

Abstract
By drawing on nature conceptually, the practice of chemical synthesis can now be broadened in scope to include the supramolecular synthesis of supermolecules and their postassembly covalent modification to form mechanically interlocked molecules, which the chemist can use to build functioning nanosystems after the style of those found in the living world.
There is absolutely no doubt, to quote from the recent Classics in Total Synthesis (1), that the “construction of nature's molecules in the laboratory from atoms and/or simple molecules, a process known as total synthesis, is one of the most demanding human practices.” Although the total synthesis of unnatural (2) as well as natural products has taxed the minds and tested the skills of chemists for more than a century, the practice has remained, until recently, a peculiarly artificial one with only occasional concessions being made to biomimetic approaches (3). The emergence of chemistry beyond the molecule (4), however, has done much to initiate a paradigm shift over the last 25 years where concepts like molecular recognition (5) and self assembly (6) are transferred from biological processes into chemical nanosystems through the medium of synthesis. What supramolecular chemistry (4) has done for synthetic chemists is to alert them to the fact that chemical synthesis does not begin and end with the making and breaking of covalent bonds. First, it has focused the attention of these key players in materials science on the importance of noncovalent and coordinative bonds in the spontaneous self organization of membranes (7) that could be used to house working artificial cells. Second, it has led to the development of the mechanical bond in molecules, such as catenanes and rotaxanes (8), providing ready access to a whole new range of actuators and switches that can be controlled chemically, electronically, or optically, and some of which are capable of receiving, storing, processing, and broadcasting information at the nano- and mesoscopic levels (9).
Transferring Concepts
There are two main areas of contemporary synthesis that come under the spell of supramolecular phenomena (10). One is supramolecular assistance to the covalent synthesis of molecular compounds, in which either (i) a temporary recyclable template dictates the outcome of a reaction to give a product that would not be nearly so efficiently made without the template; or (ii) a permanent entrapped template results in the formation of an interlocked molecule, e.g., a catenane or a rotaxane. The other is noncovalent or supramolecular synthesis, which can lead to either (i) small discrete supermolecules, or (ii) large supramolecular arrays. The guiding principles for the development of these new areas in synthesis have been molecular recognition (5) and self assembly (6), and the reaction control can be either kinetic or thermodynamic or a mixture thereof. Aside from the noncovalent self assembly of chemical systems, there is a rapidly growing interest in self assembly using dynamic covalent bonds and the creation of virtual combinatorial libraries under thermodynamic control—but that is another story (11, 12).
Concepts Exemplified
One of the most intensively studied recognition motifs, which has been exploited in numerous supramolecularly assisted covalent syntheses, is the π-donor/π-acceptor one (6), wherein [π⋅⋅⋅π] stacking is invariably greatly augmented by [C—H⋅⋅⋅O], and less so by [C—H⋅⋅⋅π], interactions (13). The template-directed synthesis (Fig. 1a) of the ubiquitous tetracationic cyclophane, cyclobis(paraquat-p-phenylene) 3⋅4PF6, from the dicationic precursor 1⋅2PF6 and the dibromide 2 provides the example (14) par excellence of a temporary recyclable template at work. It took some time to establish that the polyether chains on the π-donating templates are so absolutely crucial, if high yields are to be achieved in the kinetically controlled syntheses. Indeed, progressing outwards from the π-electron-rich 1,5-dioxynaphthalene ring system, it is the second and/or third oxygen atoms along the polyether chains that are essential for templating action to be expressed. This fact is brought into sharp focus by the x-ray diffraction analysis (Fig. 1b) carried out on the crystalline superstructure of the 2:1 complex formed between the 1,5-dioxynaphthalene-based polyether template 4 and the tetracationic cyclophane 3⋅4PF6. The interactions present in the superstructure are reflected (15) in stability constant (Ka) measurements, performed in acetonitrile solution for the 1:1 complexes formed between 3⋅4PF6 and a selection of 1,5-dioxynaphthalene-based substrates, ranging from the simple dimethoxy derivative (Ka < 100⋅M−1) to 4 (Ka > 5,000⋅M−1). This template was used to synthesize the tetracationic cyclophane 3⋅4PF6 in 81% yield when excess (3 molar equivalents) of 4 was used, and the reaction between the dicationic precursor 1⋅2PF6 and the dibromide 2 was performed under ultrahigh pressure (12 kbars) for 3 days. Although a 2:1 complex is observed in the solid state, presumably because of the formation of polar stacks of alternating donors and acceptors along the C2 axis in the crystal, neither this supermolecule nor the larger supramolecular array can be detected in acetonitrile solution. Indeed, continuous variation (Job) plots indicated that a 1:1 complex, i.e., a (2)pseudorotaxane, is the predominant species present in this solution, at least when the concentration is less than 10−3 M.
Figure 1.

(a) Template-directed synthesis of the tetracationic cyclophane 3⋅4PF6. (b) The x-ray crystal structure and superstructure of 34+/42. (c) Template-directed synthesis of the (2)catenane 5⋅4PF6. (d) The x-ray crystal structure and superstructure of 54+.
Explaining Concepts
Whereas the synthesis of the tetracationic cyclophane 3⋅4PF6 uses a template that can be reclaimed from the reaction mixture and recycled, the synthesis (16) of the (2)catenane 5⋅4PF6 in acetonitrile at room temperature from 1⋅4PF6 and 2 in the presence of 1 molar equivalent of the macrocyclic polyether, 1,5-dinaphtho (38)crown-10 (6), as a template, results (Fig. 1c) in its stoichiometric consumption, i.e., it becomes a permanent fixture of the (2)catenane. The 51% yield isolated, when all of the reactants are present in equimolar quantities, can be raised to well over 80% when the template 6 is present in the reaction mixture in 3-fold excess under ultrahigh pressures. The template-directed synthesis of 5⋅4PF6 under kinetic control constitutes a delicate interplay (Fig. 2) between covalent, noncovalent, and mechanical bonds. The formation of the first covalent bond switches on molecular recognition, such that the intermediate trication recognizes the template, inducing spontaneously the formation of a (2)pseudorotaxane. Subsequently, this 1:1 complex grasps the opportunity to switch on even more molecular recognition by forming a second covalent bond—and a mechanical bond in the bargain. The noncovalent bonds that template the formation of the (2)catenane, following their dual priming by successive covalent bond formations, live on inside the (2)catenane afterward. This important property can be revealed by, for example, dynamic NMR spectroscopy in solution and is one of the underlying fundamental reasons why such degenerate molecules, with appropriate constitutional modifications, can be transformed into bistable redox-controllable molecular switches. The other reason why suitably asymmetric analogs of 5⋅4PF6 can be switched off and on reversibly, either (i) by reducing the bipyridinium units, with the supply of up to four electrons, in the tetracationic cyclophane component; or (ii) by the oxidation of the dioxynaphthalene units, with the removal of up to two electrons, in the 1,5-dinaphtho (38)crown-10 component. In simpler language, we have a means of turning the brakes off and on across the mechanical bond of a π-donor/π-acceptor (2)catenane.
Figure 2.

Note the delicate interplay between covalent, noncovalent, and mechanical bonds in the template-directed synthesis of the (2)catenane 54+. For an explanation of the graphics, see Fig. 1c. (I) Dication 12+ dose not recognize template 6 until the reaction with dibromide 2 ensues. (II) One covalent bond is formed. (III) A recognition site is switched on in the trication. (IV) Threading is driven by noncovalent bond formation. (V) Molecular recognition is switched on partially in the 1:1 complex. (VI) Another covalent bond is formed. (VII) Yet more molecular recognition is switched on, and a mechanical bond is also formed. (VIII) The weak noncovalent bonding interactions that template the formation of 54+ live on inside the molecule afterward.
Concepts Broadened
The x-ray crystal structure (Fig. 1d) of 54+ gives a wonderful snapshot of this highly ordered molecule and the noncovalent bonding interactions that hold the crown ether ring in place with respect to the tetracationic cyclophane with which it interlocks. Moreover, it comes as no surprise that beyond the molecule in the crystal, the (2)catenane forms continuous π-donor/π-acceptor stacks. We speculated that this superstructural order, present in the solid state, might be transferrable by using the Langmuir–Blodgett (LB) technique to interfaces and surfaces. And so it transpired that, provided the hexafluorophosphate counterions are exchanged for dimyristoylphosphatidyl (DMPA−) anions (2), catenanes like 54+ can be introduced into Langmuir films and LB monolayers (17). The (2)catenane tetracations in these multilayers can be considered to reside in a liquid-crystalline-like environment, following what is essentially a simple act of supramolecular synthesis! Now that we know that superstructural order is within our grasp in both two and three dimensions with molecules that can be made to switch chemically, electrochemically, or photochemically, we have the perfect match. The marriage of these two phenomena—namely solid-state structural and superstructural order with solution-state mechanical switching at the molecular level—could constitute a significant leap in the direction of fabricating materials that might act as information storage devices.
Applying Concepts
In the event, replacement of one of the dioxynaphthalene (DNP) units in 54+ with a tetrathiafulvalene (TTF) unit gave (Fig. 3a) a (2)catenane 7⋅4PF6, which fits the bill completely. It is easy to make by template-directed synthesis, and it constitutes the desirable all-on/-off switch in solution (18). In its native state in solution, the TTF unit much prefers to reside inside the tetracationic cyclophane component until it is oxidized to the TTF+● radical cation (at E1/2 320 mV) or indeed to the TTF2+ dication (at E1/2 720 mV), although the DNP unit remains strictly neutral. In the solid state, not only does the TTF unit reside (Fig. 3b) inside the tetracationic cyclophane of 7⋅4PF6, but the molecular π-donor/π-acceptor stack extends (Fig. 3c) beyond the molecule into the supramolecular domain. Hardly surprisingly, the redox-active switchable (2)catenane 7⋅4PF6 forms (19) stable monolayers when it is cospread onto an aqueous substrate with 4 equivalents of the sodium salt of dimyristoylphosphatidyl. We speculate that the two-dimensional order, shown in Fig. 3d, is a result of the self organization of the 74+ tetracation to maximize [π⋅⋅⋅π] stacking interactions after the fashion observed in the solid-state superstructure. It would appear that supramolecular synthesis can often take its cue from the way that molecules with mutual recognition sites pack in the solid state.
Figure 3.

(a) Structural formula of the more stable translational isomer of the redox active, bistable (2)catenane 7⋅4PF4. (b) Ball-and-stick representation of the solid structure of 74+. (c) Space-filling representation of the superstructure of 74+ in the crystal. (d) Schematic representation of the monolayer of 7⋅4 dimyristoylphosphatidyl, stabilized by extensive [⋅⋅⋅] interactions.
Although most of the research on switchable catenanes and rotaxanes has been carried out in the context (20) of solution-phase mechanical processes, we have demonstrated (refs. 9 and 21; Y. Luo, C. P. Collier, J. O. Jeppesen, K. A. Nielsen, E. DeIonno, G. Ho, J. Perkins, H.-R.T., T. Yamamoto, J.F.S., and J. R. Heath, unpublished work) that relative mechanical movements between the components in interlocked molecules can be stimulated electrically within the setting of a solid-state device. Not only has reversible electronically driven switching been observed (21, 22) in devices incorporating the bistable (2)catenane 7⋅4PF6, but a crosspoint random access memory circuit and a simple logic circuit have been fabricated (23) recently by using a related amphiphilic (2)rotaxane. These demonstrations amount to a proof of principle and may be considered as positive evidence that switchable catenanes and rotaxanes can withstand simple device-processing steps allowing them to perform mechanically in a soft-matter environment.
Concepts Extended
Many would argue that the existence of polar stacks of π-donor/π-acceptor (2)catenanes in the solid state does not constitute a true supramolecular synthesis. Although good circumstantial evidence has been presented above for the self organization of the same (2)catenanes in an analogously intermolecular manner at air–water interfaces in Langmuir troughs before their being transferred to different substrates, we have uncovered, during a quest to synthesize a (3)rotacatenane (24), some clearcut examples (25) of (2)catenanes, incorporating 1,5dinaphtho (38)crown-10 interlocked with bipyridinium-based tetracationic cyclophanes that form supramolecular homodimers in solution as well as in the solid state. The (2)catenane 8⋅4PF6, shown in Fig. 4, dimerizes in hexadeuterioacetone solution at 185 K with an association constant of 20⋅M−1. Apart from being enthalpically stabilizing, this kind of self association is entropically desirable, i.e., many supermolecules are formed, while the principle (26) of maximum site occupancy is fulfilled. Indeed, the propensity of self-complementary molecules to form (27) supramolecular homodimers continues to plague many attempts to produce supramolecular polymers.
Figure 4.
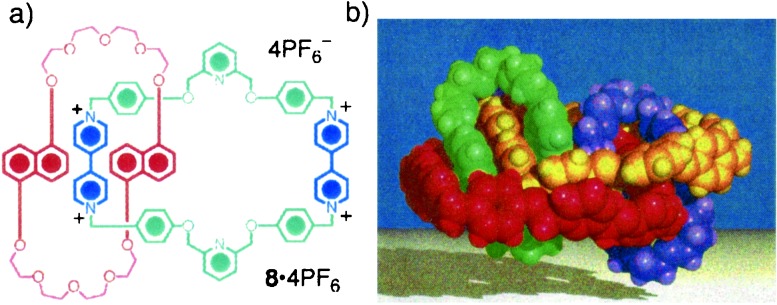
(a) Structural formula of the self-complementary (2)catenane 8⋅4PF4. (b) Space-filling representation of the supramolecular dimer formed by 84+ in the solid state. One of the (2)catenanes in made up of the components colored green and yellow, and the other of the components is colored red and mauve.
Synthetic goals will forever be to research chemists what virgin territories have been to explorers, with the possible exception that the objectives of synthesis, being often of our own making, know absolutely no boundaries other than the limits imposed by our imaginations. It was very much in this spirit that the synthetic goals summarized in Fig. 5 were reached (28). Both the (5)catenane 13⋅12PF6, also known as olympiadane, and the doubly branched (7)catenane 15⋅20PF6 provided good quality single crystals from which x-rays have shed a unique light on the exquisite order present in these molecules. The template-directed synthesis of 13⋅12PF6 and 15⋅20PF6, together with the intermediate (6)catenane 14⋅16PF6, proceeds to give roughly equal quantities of these oligocatenanes so long as the reaction of the key (3)catenane 11⋅4PF6 with 1⋅2PF6 and 2 is carried out under ultrahigh pressure. For its part, 11⋅4PF6, which emerges in sparse amounts from the use of 1,5-trisnaphtho (57)crown-15 (12) as a double template for the formation of a cyclobis(paraquat-1,4-biphenylene) link from 9⋅2PF6 and 10 at room temperature and pressure, provides the stepping stone to olympiadane and beyond. It is worthy of note that the 114+ tetracations manage to attain considerable supramolecular order when they crystallize in the presence of hexafluorophosphate counterions.
Figure 5.

(a) Template-directed synthesis of the (3)catenane 11⋅4PF6. (b) The x-ray crystal structure and superstructure of 114+. The space-filling representation of the superstructure uses yellow and green to denote the same crown ether component and black to portray the tetracationic cyclophane component. (c) Template-directed synthesis of the oligocatenanes 13⋅12PF6, 14⋅16PF6, and 15⋅20PF6. (d) The x-ray crystal structures of the (5)catenane (olympiadene) 1312+ and the doubly branched (7)catenane 1520+.
Harnessing Concepts
In the future, we can anticipate, (i) the development of new (supra)molecular motors; (ii) the design of methods to induce them to operate coherently on surfaces and within frameworks as machines (29) and devices; (iii) the elaboration of integrated power supplies to drive the machines (30) and devices; (iv) an integration of bottom-up and top-down procedures for the nano- and microfabrication of molecularly driven sensors, actuators, amplifiers, and switches; and (v) an increased understanding and appreciation of the science and engineering that lie behind nanoscale processes. All this and more is in the nature of the mechanical bond as it impacts on chemistry and beyond. In the final analysis, however, it will be the practice of chemical synthesis that relies on molecular recognition and self-assembly processes within a very much more catholic framework than is currently being contemplated by most chemists that will dictate the pace of progress in synthesis. One thing is certain: if chemists do not jump into the driving seats, then other scientists will surely hijack the juggernaut.
Acknowledgments
We are most grateful to David Williams (Imperial College, London), Vincenzo Balzani (University of Bologna, Bologna, Italy), and Jim Heath (University of California, Los Angeles) for aiding and abetting the concept transfer process in their own distinctive and highly significant ways.
References
- 1.Nicolaou K C, Sorensen E J. Classics in Total Synthesis. Weinheim, Germany: VCH; 1995. [Google Scholar]
- 2.Stoddart J F. Nature (London) 1988;334:10–11. [Google Scholar]
- 3.Breslow R, Dong S D. Chem Rev. 1998;98:1997–2011. doi: 10.1021/cr970011j. [DOI] [PubMed] [Google Scholar]
- 4.Lehn J-M. Supramolecular Chemistry. Weinheim, Germany: VCH; 1995. [Google Scholar]
- 5.Gellman S H, editor. Chem Rev. 1997;97:1231–1734. doi: 10.1021/cr970328j. [DOI] [PubMed] [Google Scholar]
- 6.Philp D, Stoddart J F. Angew Chem Int Ed Engl. 1996;35:1154–1196. [Google Scholar]
- 7.Fuhrhop J-H, Köning J. Membranes and Molecular Assemblies: The Synkinetic Approach. Cambridge, U.K.: The Royal Society of Chemistry; 1994. [Google Scholar]
- 8.Sauvage J-P, Dietrich-Buchecker C, editors. Molecular Catenanes, Rotaxanes and Knots. Weinheim, Germany: Wiley–VCH; 1999. [Google Scholar]
- 9.Pease A R, Jeppesen J O, Stoddart J F, Luo Y, Collier C P, Heath J R. Acc Chem Res. 2001;34:433–444. doi: 10.1021/ar000178q. [DOI] [PubMed] [Google Scholar]
- 10.Fyfe M C T, Stoddart J F. Acc Chem Res. 1997;30:393–401. [Google Scholar]
- 11.Greig L M, Philp D. Chem Soc Rev. 2001;30:287–302. [Google Scholar]
- 12. Rowan, S. J., Cantrill, S. J., Cousins, G. R. L., Sanders, J. K. M. & Stoddart, J. F. (2002) Angew. Chem. Int. Ed.41, in press. [DOI] [PubMed]
- 13.Houk K N, Menzer S, Newton S P, Raymo F M, Stoddart J F, Williams D J. J Am Chem Soc. 1999;121:1479–1487. [Google Scholar]
- 14.Asakawa M, Dehaen W, L'abbé G, Menzer S, Nouwen J, Raymo F M, Stoddart J F, Williams D J. J Org Chem. 1996;61:9591–9595. [Google Scholar]
- 15.Gillard R E, Raymo F M, Stoddart J F. Chem Eur J. 1997;3:1933–1940. [Google Scholar]
- 16. Ashton, P. R., Brown, C. L., Chrystal, E. J. T., Goodnow, T. T., Kaifer, A. E., Parry, K. P., Philp, D., Slawin, A. M. Z., Spencer, N., Stoddart, J. F. & Williams, D. J. (1991) J. Chem. Soc., Chem. Commun. 634–639.
- 17.Brown C L, Jonas U, Preece J A, Ringsdorf H, Seitz M, Stoddart J F. Langmuir. 2000;16:1924–1930. [Google Scholar]
- 18.Balzani V, Credi A, Mattersteig G, Matthews O A, Raymo F M, Stoddart J F, Venturi M, White A J P, Williams D J. J Org Chem. 2000;65:1924–1936. doi: 10.1021/jo991781t. [DOI] [PubMed] [Google Scholar]
- 19.Asakawa M, Higuchi M, Mattersteig G, Nakamura T, Pease A R, Raymo F M, Shimizu T, Stoddart J F. Adv Mater. 2000;12:1099–1102. [Google Scholar]
- 20.Balzani V, Credi A, Raymo F M, Stoddart J F. Angew Chem Int Ed. 2000;39:3348–3391. doi: 10.1002/1521-3773(20001002)39:19<3348::aid-anie3348>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 21.Collier C P, Mattersteig G, Wong E W, Luo Y, Beverly K, Sampaio J, Raymo F M, Stoddart J F, Heath J R. Science. 2000;289:1172–1175. doi: 10.1126/science.289.5482.1172. [DOI] [PubMed] [Google Scholar]
- 22.Collier C P, Jeppesen J O, Luo Y, Perkins J, Wong E W, Heath J R, Stoddart J F. J Am Chem Soc. 2001;123:12632–12641. doi: 10.1021/ja0114456. [DOI] [PubMed] [Google Scholar]
- 23. Luo, Y., Collier, C. P., Ho, G., Diehl, M. R., Nielsen, K. A., Mattersteig, G., Jeppesen, J. O., Pease, A. R., Stoddart, J. F. & Heath J.R. (2002) IBM J. Res. Dev., in press.
- 24. Amabilino, D. B., Ashton, P. R., Bravo, J. A., Raymo, F. M., Stoddart, J. F., White, A. J. P. & Williams, D. J. (1999) Eur. J. Org. Chem. 1295–1302.
- 25.Cabezon B, Cao J, Raymo F M, Stoddart J F, White A J P, Williams D J. Chem Eur J. 2000;6:2262–2273. doi: 10.1002/1521-3765(20000616)6:12<2262::aid-chem2262>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 26.Krämer R, Lehn J-M, Marquis-Rigault A. Proc Natl Acad Sci USA. 1993;90:5394–5398. doi: 10.1073/pnas.90.12.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cantrill S J, Youn G Y, Stoddart J F, Williams D J. J Org Chem. 2001;66:6857–6872. doi: 10.1021/jo010405h. [DOI] [PubMed] [Google Scholar]
- 28.Amabilino D B, Ashton P R, Balzani V, Boyd S E, Credi A, Lee J Y, Menzer S, Stoddart J F, Venturi M, Williams D J. J Am Chem Soc. 1998;120:4295–4307. [Google Scholar]
- 29.Chia S, Cao J, Stoddart J F, Zink J I. Angew Chem Int Ed. 2001;40:2447–2451. [PubMed] [Google Scholar]
- 30. Balzani, V., Credi, A. & Venturi, M. (2002) Proc. Natl. Acad. Sci. USA99, in press. [DOI] [PMC free article] [PubMed]