

This perspective has a simple message: Covalent synthesis under thermodynamic control can be a powerful tool for preparing mechanically interlocked structures and specific macrocycles in high yield and at the same time serve as an engine for the creation of dynamic combinatorial libraries (DCLs), which are responsive to their environment. Here we will highlight some examples that illustrate the potential of thermodynamically controlled covalent chemistry and we will point out some exciting directions for the future. A more comprehensive review of the area has been provided elsewhere (1).
Most of the reactions used in organic synthesis are irreversible in practice. The outcome of a given procedure is determined by the kinetics of the desired reaction as well as the kinetics of the undesired competing processes. Hence, yields and product distributions reflect the relative differences between the various initial and transition-state Gibbs energies. Optimization of such kinetically controlled processes involves changing the Gibbs energies of initial states or, more frequently, transition states. Impressive achievements have been made following this approach mainly through the use of catalysts. Nevertheless, improving kinetically controlled reactions has been and still remains a challenge, largely as a result of the elusive nature of the activated complex, which does not lend itself to direct study. The situation is further complicated by the fact that effects on the Gibbs energy of the initial state need to be taken into consideration.
For thermodynamically controlled reactions the situation is much less complex. Product distributions now depend on only the relative Gibbs energies (stabilities) of the products themselves. Of course, chemists have long made use of the fact that product distributions of some reactions are under kinetic control under one set of conditions, whereas the thermodynamic product prevails under other conditions. But why not go one step further and actively manipulate the Gibbs energies of the products so as to shift the equilibria in any desired direction? With the increased understanding of supramolecular interactions, molecular recognition has become a versatile tool that enables control over equilibrium system. Selective recognition of one of the components in an equilibrium system will result in the creation of an additional well in the Gibbs energy landscape (Fig. 1). If this well is deep enough, the recognized compound will dominate the mixture at the expense of undesired products. In practice this outcome requires addition of a species (a template) that selectively recognizes, binds, and stabilizes the desired compound. In a sense, this template plays a role similar to that of a catalyst in kinetically controlled synthesis.
Figure 1.
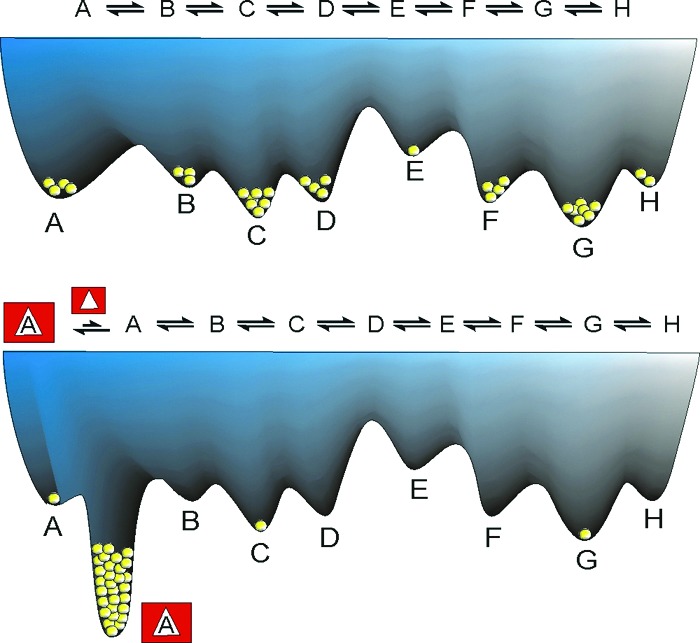
Free energy landscape of an equilibrium mixture showing the effect of adding a template that strongly and selectively binds to one of the equilibrating species.
Despite the fact that equilibrium systems seem much more tractable than kinetically controlled transformations, relatively little use has been made of them so far in synthesis. Although there are good reasons not to strive for equilibrium distributions in many instances, there are other cases where thermodynamically controlled synthesis is the way forward. In particular, thermodynamic control is highly advantageous in the synthesis of macrocycles and mechanically interlocked compounds. Also, combinatorial chemistry is more versatile when operated under thermodynamic control. As we will show below, dynamic combinatorial chemistry enables high-yield synthesis of, for instance, a host molecule in one step without prior knowledge of its exact structure.
Reversible Covalent Chemistry
A major reason why the use of reversible covalent chemistry is not more widespread is the rather limited number of reversible reactions available. Furthermore, many reactions are reversible only under conditions that are not compatible with the subtle noncovalent interactions involved in templating. Finally, after the equilibrium distribution has been reached it must be possible to turn off the reversible reaction to isolate and handle the desired product. In practice this is most often accomplished by removal of the catalyst responsible for reversibility or by altering the pH of the solution.
The reversible reactions that have thus far been used successfully in templated thermodynamically controlled synthesis are summarized in Fig. 2. They can be divided into two categories, depending on the symmetry of the reversible linkage.
Figure 2.
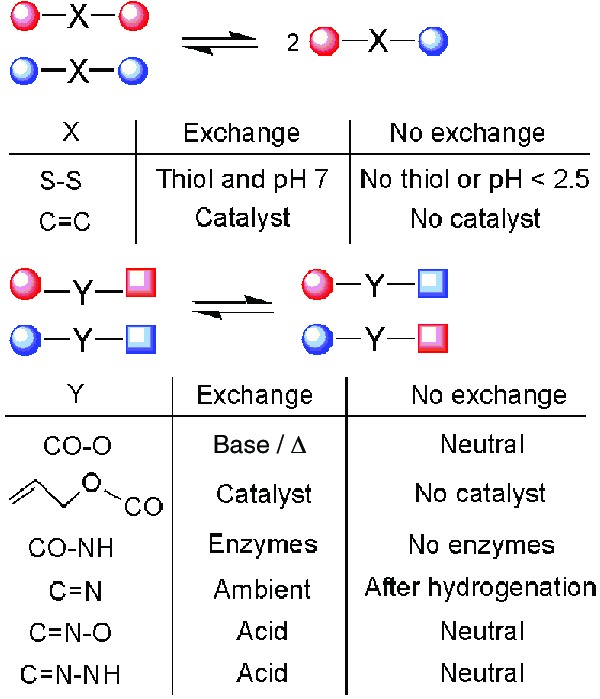
Reversible reactions that have been used successfully in thermodynamically controlled synthesis.
Disulfide exchange (2, 3) and alkene metathesis (4) are symmetrical. Disulfide exchange is mediated by a catalytic amount of thiolate anion attacking the existing disulfide bonds. Exchange proceeds readily under neutral to mildly basic conditions, whereas acidifying the medium causes protonation of the thiolate anion and turns off exchange (2, 3).
Alkene metathesis (4) is controlled through addition or removal of a catalyst. The usefulness of this reaction has been increasing rapidly, as new, more efficient, and general catalysts become available.
Reversible reactions that feature unsymmetrical linkages include amide exchange (5), transesterification (6–8), as well as exchange of imines (9, 10) and derivatives thereof. Only one example of amide exchange has been reported (5), making use of enzymes to cleave and reform the amide bonds. Transesterification has received more attention. Unfortunately, this reaction generally requires harsh conditions that are likely to interfere with the weak interactions underlying recognition by a template (6, 7). In the special case of allyl esters, Pd-catalyzed transesterification proceeds under mild conditions (8).
Reactions based on exchange around the C = N bond are more versatile. The parent imines can be used, but they suffer from an inherent instability. Reduction of the imines to amines is generally required to stop the equilibration process and allow for isolation of the product (9, 10). These final amines are geometrically and electronically different from the imines that are recognized by the template, potentially leading to loss of template recognition. Oximes (11) and hydrazones (12) do not suffer from this disadvantage. In these cases, exchange requires acid and is switched off after neutralization to yield a robust product.
Manipulating Equilibria
Reactions that are most likely to benefit from being controlled by thermodynamics are processes such as macrocyclizations where several products are accessible that differ in size rather than connecting chemistry. Ring-closure reactions performed under kinetic control are notorious for giving relatively large amounts of unwanted oligomeric side products even under high-dilution conditions. Under thermodynamic control, the product distribution of the same reaction is governed only by the stabilities of the different macrocycles. These stabilities are partly determined by interactions within the macrocycles (e.g., ring strain) and partly by the concentration at which the reaction is carried out. Disregarding ring strain, relatively dilute conditions (less than 10 mM) will favor smaller ring sizes (13). Higher oligomers might still be formed in the initial stages of the reaction but they will be consumed as equilibrium is approached. As concentrations are raised, the product distribution will gradually shift toward higher oligomers, culminating in polymer formation for very concentrated solutions (for a review of thermodynamically controlled polymerizations, see ref. 1).
Equilibrium systems are adaptive; apart from a concentration dependency they will also respond to exterior influences. The more fundamental of those influences, such as temperature, pressure, and effects of solvents and extents of ionization of the molecules, has received relatively little attention in the area of small molecule synthetic chemistry. Yet these parameters might well prove to be useful handles in optimizing the proportion of desired product in equilibrium mixtures.
More specific manipulation of an equilibrium composition is possible by exposing the system to templates. Two types of templating approaches can be distinguished. In the synthesis of mechanically interlocked structures the template will usually be part of the final product.§ Alternatively the template can be used simply as a mould, in which case it will be removed once the equilibrium is frozen, giving access to the free receptor molecule. Even though these approaches lead to topologically different products, they share two necessary features: (i) a template effect that stabilizes the desired product via supramolecular interactions and (ii) one or more reversible connections allowing for proofreading and editing of undesired products (Fig. 3).
Figure 3.

Overview of different structures that benefit from thermodynamic control.
Thermodynamically Controlled Synthesis of Mechanically Interlocked Structures.
The interest in mechanically interlocked structures like catenanes (interlocked rings) and rotaxanes (one or more rings mechanically locked onto a linear thread by bulky stoppers) has been growing rapidly because of their potential for the development of nanoscale devices or molecular machines (14–16).
The current strategies for the construction of these mechanically interlocked structures make use of noncovalent interactions that bring together the components of the superstructure in a specific orientation. The final step in the synthesis is the formation of one or more covalent bonds that fix the product in the interlocked orientation. In most of the syntheses reported to date, this final reaction is carried out under kinetic conditions. As a result, if the reaction does not proceed via a threaded precursor, undesired noninterlocked products are produced that cannot be rescued, reducing the yield of the target molecule. As a thermodynamically controlled approach includes bond breaking and reformation, it permits recycling of these side products to produce the correct structure.
We explored the use of alkene metathesis under thermodynamic conditions for the preparation of [2]catenane 4 (Fig. 4) (17), exploiting π-donor/π-acceptor interactions between π-deficient aromatic diimides and π-rich aromatic diethers to stabilize the desired product. The ring-closing metathesis of dialkene 1 leads to the formation of macrocycle 2, accompanied presumably by a variety of linear and cyclic oligomeric species (Fig. 4). When half a molar equivalent of crown 3 and additional catalyst were added, [2]catenane 4 was produced as a mixture of cis/trans isomers in 50% isolated yield.
Figure 4.

Thermodynamically controlled preparation of a [2]catenane by using π-donor/π-acceptor interactions and alkene metathesis.
A more subtle example of the thermodynamic approach has been reported by Leigh and coworkers (18) who prepared [2]catenane 6. The amide macrocycle 5 is self-complementary and can dimerize by forming four hydrogen bonds between the amide hydrogen atoms and the ester carbonyls (Fig. 5). In addition, it contains an internal alkene available for metathesis under reversible thermodynamic control. Under appropriate ring-opening and ring-closing alkene metathesis conditions, macrocycle 5 self-assembles to give [2]catenane 6 in >95% yield. The amount of catenane formed depends on the initial concentration of the macrocycle used. At low concentration (0.0002 M), only the noninterlocked macrocycle is present in the reaction mixture, whereas above 0.2 M higher cyclic oligomers become increasingly abundant.
Figure 5.
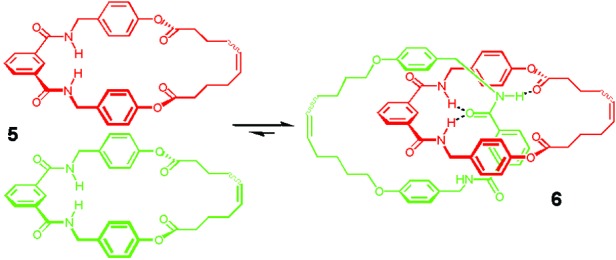
Thermodynamically controlled preparation of a [2]catenane by using hydrogen bonds and alkene metathesis.
That the reactions are under thermodynamic control was demonstrated by taking the product mixture obtained at one concentration and reexposing it to the catalyst at a different concentration. In all cases, the product distribution adjusts to that obtained if only the macrocycle had been exposed to the catalyst at that concentration.
The thermodynamic preference of this system can be drastically affected by switching off the hydrogen-bonding ability of the macrocycle by trifluororoacetylation of the amide functionality. In the absence of the catalyst, the trifluoroacetylated [2]catenane is stable while in its presence the catenane unthreads to form the noninterlocked macrocycles.
The saturated analog of [2]catenane 6 had been previously prepared by using kinetically controlled condensation of acid chlorides and masked phenols, but as undesired products could not be recycled the highest yield obtained was 18% (19).
Stoddart and colleagues (10) reported the template-directed preparation of [2]rotaxane 12 under thermodynamic conditions. In this case, the interaction between a protonated dialkylammonium center and a crown ether was used as recognition motif for the stabilization of the rotaxane, whereas imine formation/breaking was used as “correcting” reversible reaction (Fig. 6).
Figure 6.
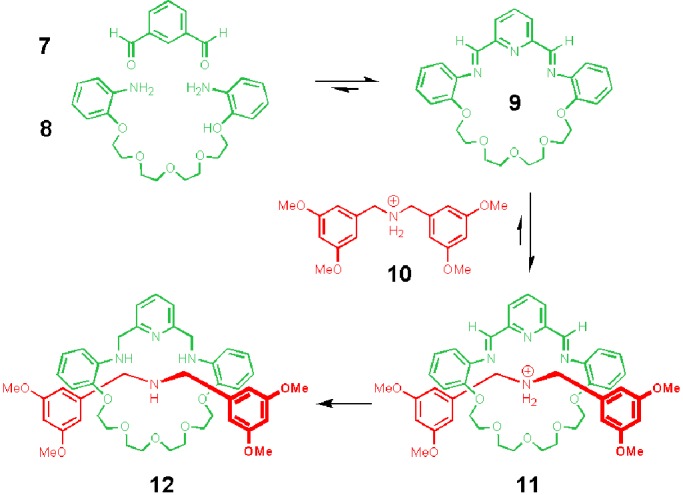
Thermodynamically controlled preparation of a [2]rotaxane by using ammonium/crown ether interaction and transimination.
On mixing of dialdehyde 7 and diamine 8 in chloroform, an interconverting mixture of macrocycle 9 and higher cyclic and linear oligomers is obtained. Addition of the thread (10) that binds 9 with a binding constant of 300 M−1, shifts the equilibrium of the interconverting imines toward the formation of the [2]rotaxane 11. Because imine bonds are kinetically labile, a fixation step by reduction is necessary to obtain a stable interlocked product. The attractive feature in Stoddart's system is that reduction of the imine bonds is faster in the desired rotaxane than in the nonthreaded macrocycles (like 9); this results in a further constructive shift in the equilibrium between the threaded and unthreaded macrocycles leading to the formation of the stable [2]rotaxane 12 in 70% isolated yield.
Templating Dynamic Combinatorial Libraries (20–22).
Allowing thermodynamics to control the composition of combinatorial libraries can give truly remarkable results and provides the most compelling demonstration of the potential of reversible covalent chemistry. We have developed dynamic combinatorial libraries (6) with a view to generating macrocyclic receptors. The libraries are generated by linking bifunctional building blocks together through reversible bonds. A guest molecule is then introduced and allowed to select its ideal host from among the available macrocycles in the mixture. Binding of the guest to its preferred host leads to stabilization and concomitant shift of the equilibrium toward this species: the best binder will be amplified. Thus, introduction of the guest will convert an initially diverse library into the desired molecule, ideally in high purity, thereby combining identification and isolation of the target receptor into one step.
Our initial attempts to generate dynamic combinatorial libraries focused on transesterification (6, 7, 23) but we have since moved on to the more practical hydrazone and disulfide exchange reactions. Generating diverse dynamic combinatorial libraries based on these chemistries is straightforward. Hydrazone libraries form spontaneously after adding acid to a solution of building blocks such as 13 containing a hydrazide and an aldehyde (protected as an acetal) functionality (12). Dynamic libraries of macrocyclic disulfides form readily simply after dissolving dithiol building blocks in water at pH 7 and stirring the solution in an open flask. Oxygen from the air is sufficient to convert the thiols to disulfides. While oxidation is taking place the remaining thiol mediates the exchange process responsible for equilibration (2).
Exposure of these libraries to templates has resulted in dramatic shifts in the distribution of the library in a number of cases (6, 24–27). In one instance (26), cyclization of building block 13 derived from l-proline-l-phenylalanine in chloroform solution initially produced a mixture of at least 10 interconverting macrocycles. When the dynamic mixture was exposed to LiI it transformed to essentially pure trimer 14 (Fig. 7). Binding studies demonstrated that the selected trimer forms a 1:1 complex with Li+ ion with a binding constant of 4 × 104 M−1. This example clearly illustrates the power of dynamic combinatorial chemistry: a new receptor molecule was identified while at the same time the thermodynamically controlled chemistry immediately provided an efficient synthetic route toward an otherwise difficult to access molecule. Furthermore, it is unlikely that the selected receptor would have been the outcome of any rational design. In fact, even knowing that 1 binds strongly to Li+ we are still rather puzzled as to how it actually manages to do so.
Figure 7.
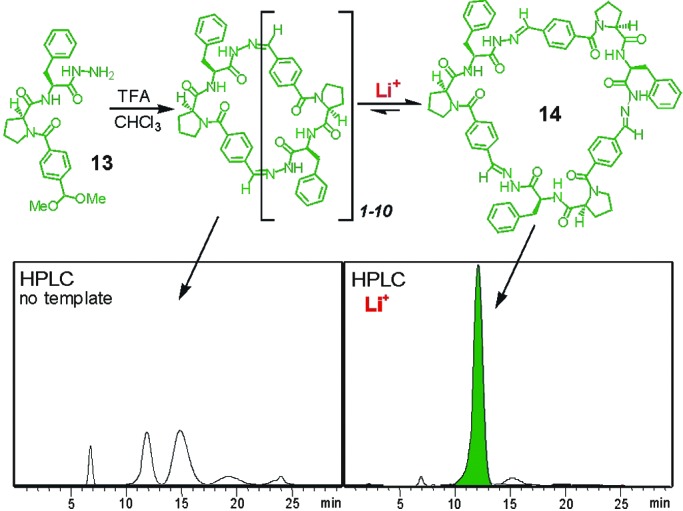
A dynamic combinatorial library of hydrazone-based pseudopeptide macrocycles. Shown are the HPLC traces of the library before (Left) and after (Right) addition of Li+ template.
Complementary to our work, others have studied dynamic combinatorial libraries by using hosts to template the formation of guest molecules (9, 28). When using proteins or nucleic acids as the host molecules, this approach has considerable potential for drug development (for a recent review, see ref. 22).
Outlook
The attractions of the use of reversible covalent chemistry under thermodynamic conditions have been illustrated by the preparation of several supramolecular architectures obtained through rational design or with a combinatorial approach. In these examples a bond breakage process is part of the synthetic strategy. Although bond breakage is an intrinsic part of thermodynamically controlled chemistry, it would be challenging to use this strategy in a kinetically controlled system.
Although the thermodynamic approach has in a short time earned its place among the more useful tools available to supramolecular chemists, we believe that it is not yet used to its full potential. For instance, thermodynamically controlled chemistry seems the method of choice for the synthesis of capsule-like molecules from relatively easily prepared trifunctional building blocks. First steps on this road have recently been made (29, 30). Furthermore, dynamic combinatorial chemistry seems a highly promising method for the identification of new supramolecular catalysts. Analogous to the catalytic antibody approach, using a transition state analogue as a template it should be possible to identify catalytically active receptors. These targets are now being actively pursued.
Acknowledgments
We are grateful for support from the Marie Curie Individual Fellowships Program (HPMF-CT-1999-00069) and the Royal Society University Research Fellowships Program (to S.O.), the Fundación Antorchas and the Consejo Nacional de Investigaciones Científicas y Tecnicas (Argentina) (to R.L.E.F.), and the Engineering and Physical Sciences Research Council (to J.K.M.S.).
Footnotes
Synthesis of mechanically interlocked structures can also be performed by using removable templates like metal ions. See ref. 14.
References
- 1. Rowan, S. J., Cantrill, S. J., Cousins, G. R. L., Sanders, J. K. M. & Stoddart, J. F. (2002) Angew. Chem. Int. Ed. Engl.41, in press. [DOI] [PubMed]
- 2.Otto S, Furlan R L E, Sanders J K M. J Am Chem Soc. 2000;122:12063–12064. [Google Scholar]
- 3.Ramström O, Lehn J-M. ChemBioChem. 2000;1:41–48. doi: 10.1002/1439-7633(20000703)1:1<41::AID-CBIC41>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 4.Grubbs R H, Chang S. Tetrahedron. 1998;54:4413–4450. [Google Scholar]
- 5.Swann P G, Casanova R A, Desai A, Frauenhoff M M, Urbancic M, Slomczynska U, Hopfinger A J, Le Breton G C, Venton D L. Biopolymers. 1996;40:617–625. doi: 10.1002/(sici)1097-0282(1996)40:6<617::aid-bip3>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 6. Brady, P. A. & Sanders, J. K. M. (1997) J. Chem. Soc. Perkin Trans. 1, 3237–3253.
- 7.Rowan S J, Sanders J K M. J Org Chem. 1998;63:1536–1546. [Google Scholar]
- 8. Kaiser, G. & Sanders, J. K. M. (2000) Chem. Commun., 1763–1764.
- 9.Huc I, Lehn J-M. Proc Natl Acad Sci USA. 1997;94:2106–2110. doi: 10.1073/pnas.94.6.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glink P T, Oliva A I, Stoddart J F, White A J P, Williams D J. Angew Chem Int Ed Engl. 2001;40:1870–1874. [PubMed] [Google Scholar]
- 11.Polyakov V A, Nelen M I, Nazarpack-Kandlousy N, Ryabov A D, Eliseev A V. J Phys Org Chem. 1999;12:357–363. [Google Scholar]
- 12. Cousins, G. R. L., Poulsen, S.-A. & Sanders, J. K. M. (1999) Chem. Commun., 1575–1576.
- 13.Ercolani G, Mandolini L, Mencarelli P, Roelens S. J Am Chem Soc. 1993;115:3901–3908. [Google Scholar]
- 14.Sauvage J-P. Acc Chem Res. 1990;23:312–327. [Google Scholar]
- 15.Amabilino D B, Stoddart J F. Chem Rev. 1995;95:2725–2828. [Google Scholar]
- 16.Balzani V, Credi A, Raymo F M, Stoddart J F. Angew Chem Int Ed Engl. 2000;39:3348–3391. doi: 10.1002/1521-3773(20001002)39:19<3348::aid-anie3348>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 17.Hamilton D G, Feeder N, Teat S J, Sanders J K M. New J Chem. 1998;22:1019–1021. [Google Scholar]
- 18.Kidd T J, Leigh D A, Wilson A J. J Am Chem Soc. 1999;121:1599–1600. [Google Scholar]
- 19.Leigh D A, Moody K, Smart J P, Watson K J, Slawin A M Z. Angew Chem Int Ed Engl. 1996;35:306–310. [Google Scholar]
- 20.Karan C, Miller B L. Drug Discov Today. 2000;5:67–75. doi: 10.1016/s1359-6446(99)01431-2. [DOI] [PubMed] [Google Scholar]
- 21.Lehn J-M, Eliseev A V. Science. 2001;291:117–125. doi: 10.1126/science.1060066. [DOI] [PubMed] [Google Scholar]
- 22.Otto S, Furlan R L E, Sanders J K M. Drug Discov Today. 2002;7:24–32. doi: 10.1016/s1359-6446(01)02086-4. [DOI] [PubMed] [Google Scholar]
- 23. Brady, P. A., Bonar-Law, R. P., Rowan, S. J., Suckling, C. J. & Sanders, J. K. M. (1996) Chem. Commun., 319–320.
- 24. Furlan, R. L. E., Cousins, G. R. L. & Sanders, J. K. M. (2000) Chem. Commun.1761–1762.
- 25.Cousins G R L, Furlan R L E, Ng Y-F, Redman J E, Sanders J K M. Angew Chem Int Ed Engl. 2001;40:423–428. [PubMed] [Google Scholar]
- 26.Furlan R L E, Ng Y-F, Otto S, Sanders J K M. J Am Chem Soc. 2001;123:8876–8877. doi: 10.1021/ja0160703. [DOI] [PubMed] [Google Scholar]
- 27.Furlan R L E, Ng Y-F, Cousins G R L, Redman J E, Sanders J K M. Tetrahedron. 2002;58:771–778. [Google Scholar]
- 28.Karan C, Miller B L. J Am Chem Soc. 2001;123:7455–7456. doi: 10.1021/ja010325v. [DOI] [PubMed] [Google Scholar]
- 29.Tam-Chang S-W, Stehouwer J S, Hao J. J Org Chem. 1999;64:334–335. [Google Scholar]
- 30.Ro S, Rowan S J, Pease A R, Cram D J, Stoddart J F. Org Lett. 2000;2:2411–2414. doi: 10.1021/ol005962p. [DOI] [PubMed] [Google Scholar]