

Abstract
Synthetic molecular receptors that completely surround their target molecules can be created through the use of noncovalent interactions. These molecular capsules selectively sequester guest molecules from the influence of bulk solvent and other molecules on the basis of size, shape, and chemical complementarity. This reversible isolation spawns unique behavior within the confines of the host; the catalysis of chemical reactions and the stabilization of reactive species are possible outcomes that have been recently demonstrated. Compartmentalization of reagents can also have a dramatic effect on reactions that take place outside of the capsule, producing nonlinear kinetics in relatively simple reaction systems.
The centennial of Emil Fischer's “lock-and-key” description of biochemical interactions (1), commemorated a few years ago, reminded chemists of the long history of molecular recognition. How and why molecules fit together, and the weak intermolecular forces that act on them, are questions of complementarity—of size, shape, and chemical surface. In biology, nowhere is the answer more apparent than in the structure of the DNA double helix. In chemistry, no single structure has had as much impact, but Charles Pedersen's discovery of crown ethers (2) in the 1960s established the “ion-in-the-hole” formula for recognition. Syntheses of macrocyclic polyethers to encircle metal and ammonium ions proliferated and persist to this day. Additionally, synthetic receptors with different concave shapes—clefts (3), armatures (4), tweezers (5, 6), bowls (7), and others (8, 9)—were devised. Their inner surfaces could be tailored to recognize the outer surfaces of more complex target structures. As these synthetic receptors became more sophisticated, it was manifest that structures could be created that completely surrounded their targets: molecules within molecules.
The first of these structures consisted of networks of covalent bonds that acted as roughly spherical hosts to seal in their guests. They were invented by Collet and coworkers (10) (cryptophanes) and Cram et al. (11, 12) (carcerands), and the ominous names reflect the irreversible nature of their binding: mechanical forces rather than chemical attractions held the guest within the host. Subsequently, organic frameworks could be gathered with noncovalent forces; they self-assembled into capsular structures that more or less completely surrounded their guests but bound them reversibly. Our first examples made use of hydrogen bonds (13) (two more recent structures are shown in Fig. 1 a and d). Alternative host assemblies using stronger metal/ligand interactions were devised by Fujita et al. (14) and Raymond and coworkers (15). They provide spectacular architectures, examples of which are shown in Fig. 1 b and c. We term all of these structures “encapsulation complexes.”
Figure 1.

Representative self-assembling molecular capsules (some protons and substituents have been omitted from the models for clarity).
Encapsulated molecules are removed from a solvation cage and placed in enforced proximity to the host and, when space permits, to other guests. This constrained environment governs the guest's encounters with potential reactive partners and fundamentally alters their concentrations (molecules per volume). This influence is exerted for the duration of the lifetime of the encapsulation complex, which may be from milliseconds to hours. The intimate relationship that is implicit in the existence of molecules within molecules gives rise to a number of intensified properties, but the host's control over guest reactivity is perhaps the most tangible illustration of this connection.
Encapsulation complexes have been used to accelerate, even catalyze, chemical reactions within their cavities and are proving increasingly useful for the isolation of reactive species. They provide alternatives to inert solid matrices and near-vacuum gas phases, because encapsulation takes place at ambient temperatures in solution. The control of molecular reactivity by a self-assembled encapsulation complex was first observed within supramolecular capsule 2 (Fig. 1a) (16–18). Two reactive partners, 9 and 10, are encapsulated simultaneously within the host. During this event—transient on the human timescale but interminably protracted on the timescale of diffusion complexes in bulk solvent—they are held in enforced proximity to each other, and acceleration of the bimolecular Diels–Alder reaction is the result (Fig. 2a). Adduct 11 is ejected from the capsule in favor of two molecules of starting material 9, and catalytic turnover is observed (19). A recent example allows the direct observation of the encapsulated “Michaelis complex” (not shown) (20).
Figure 2.
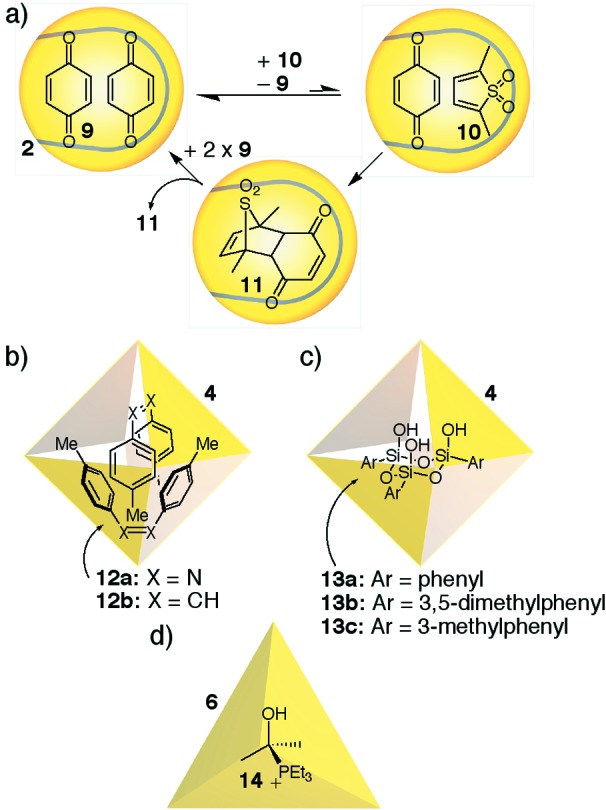
Control over reactivity through encapsulation.
Control of reactivity by supramolecular capsules includes the stabilization of reactive species that are not otherwise observed in free solution. Fujita showed that tennis-ball-shaped noncovalent dimers of molecules such as 12 are formed within the cavity of capsule 4a (Fig. 2b) (21, 22). Free in solution, such a dimer would have little reason to form. Within the confined environment of the capsule, however, a variety of such elongated molecules are encouraged to associate.
The analogous Pt-based capsule 4b demonstrates an even more impressive feat: aryltrimethoxysilanes undergo a sol–gel condensation to form polymeric species, but in the presence of host 4b, this reaction follows a different course. The process is halted after the formation of cyclic silanol trimers (13) within the cavity of the supramolecular capsule (Fig. 2c) (23). Cyclic silanol oligomers of this type have long been considered intermediates in the sol–gel condensation, but the highly reactive cyclic trimer had never been directly observed. Trapping the intermediate within an encapsulation complex allowed the examination of this species using NMR, mass spectrometry, and even x-ray crystallography under ambient conditions (22, 24).
The relative concentration of species within a supramolecular capsule is not easily defined but can be expressed in terms of the volume available to a single isolated molecule. Recent work by Raymond and coworkers has demonstrated that this concept has very real consequences (25). Capsule 6 exists in aqueous solution but, in the absence of a suitable guest, the “empty” cavity is filled with adventitious molecules of acetone remaining from the preparation. Treatment of the “empty” capsule with triethylphosphine results in a new encapsulated species, identifiable as the cationic adduct of triethylphosphine and acetone (Fig. 2d). This adduct (14) is a known species, but it is unstable in aqueous solution. Segregating the components of this equilibrium within encapsulation complex 6 results in an encapsulated reactive species that experiences negligible concentration of water in aqueous solution.
The isolation of encapsulated compounds from solution can have profound effects on the reactions they undergo. But what about systems in which molecular capsules are simply used to bind and segregate different components of a reaction? The simple compartmentalization of reagents by molecular capsules within a single solution can give rise to deceptively complex kinetic behavior. Reversible encapsulation of dicyclohexylcarbodiimide (DCC), a dehydrating agent for coupling amines with carboxylic acids, is accomplished by cylindrical host 8 (26). Addition of benzoic acid 15 and aniline 16 to the solution of DCC encapsulated within capsule 8 sets off a reaction cascade (Fig. 3) (27). Trace DCC that is free in solution promotes the formation of an amide bond between the acid and aniline reactants. The products are anilide 17 and dicyclohexylurea, both of which are better guests for capsule 8 than DCC. As the new products are generated, they displace increasing amounts of DCC from the segregated environment provided by the capsule, and the rate of the reaction increases as the reaction proceeds. The rate profiles are sigmoidal in character and are reminiscent of autocatalysis: the product accelerates its own formation. However, this is not an example of classical autocatalysis, as no catalyst is present in the system. The behavior that arises is based on recognition, the earmark of self-replication. The nonlinear kinetics can be viewed as an emergent property of the system as a whole, a partnership of compartmentalization and molecular recognition that gives rise to chemical amplification. This enigmatic result highlights the role that compartmentalization may play in the creation and maintenance of complex systems.
Figure 3.
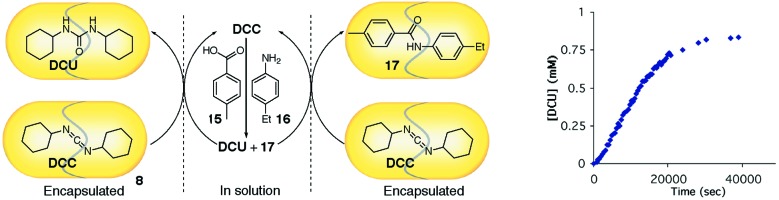
Compartmentalization of reagents gives rise to sigmoidal kinetics reminiscent of autocatalysis.
Molecules can behave in extraordinary ways when specific encounters with neighboring molecules are controlled and the influence of the solvent is eliminated. Nature achieves this feat by cloaking small molecules in proteins and nucleic acids, rigorously controlling the collisions and concentration of the guest molecule, but at the same time denying chemists the opportunity to carry out simple physical organic studies on reactive components. In contrast, encapsulation complexes are readily accessible systems operating in homogenous media at room temperature. Encapsulation complexes are not intended as models for biological receptors, but their aptitude for encapsulating transition states (the moving targets of reaction processes) and reactive intermediates (the moving targets of physical chemists) has made them a promising venue for the creation and study of unique molecules and unique molecular behavior.
Acknowledgments
We thank Dr. Lubomir Sebo for expert assistance with graphics.
References
- 1.Fischer E. Ber Dt Chem Ges. 1894;27:2985. [Google Scholar]
- 2.Pedersen C J. J Am Chem Soc. 1967;89:2495–2496. [Google Scholar]
- 3.Rebek J., Jr Science. 1987;235:1478–1484. doi: 10.1126/science.3823899. [DOI] [PubMed] [Google Scholar]
- 4.Adrian J C, Jr, Wilcox C S. J Am Chem Soc. 1989;111:8055–8057. [Google Scholar]
- 5.Zimmerman S, Wu W. J Am Chem Soc. 1989;111:8054–8055. [Google Scholar]
- 6.Brown S P, Schaller T, Seelbach U P, Koziol F, Ochsenfeld C, Klarner F-G, Spiess H W. Angew Chem, Int Ed. 2001;40:717–720. [PubMed] [Google Scholar]
- 7.Sanderson P G E, Kilburn J D, Still W C. J Am Chem Soc. 1989;111:8314–8315. [Google Scholar]
- 8.Dixon R P, Geib S J, Hamilton A D. J Am Chem Soc. 1992;114:365–366. [Google Scholar]
- 9.Paliwal S, Geib S, Wilcox C S. J Am Chem Soc. 1994;116:4497–4498. [Google Scholar]
- 10.Canceill J, Lacombe L, Collet A. J Am Chem Soc. 1986;108:4230–4232. [Google Scholar]
- 11.Cram D J, Tanner M E, Thomas R. Angew Chem Int Ed Engl. 1991;30:1024–1027. [Google Scholar]
- 12.Sherman J C, Knobler C B, Cram D J. J Am Chem Soc. 1991;113:2194–2204. [Google Scholar]
- 13.Branda N, Wyler R, Rebek J., Jr Science. 1994;263:1267–1268. doi: 10.1126/science.8122107. [DOI] [PubMed] [Google Scholar]
- 14.Fujita M, Oguro D, Miyazawa M, Oka H, Yamaguchi K, Ogura K. Nature (London) 1995;378:469–471. [Google Scholar]
- 15.Parac T N, Caulder D L, Raymond K N. J Am Chem Soc. 1998;120:8003–8004. [Google Scholar]
- 16.Kang J, Rebek J., Jr Nature (London) 1996;382:239–241. doi: 10.1038/382239a0. [DOI] [PubMed] [Google Scholar]
- 17.Kang J, Rebek J., Jr Nature (London) 1997;385:50–52. doi: 10.1038/385050a0. [DOI] [PubMed] [Google Scholar]
- 18.Kang J, Hilmersson G, Santamaria J, Rebek J., Jr J Am Chem Soc. 1998;120:3650–3656. [Google Scholar]
- 19.Kang J, Santamaria J, Hilmersson G, Rebek J., Jr J Am Chem Soc. 1998;120:7389–7390. [Google Scholar]
- 20. Chen, J. & Rebek, J., Jr. (2002) Org. Lett., in press. [DOI] [PubMed]
- 21.Kusukawa T, Fujita M. J Am Chem Soc. 1999;121:1397–1398. [Google Scholar]
- 22.Kusukawa T, Yoshizawa M, Fujita M. Angew Chem Int Ed Engl. 2001;40:1879–1884. [PubMed] [Google Scholar]
- 23.Yoshizawa M, Kusukawa T, Fujita M, Yamaguchi K. J Am Chem Soc. 2000;122:6311–6312. doi: 10.1021/ja010875t. [DOI] [PubMed] [Google Scholar]
- 24.Yoshizawa M, Kusukawa T, Fujita M, Sakamoto S, Yamaguchi K. J Am Chem Soc. 2001;123:10454–10459. doi: 10.1021/ja010875t. [DOI] [PubMed] [Google Scholar]
- 25.Ziegler M, Brumaghim J L, Raymond K N. Angew Chem Int Ed Engl. 2000;39:4119–4121. doi: 10.1002/1521-3773(20001117)39:22<4119::aid-anie4119>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 26.Heinz T, Rudkevich D M, Rebek J., Jr Nature (London) 1998;394:764–766. [Google Scholar]
- 27. Chen, J., Craig, S. L. & Rebek, J., Jr. (2002) Nature (London), in press.