholar]
- 54.Newell W R, van Amerongen H, Barber J, van Grondelle R. Biochim Biophys Acta. 1991;1057:232–238. [Google Scholar]
- 55.Montoya G, Cases R, Rodrígues R, Aured M, Picorel R. Biochemistry. 1994;33:11798–11804. doi: 10.1021/bi00205a016. [DOI] [PubMed] [Google Scholar]
- 56.Oren-Shamir M, Maruthi Sai P S, Edelman M, Scherz A. Biochemistry. 1995;34:5523–5526. doi: 10.1021/bi00016a025. [DOI] [PubMed] [Google Scholar]
- 57.Gall B, Zehetner A, Scherz A, Scheer H. FEBS Lett. 1998;434:88–92. doi: 10.1016/s0014-5793(98)00956-9. [DOI] [PubMed] [Google Scholar]
- 58.Tang D, Jankowiak M, Seibert M, Yocum C F, Small G J. J Phys Chem. 1990;94:6519–6522. [Google Scholar]
- 59.Finzi L, Elli G, Cucchelli G, Garlaschi F M, Jennings R C. Biochim Biophys Acta. 1998;1366:256–264. [Google Scholar]
- 60.Finzi L, Zucchelli G, Garlaschi F M, Jennings R C. Biochemistry. 1999;38:10627–10631. doi: 10.1021/bi990568o. [DOI] [PubMed] [Google Scholar]
- 61.Hong Q, Durrant J, Hastings G, Porter G, Klug D R. Chem Phys Lett. 1993;202:183–185. [Google Scholar]
- 62.Danielius R V, Satoh K, van Kan P J M, Plijter J J, Nuijs A M, van Gorkom H J. FEBS Lett. 1987;213:241–244. [Google Scholar]
- 63.den Hartog F T H, Vacha F, Lock A J, Dekker J P, Völker S. J Phys Chem. 1998;102:9174–9180. [Google Scholar]
- 64.Rigby S E J, Nugent J H A, O'Malley P J. Biochemistry. 1994;33:10043–10050. doi: 10.1021/bi00199a031. [DOI] [PubMed] [Google Scholar]
- 65.Vasil'ev S, Orth P, Zouni A, Owens T G, Bruce D. Proc Natl Acad Sci USA. 2001;98:8602–8607. doi: 10.1073/pnas.141239598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Merry S A P, Kumazaki S, Tachibana Y, Joseph D M, Porter G, Yoshohara K, Barber J, Durrant J R, Klug D R. J Phys Chem. 1996;100:10469–10478. [Google Scholar]
- 67.Durrant J R, Hastings G, Joseph D M, Barber J, Porter G, Klug D R. Proc Natl Acad Sci USA. 1992;89:11632–11636. doi: 10.1073/pnas.89.23.11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klug D R, Rech T, Joseph D M, Barber J, Durrant J R, Porter G. Chem Phys. 1995;194:433–442. [Google Scholar]
- 69.Mimuro M, Tomo T, Nishimura Y, Yamazaki I, Satoh K. Biochim Biophys Acta. 1995;1232:81–88. [Google Scholar]
- 70.Tomo T, Mimuro M, Iwaki M, Kobayashi M, Itoh S, Satoh K. Biochim Biophys Acta. 1997;1321:21–30. [Google Scholar]