

For some time now (1–5), we have worked on the development of a “molecular construction kit,” analogous to the children's Tinkertoy† construction set, which permits the assembly of complicated objects from a limited set of rods, connectors, and other simple building elements. The idea is to do civil engineering with individual molecular components ranging in size from a few to several dozen Å. A somewhat related concept of “molecular Lego,” based on a different set of structural elements, was proposed (7) and developed (8) independently by Stoddard. Both belong to the wider category of “modular chemistry,” in which a small number of mid-sized rigid molecular structural elements are combined into complex structures (9).
At the start of our project, a very limited selection of types and lengths of straight terminally functionalized molecular rods and connectors was available. We synthesized additional ones based on the oligomers of [1.1.1]propellane ([n]staffanes) (10) and on oligomeric 10-vertex and 12-vertex p-carboranes (11). Other laboratories produced polycubyls (12), oligomers of [2.2.2]propellane (13), additional oligomers of 12-vector carboranes (14), and many rods constructed from the more common p-phenylene and acetylene subunits. A combination of several types of such structural elements permits one to achieve an accurate match to a desired rod length. A recent comprehensive review (15) makes it abundantly clear that after a decade of effort, considerable flexibility exists in the choice of molecular rod lengths and properties.
After some initial experiments with point (4) connectors, which function by forming bonds from a central atom to the termini of several rods, we turned our attention to star (4) connectors, which function by forming a bond between each of several star arm termini to the terminus of a rod or of another star arm. Various suitable star connectors such as 1,3,5-trisethynylbenzene (16) and hexaethynylbenzene (17) were already known and our laboratory provided a few additional terminally functionalized trigonal (18) and tetragonal (19) structures of this type, as did others.
Early on, we were faced with several decisions concerning the nature of the porous objects to be built from our rods and connectors: (i) Should they be free-floating or attached to a surface? (ii) Should they be limited in size (“zero-dimensional”) or “infinitely” periodic in two or three dimensions? (iii) If periodic, should they be single giant covalent molecules, or should they be supramolecular—i.e., formed from repeated units held together by weak intermolecular interactions? (iv) Most important, toward which purpose should their production be directed?
Objective
After producing some dumb-bell-shaped objects from our [n]staffanes for fun (20), we soon decided to discontinue work with free-floating structures. Surface-anchored structures, although harder to make and characterize, seemed more intriguing. A beautiful collection of free-floating porous polygonal and polyhedral molecules built from rods and connectors has since resulted from work at other laboratories (21–26).
Our initial decision concerning the size of the objects to be built was in favor of two-dimensionally periodic structures. The engineering of nanoporous three-dimensional crystals from molecular constituents was already underway elsewhere (27) and has since made great strides (28, 29), whereas truly two-dimensional monolayer grids and networks built from molecular rods and connectors were unknown and developing a general method for their controlled production represented a challenge that did not appear to be addressed in any other laboratory. Later, we thought, it would be possible to use epitaxy to go into the third dimension in an aperiodic fashion, by adding several different layers on top of each other in register. The resulting “designer solids” would be quite distinct from ordinary three-dimensional crystals.
We decided to make our two-dimensional structures as sturdy as possible, and given their extreme thinness, the choice of covalent rather than supramolecular structures seemed logical. It carried a significant penalty in that under most common conditions the formation of strong covalent bonds is irreversible. This fact prevents the correction of random errors in the synthesis and generates structures with high defect density. The two-dimensional synthesis would therefore be limited to the relatively few synthetic methods that form strong covalent bonds reversibly, or else random errors would have to be avoided by supramolecular preorganization of the reacting components performed under reversible conditions.
Assuming we could make sturdy two-dimensional grids of controlled structure, dimensions, and chemical functionalization, what would they be good for? Following up on earlier ideas (2–5) we are now concentrating on two options. A simple one is to use the grids as ultrathin separation barriers, more regular and thinner than those that have been described to date (30). A more challenging one is related to our interest in surface-mounted dipolar molecular rotors and propellers (31, 32): the grids could be used as scaffolds for the fabrication of regular planar arrays of interacting dipolar electrical rotors. Such arrays, assembled in a controlled fashion, would be quite interesting. They could be ferroelectric (hexagonal or trigonal grids) or antiferroelectric (square grids) (33), could support slowly propagating waves of rotational excitation (34–36), and might exhibit other interesting dielectric and optical properties.
General Considerations
The way in which we decided to go about producing two-dimensional grids was by linear coupling of arm-ends of star-shaped monomers forced to adhere to a surface with their arms parallel to the surface (4, 5, 37). The coupler is brought by diffusion from a solution contacting the surface. A schematic representation of the intended synthesis is shown in Fig. 1. For this purpose, the arms themselves, or alternatively, tentacles attached to the connector especially for the purpose, have to contain chemical functionalities with a large affinity for a surface, and their adhesion has to be essentially irreversible to avoid three-dimensional cross-linking in solution during the coupling step. Yet, the monomers have to be free to rotate and translate in the surface if they are to organize into a perfect grid. This condition suggested the use of a liquid–liquid or air–liquid interface. These interfaces also offer the advantages of no lasting imperfections such as steps and dislocations, and no permanent surface structure that would dictate a repeat period—this was important because we were interested in general synthetic procedures applicable to all rod lengths and connector sizes. An additional advantage of liquid–liquid and air–liquid interfaces is the promise of permitting grids to be harvested by fishing with a metal grid of the kind used in electron microscopy, in addition to other methods of transfer applicable to both solid and liquid surfaces.
Figure 1.

Interfacial synthesis of two-dimensional square (a) and hexagonal (b) grids from star-shaped monomers. The freely floating monomers are first adsorbed on a surface and then coupled into a grid.
Our main concern about using air–liquid and, particularly, liquid–liquid interfaces was that they are not as sharply defined on atomic scale as solid–liquid interfaces, and might permit excessive vertical excursions of the interfacially adsorbed material, which would result in the formation of irregular multilayered three-dimensionally cross-linked structures. Two-dimensional covalent coupling of molecules organized in a Langmuir–Blodgett (LB) film at an air–water interface had been described and yielded sturdy insoluble films, but no evidence for two-dimensional order was detected (38). We did not want to encumber our monomers with long alkyl chains that might be necessary to force them to stay at an air–water interface.
In the end, we decided to use mercury as the subphase and to rely on chemisorption rather than physisorption of the connectors to its well defined surface. Polarographers have investigated the adsorption of organic molecules in the mercury–water interface for many years (39), and firmly adhering monolayers of anions, including organic ones, such as oxalate (40), have been long known. Among uncharged species, compounds of the pyridine family (41) and those containing sulfur (42) seemed particularly promising as candidates for functionalities that would assure firm chemical bonding of our tentacles to the mercury surface.
The use of a metal as one of the phases has other advantages. It facilitates in situ use of grazing incidence spectroscopy, and offers good control of adsorption by adjustment of the surface potential. This could be relevant for the ultimate removal of the grid from the surface on which it was synthesized. Another possibility is to sever the tentacles chemically after they have served their purpose, and we demonstrated this successfully with trigonal connectors adsorbed on gold (18).
A Covalent Two-Dimensional Grid
The very first cross-shaped monomer tested, the anionic lanthanum sandwich complex of tetrapyridylporphyrin that we prepared for the purpose (ref. 37; Fig. 2), adsorbed firmly on mercury under open circuit conditions and was not removed even by boiling organic solvents, which merely exchanged the counterion. This struck us as fortunate, because under identical conditions, tetrapyridylporphyrin itself did not adsorb firmly. We assumed that the difference is related to the presence of negative charge on the sandwich complex and that at a suitable imposed potential the latter would adsorb firmly as well. We proposed that the strong adsorption might be due to mercury ions binding the connectors and preorganizing them into a continuous network. Because in our apparatus the mercury was in contact with copper, it was also conceivable that the binding ions were copper. It was clear from IR spectra that the porphyrin rings of the adsorbed lanthanum complex were parallel to the surface, suggesting that the four pyridine rings of the bottom deck of the sandwich acted as tentacles and were adsorbed on the surface, whereas those of the other would be available for linking.
Figure 2.

A quasilinear coupler (a, p-xylylene dibromide), a cross shaped monomer (b, lanthanum sandwich complex of tetrapyridylporphyrin), an idealized structure (c), and an STM image (d) of a square grid.
Treatment with p-xylylene dibromide as a quasilinear coupling agent yielded a product that still adhered well to mercury (43, 44). IR spectra showed that it contained the p-xylylene units and pyridinium rings as expected, and that the porphyrin rings remained parallel to the surface. For an examination of long-range order, we chose scanning tunneling microscopy (STM). Unfortunately, STM cannot be done in situ on liquid mercury (45, 46), and we decided to transfer the product to highly ordered pyrolytic graphite (HOPG) for imaging. This involved boiling in hydrochloric acid to remove all mercury oxide impurities, casting a very thin film of polystyrene over the surface, transferring the film to an HOPG surface, and dissolving the polystyrene. An examination by atomic force microscopy showed that the bottom surface of the polystyrene film that carried the grid was rough, and the transfer undoubtedly caused folding and mechanical damage. Nevertheless, the images (Fig. 2) revealed a series of product molecules in the form of flakes about 100–150 nm across and 0.7 nm thick. Each flake was composed of squares of the anticipated size, arranged into a grid that was locally ordered to a surprising degree, although the overall order was poor and there were many defects.
Two aspects of the result called for a closer examination. First, the IR spectra of the product were more intense than expected for a monolayer, and we were not sure that the STM tip did not image merely the bottom layer of several. The presence of multilayers could be an artifact introduced by the transfer to HOPG, but it could also be an indication of problems with the initial coupling on mercury. A better transfer procedure was needed, and it seemed best to enlarge the size of the product molecules sufficiently to permit fishing with a metal net. Second, the local regularity within the grid was striking, considering that the coupling conditions were irreversible. We felt that this supported the tentative proposal (37) that mercury ions formed by oxidation of the elemental liquid weakly bind to pairs of pyridine nitrogen atoms, preorganizing the cross-shaped connectors into a supramolecular grid before the treatment with p-xylylene dibromide. The pyridine arms in the upper deck appear to be a little too long compared with the N–Hg2+–N distance, but a rotation of the upper deck relative to the lower deck should permit the p-xylylene unit to bridge them comfortably. There seemed to be no close precedent to such formation of a metal-ion-bound open grid on mercury, but the formation of compact insoluble layers was well known as mentioned above, and somewhat related metal-ion-bound supramolecular grid formation on a graphite electrode had been proposed (47).
Both an increase in the size of the product molecules and an improvement in their regularity required an optimization and annealing of the supramolecular grid present before the coupling procedure. We therefore postponed further work on the covalent grid and the full publication of our results and decided to first examine the putative metal-ion-bound supramolecular grids in more detail.
Cation-Bonded Supramolecular Two-Dimensional Grids
We started by securing the collaboration of an electrochemist from an institution with a long tradition in polarography on mercury electrodes, Lubomír Pospíšil from the Heyrovský Institute in Prague, Czech Republic. The work done so far has used star-shaped molecules with tentacles containing one or more thioether sulfur atoms. Four-armed sandwich complexes of tetraarylcyclobutadienecyclopentadienylcobalt carrying five tentacles on the cyclopentadienyl deck (48; L. Pospíšil, N. Varaksa, T. F. Magnera, T. Brotin, B. Noll & J. Michl, unpublished results) and three-armed benzene derivatives with three tentacles at the ends of the arms (N. Varaksa, L. Pospíšil, Z. Janoušek, B. Grüner, B. Wang, J. Pecka, R. Harrison, B. Noll, and J. Michl, unpublished results, and refs. 49 and 50) were all found to promote the anodic dissolution of mercury at relatively negative potentials, forming chemisorbed surface layers characterized by low electrode capacitance. At somewhat more negative potentials, capacitance increased, suggesting that the solute was then merely physisorbed, and at much more negative potentials, it was the same as in a pure supporting electrolyte, showing no evidence for adsorption at all. Simple thioethers did not form such chemisorbed layers, suggesting that the presence of multiple thioether functionalities in a single molecule indeed led to network formation.
As a first step in structural characterization of the chemisorbed
layer, we decided to determine the surface area per redox center and
per connector molecule. Their mutual relation would provide a
convolution of information on the number of redox centers per molecule
and on the number of electrons exchanged by each. In perfect monolayer
grids, a tetratentacled connector would half-own four metal ions, and a
trigonal one, three. Because each ion could exchange one
(Hg+) or two (Hg2+ or
Hg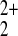 ) electrons, two or four electrons could be
exchanged per a tetragonal connector, and three halves or three per a
trigonal one. A pentatentacled monomer would not be able to form a
regular periodic grid, and would perhaps be less likely to use all of
its thioether sulfur atoms for metal binding efficiently.
Electrochemical techniques gave results that were of the right order of
magnitude, but they were not entirely satisfactory because of practical
limitations imposed by solubility and related factors.
) electrons, two or four electrons could be
exchanged per a tetragonal connector, and three halves or three per a
trigonal one. A pentatentacled monomer would not be able to form a
regular periodic grid, and would perhaps be less likely to use all of
its thioether sulfur atoms for metal binding efficiently.
Electrochemical techniques gave results that were of the right order of
magnitude, but they were not entirely satisfactory because of practical
limitations imposed by solubility and related factors.
For an independent determination of the mercury surface area per connector molecule under controlled potential conditions at an interface with a liquid electrolyte, we built (50) a trough that modified an LB design developed for work on a mercury–air interface (51) by adding a conducting superphase (acetonitrile with a supporting electrolyte) and by employing the mercury pool as the working electrode in a three-electrode electrochemical cell. This “electrochemical LB trough” turned out to be a very valuable tool.
LB isotherms were obtained first for two pentatentacled molecules, one whose tentacles were short and contained a single thioether sulfur atom each, and another whose tentacles were long and contained two such sulfur atoms separated by two methylene groups (L. Pospíšil, N. Varaksa, T. F. Magnera, T. Brotin, B. Noll & J. Michl, unpublished results). The former formed a firmly chemisorbed surface layer at potentials less negative than that of the mercury redox peak, in agreement with electrochemical capacitance measurements. The surface area per molecule corresponded well to tight packing of the larger of the two decks of the sandwich complex, the tetrasubstituted cyclobutadiene. At more negative potentials, where capacitance measurements suggest mere physisorption, the LB isotherms indicated nearly no resistance to compression, which apparently causes the adsorbed molecules to move into the superphase. The LB results for the molecule with five tentacles containing two sulfur atoms apiece were more intriguing (Fig. 3). Here, compression met with significant resistance both at potentials less negative and those more negative than the mercury redox peak, but the extrapolated surface area per molecule was much larger (∼4.5 nm2) in the former region than in the latter (∼3.3 nm2). Again, this was in agreement with electrochemical data. It suggested first, that the adsorption can remain firm even when the mercury ions are reduced to elemental mercury, and second, that the tentacles then spread much less on the surface, permitting a tight packing of the upper decks. The mental image that we have of these molecules is that of five-legged “daddy longlegs” spider resting on its belly with legs stretched at less negative potentials and standing up at more negative potentials, but this is yet to be proven.
Figure 3.

Surface areas (a) for tetragonal connectors with five tentacles (b) on mercury–acetonitrile interface determined from LB isotherms as a function of surface potential.
Especially clear-cut results were obtained with trigonal
connectors that contained one thioether sulfur atom in each of the
three tentacles (N. Varaksa, L. Pospíšil, Z.
Janoušek, B. Grüner, B. Wang, J. Pecka, R. Harrison, B.
Noll, and J. Michl, unpublished results, and refs. 49 and 50). At
potentials more positive than −0.05 V, a value located in the rising
part of the mercury oxidation wave (−0.1 V), electrode capacitance is
extraordinarily low and demonstrates the presence of a firmly adsorbed
and highly organized surface layer. The LB isotherms measured in this
region are potential-independent and yield an area of about 6.3
nm2 per molecule. At potentials more negative
than −0.1 V, where capacitance values correspond to physisorption, the
LB isotherms show very little resistance and yield a zero area per
molecule—i.e., compression easily removes the physisorbed solute into
bulk solution. The results of electrochemical measurements show that
the bridging mercury cation is Hg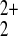 , or less likely
Hg2+, and not
Hg
, or less likely
Hg2+, and not
Hg , as initial results indicated
(49), but we are still performing additional experiments.
, as initial results indicated
(49), but we are still performing additional experiments.
Further in situ characterization of the structure of the chemisorbed supramolecular grid is to be done by grazing incidence IR spectroscopy. Ex situ characterization by transfer to HOPG and STM imaging is presently being attempted. Until these results become available, we cannot feel confident about the structure of the grid. Nevertheless, it is reasonable to ask whether it is possible to propose a structure that is realistic and would fit the area per molecule deduced from the LB isotherms.
If we assume a regular hexagonal grid without defects, the observed molecular surface area corresponds to a structure composed of hexagons with an ∼2.2-nm edge length between centers of benzene rings. This is entirely reasonable for the structure of the connector, considering the flexibility of the tentacles. Molecular models suggested two likely conformations of the tentacle chain, differing strikingly in the orientation of the S–Hg2+–S or S–Hg+–Hg+–S link relative to the edge of the hexagon. In one of these, the two are nearly parallel and in the other, nearly perpendicular. Optimization of the two geometries with the AM1 method yielded planar hexagonal structures with edge lengths of 2.7 and 1.9 nm, respectively. Pending further investigation, we propose the latter structure for the observed grid (ref. 50; Fig. 4), and attribute the difference between 2.2 and 1.9 nm to several factors: the uncertainty in the nature of the mercury ion (Hg2+ was assumed in the modeling), errors in the modeling, which used a semiempirical Hamiltonian and neglected the presence of the mercury surface and of counterions, and imperfections in the grid, which undoubtedly reduce the density of surface packing.
Figure 4.

Elementary unit of the hexagonal grid structure proposed for a mercury–ion connected trigonal connectors chemisorbed on mercury–acetonitrile interface.
Our original optimism with regard to facile equilibration and annealing of the supramolecular grid simply by keeping the potential near −0.1 V may have been unfounded, because of an unexpected discovery of very remarkable substantial hysteresis in the oxidative formation and reductive removal of the grid. When the adsorbed monolayer is first formed at a potential more positive than −0.05 V and the potential is subsequently swept to more negative values, reduction of the mercury ions is observed at −0.1 V, but the adsorbed layer survives intact until the potential reaches −0.85 V. This is clear both from the capacitance curve and from the LB isotherms, both of which remain entirely unchanged until then. Yet, the monolayer does not form spontaneously from solution at these negative potentials. Its enormous metastability, presumably due to slow hole nucleation, is puzzling, as one would not expect neutral Hg atoms to hold the thioether sulfur atoms of two tentacles together particularly well. However, it is not entirely unprecedented in that the “five-legged spider” molecules mentioned above are also chemisorbed even after the mercury ions are reduced.
Currently, we are examining connectors whose tentacles contain pyridine rings. We now expect to find that the lanthanum sandwich complex of tetrapyridylporphyrin indeed binds so well to the surface of mercury because it builds a mercury ion bound square grid already at open circuit potential. It is reasonable that the negative charge of this anion would facilitate the oxidation process relative to the electroneutral tetrapyridylporphyrin itself, and the latter will presumably form a similar grid at potentials more positive than that of the open circuit.
In general, the hybrid supramolecular-covalent approach to covalent grid formation seems promising, although it will be necessary to adjust the length of the supramolecular grid forming tentacles to conform to that of the covalent grid forming arms separately in each case. For some purposes, the ion-bonded supramolecular grids may be adequate in themselves, particularly if they can be transferred to other surfaces intact.
Hydrogen-Bonded Supramolecular Two-Dimensional Grids
Along with metal ion coordination, hydrogen bonding is a favorite in the construction of supramolecular structures under reversible conditions. We felt that if we could construct a weakly bound supramolecular grid from connectors and hydrogen-bonding linkers, perhaps the latter could be later exchanged gradually for irreversibly bound covalent ones of a similar size and shape. If this were feasible without ever taking more than a small fraction of the linkers out of the grid, the regular structure might survive intact until the grid is fully covalent. The first question was, can the initial hydrogen-bonded two-component supramolecular grids be formed?
An examination of the effect of the addition of hydroquinone and 4,4′-dihydroxybiphenyl on the LB isotherms of tetrapyridylporphyrin on a water–air interface encouraged us to believe that they perhaps indeed form a regular square grid in which the pyridine arms of neighboring porphyrins are tied together by the dihydroxyarene linkers. When such water surfaces were formed on HOPG and the water was evaporated, STM showed very regular and very large domains of deposit on the HOPG surface (Fig. 5). However, they did not have the expected tetragonal symmetry, and the best structural interpretation that we could offer was that the porphyrins were bound together by hydroquinones as intended, but were arranged in alternating double rows of macrocycles lying flat and standing perpendicular (52). The orientations in which the double rows ran were presumably dictated by the direction in which the water edge withdrew at the end of the evaporation process. We are presently attempting to avoid this distortion by the use of a horizontal (Schäfer) transfer from water surface to HOPG, and we are also working with similar grids on a mercury–benzene interface.
Figure 5.

An STM image of a grid formed from tetrapyridylporphyrin and hydroquinone after transfer to HOPG.
Perspective
We now appear to be tantalizingly close to being able to synthesize both metal-bonded and hydrogen-bonded regular two-dimensional grids of arbitrary and controlled square and hexagonal structure. Once their domain size and defect density are acceptable, we plan to probe their utility in the fabrication of regular surface-mounted arrays of dipolar molecular rotors. We also hope to proceed with efforts to convert these supramolecular structures into regular free-standing covalent grids of potential use as dipolar rotor carriers or separation membranes.
Acknowledgments
We are grateful to a large number of enthusiastic collaborators, whose names are given in the references quoted. Over the years, our work on molecular Tinkertoys was supported by the National Science Foundation, the Department of Energy, and the U.S. Army Research Office.
Footnotes
Tinkertoy is a trademark of Playskool, Inc., Pawtucket, RI, and designates a children's toy construction set consisting of straight wooden sticks and other simple elements insertable into spool-like connectors. The assembly of triangular trinuclear metal cluster units into polyhedra and stacks has also been referred to as “Tinker-Toy” construction (6).
References
- 1.Kaszynski P, Michl J. J Am Chem Soc. 1988;110:5225–5226. [Google Scholar]
- 2.Michl J, Kaszynski P, Friedli A C, Murthy G S, Yang H-C, Robinson R E, McMurdie N D, Kim T. In: Strain and Its Implications in Organic Chemistry, NATO Advanced Science Institute Series. de Meijere A, Blechert S, editors. Vol. 273. Dordrecht, The Netherlands: Kluwer; 1989. pp. 463–482. [Google Scholar]
- 3.Kaszynski P, Friedli A C, Michl J. J Am Chem Soc. 1992;114:601–620. [Google Scholar]
- 4.Michl J. In: Applications of Organometallic Chemistry in the Preparation and Processing of Advanced Materials. Harrod J F, Laine R M, editors. Dordrecht, The Netherlands: Kluwer; 1995. pp. 243–267. [Google Scholar]
- 5.Harrison R M, Magnera T F, Vacek J, Michl J. In: Modular Chemistry. Michl J, editor. Dordrecht, The Netherlands: Kluwer; 1997. pp. 1–16. [Google Scholar]
- 6.Underwood D J, Hoffman R, Tatsumi K, Nakamura A, Yamamoto Y. J Am Chem Soc. 1985;107:5968–5980. [Google Scholar]
- 7.Stoddard F. Chem Br. 1988;24:1203–1208. [Google Scholar]
- 8.Raymo F M, Stoddart J F. Chem Rev. 1999;99:1643–1664. doi: 10.1021/cr970081q. [DOI] [PubMed] [Google Scholar]
- 9.Michl J, editor. Modular Chemistry, NATO Advanced Science Institute Series. Dordrecht, The Netherlands: Kluwer; 1997. 499. [Google Scholar]
- 10.Levin M, Kaszynski P, Michl J. Chem Rev. 2000;100:169–234. doi: 10.1021/cr990094z. [DOI] [PubMed] [Google Scholar]
- 11.Müller J, Baše K, Magnera T F, Michl J. J Am Chem Soc. 1992;114:9721–9722. [Google Scholar]
- 12.Eaton P E, Maggini M. J Am Chem Soc. 1988;110:7230–7232. [Google Scholar]
- 13.Zimmerman H E, Goldman T D, Hirzel T K, Schmidt S P. J Org Chem. 1980;45:3933–3951. [Google Scholar]
- 14.Yang X, Jiang W, Knobler C B, Hawthorne M F. J Am Chem Soc. 1992;114:9719–9722. [Google Scholar]
- 15.Schwab P F H, Levin M D, Michl J. Chem Rev. 1999;99:1863–1934. doi: 10.1021/cr970070x. [DOI] [PubMed] [Google Scholar]
- 16.Huebel W, Mereny R. Angew Chem. 1962;74:781. [Google Scholar]
- 17.Diercks R, Armstrong J C, Boese R, Vollhardt K P C. Angew Chem Int Ed Engl. 1986;25:268–269. [Google Scholar]
- 18.Schöberl U, Magnera T F, Harrison R, Fleischer F, Pflug J L, Schwab P F H, Meng X, Lipiak D, Noll B C, Allured V S, et al. J Am Chem Soc. 1997;119:3907–3917. [Google Scholar]
- 19.Harrison R M, Brotin T, Noll B C, Michl J. Organometallics. 1997;16:3401–3412. [Google Scholar]
- 20.Janecki T, Shi S, Kaszynski P, Michl J. Collect Czech Chem Commun. 1993;58:89–104. [Google Scholar]
- 21.Zhang J, Moore J S, Xu Z, Aguirre R A. J Am Chem Soc. 1992;114:2273–2274. [Google Scholar]
- 22.Drain C M, Nifiatis F, Vasenko A, Batteas J D. Angew Chem Int Ed Engl. 1998;37:2344–2346. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2344::AID-ANIE2344>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 23. Fujita, M., Umemoto, K., Yoshizawa, M., Fujita, N., Kusukawa, T. & Biradha, K. (2001) Chem. Commun. 509–518.
- 24.Leininger S, Olenyuk B, Stang P J. Chem Rev. 2000;100:853–908. doi: 10.1021/cr9601324. [DOI] [PubMed] [Google Scholar]
- 25.Caulder D L, Raymond K N. Acc Chem Res. 1999;32:975–982. [Google Scholar]
- 26.Cotton F A, Lin C, Murillo C A. Acc Chem Res. 2001;34:759–771. doi: 10.1021/ar010062+. [DOI] [PubMed] [Google Scholar]
- 27.Hoskins B F, Robson R. J Am Chem Soc. 1989;111:5962–5964. [Google Scholar]
- 28.Zaworotko M J. Chem Soc Rev. 1994;23:283–288. [Google Scholar]
- 29.Eddaoudi M, Moler D B, Li H, Chen B, Reineke T M, O'Keeffe M, Yaghi O M. Acc Chem Res. 2001;34:319–330. doi: 10.1021/ar000034b. [DOI] [PubMed] [Google Scholar]
- 30.Lee W, Hendel R A, Dedek P, Janout V, Regen S L. J Am Chem Soc. 1995;117:10599–10600. [Google Scholar]
- 31.Vacek J, Michl J. New J Chem. 1997;21:1259–1267. [Google Scholar]
- 32.Vacek J, Michl J. Proc Natl Acad Sci. 2001;98:5481–5486. doi: 10.1073/pnas.091100598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rozenbaum V M, Ogenko V M, Chuiko A A. Sov Phys (Uspekhi) 1991;34:883–902. [Google Scholar]
- 34.Zorski H, Infeld E. Phys Rev Lett. 1992;68:1180–1183. doi: 10.1103/PhysRevLett.68.1180. [DOI] [PubMed] [Google Scholar]
- 35.DeLeeuw S W, Solvaeson D, Ratner M A, Michl J. J Phys Chem B. 1998;102:3876–3885. [Google Scholar]
- 36.Sim E, Ratner M A, deLeeuw S W. J Phys Chem B. 1999;103:8663–8670. [Google Scholar]
- 37.Magnera T F, Peslherbe L M, Körblová E, Michl J. J Organomet Chem. 1997;548:83–89. [Google Scholar]
- 38.Palacin S, Porteu F, Ruaudel-Teixier A. Thin Films. 1995;20:69–82. [Google Scholar]
- 39.Zuman P, Rusling J F. Encyclopedia of Surface and Colloid Science. New York: Dekker; 2002. , in press. [Google Scholar]
- 40.Müller C, Claret J, Sarret M. J Electroanal Chem. 1986;207:263–278. [Google Scholar]
- 41.Andreoli R, Battistuzzi Gavioli G, Benedetti L, Borsari M, Fontanesi C. J Electroanal Chem. 1990;293:209–218. [Google Scholar]
- 42.Philipp R, Retter U. Electrochim Acta. 1995;40:1581–1585. [Google Scholar]
- 43.Magnera T F, Pecka J, Vacek J, Michl J. Nanostructural Materials: Clusters, Composites, and Thin Films. 1997. , ACS Symposium Series 679, eds. Moskovits, M. & Shalaev, V. (Am. Chem. Soc., Washington, DC), pp. 213–220. [Google Scholar]
- 44.Magnera T F, Pecka J, Michl J. In: Science and Technology of Polymers & Advanced Materials. Prasad P N, Mark J E, Kandil S H, Kafafi Z H, editors. New York: Plenum; 1998. pp. 385–391. [Google Scholar]
- 45.Bruckner-Lea C, Janata J, Conroy J, Pungor A, Caldwell K. Langmuir. 1993;9:3612–3617. [Google Scholar]
- 46.Conroy J F T, Caldwell K, Bruckner-Lea C, Janata J. Electrochim Acta. 1995;40:2927–2934. [Google Scholar]
- 47.Shi C, Anson F C. Inorg Chim Acta. 1994;225:215–227. [Google Scholar]
- 48.Brotin T, Pospíšil L, Fiedler J, King B T, Michl J. J Phys Chem B. 1998;102:10062–10070. [Google Scholar]
- 49.Pospíšil L, Heyrovský M, Pecka J, Michl J. Langmuir. 1997;13:6294–6301. [Google Scholar]
- 50.Varaksa N, Pospíšil L, Magnera T F, Michl J. Proc Natl Acad Sci. 2002;99:5012–5017. doi: 10.1073/pnas.082098299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith T. J Coll Interface Sci. 1968;26:509–517. [Google Scholar]
- 52. Magnera, T. F. & Michl, J. (1998) Atual. Fís Quim. Org, 50–55.