

Abstract
Supramolecular systems—their successes, problems, and potential—are discussed with allusion to specific examples.
Supramolecular chemistry, broadly speaking, entails the study of intermolecular bonding. The discipline blossomed two decades ago with host–guest systems in which (i) metal ions were complexed to crown ethers and (ii) small molecules (such as urea) were bound to larger hosts by means of multiple hydrogen bonds. One of my favorite examples is cited below.
Physicians need to continuously monitor the blood levels of O2, glucose, etc., of their bedridden patients. In the recent past, no good method for assaying blood K+ (which exists in the presence of excess Na+) was available. This problem in clinical analysis was solved by J. E. Trend et al. (of the 3M Co.; ref. 1) with the compound drawn below. When a K+ ion binds to the cryptand portion of the molecule, the attached chromophore emits a fluorescence, the intensity of which is proportional to the K+ concentration.
Because even a Na+ concentration of 145 mM hardly affects the fluorescence, the selectivity for K+ is satisfactory. Anchoring the molecule to a polymer allowed the construction of a flow-through device that continuously and quantitatively monitors, by means of fluorescence, the K+ in the blood. This construction is an excellent example of supramolecular chemistry reaching the marketplace to the benefit of the man in the street.
Early on in the history of supramolecular chemistry, people realized that an enzyme and its substrate were a type of host–guest system. Thus, with the aid of the post-World War II advances in synthetic and physical organic chemistry, attempts were made to simulate enzymes by constructing catalytic hosts. Success in this “biomimetic” branch of supramolecular chemistry has, unfortunately, been modest at best. We lack, for example, a host molecule that selectively binds two aldehydes and subsequently catalyzes their aldol condensation with an enzyme-like efficiency (i.e., a l08 to 1010 acceleration). It is instructive to inquire into the reasons for our difficulties because they are relevant to the entire field of supramolecular science.
Enzyme catalysis depends, in my opinion (2), on multiple catalytic groups being held rigidly at van der Waals distances from the labile substrate atoms. Accept (for the moment at least) the premise that motional freedom among the reactive entities is deadly to catalysis. In this light, the difficulties with designing a catalytic host for an aldol condensation are seen to be considerable. The host cavity must fix the position of two aldehydes in such a manner that the α-carbon of one aldehyde lies immobilized within contact distance of the other's carbonyl carbon. Catalytic groups attached to the host must be held, with little probability of “escape,” at contact distances from the enolizable proton, carbonyl oxygens, etc. Yet for reasons given in the next paragraph, we are not yet sufficiently sophisticated to synthesize such a “wonder host.”
We do not at present possess a sufficient handle on intermolecular forces, the heart and soul of supramolecular chemistry, to predict reliably the structure of many host–guest complexes. Noncovalent bonds are, for one thing, not as stable and directional as covalent bonds. Hydrogen bonds display only weakly preferred orientations and, in addition, hydrogen-bonding sites are often swamped by water should this be the solvent of interest (as it often is). It is hardly a surprise that most host–guest studies have been carried out, reluctantly perhaps, in aprotic solvents such as chloroform. Hydrophobic forces, although operative in water, are even more uncontrollable because they lack simple rules for directionality. It is very difficult to predict, for example, the orientation of a water-insoluble guest within the hydrophobic cavity of a water-soluble macrocyclic host. In summary, the ability to predict supramolecular geometries, similar to what is done routinely with covalent structures, remains one of the great challenges in the field. In the absence of this skill, success with catalytic hosts must often rely on intuition and good fortune.
There is a second problem confronting the chemistry of host–guest systems that I should mention here as well. It might be quite easy to design on paper a host bearing a “wish list” of optimally oriented functionalities. One can imagine, for example, an “aldolase” host molecule that is three-dimensionally speckled with a half dozen exquisitely arranged catalytic and binding groups. The problem, of course, is to synthesize such a molecule. Is it worthwhile to spend many person-years preparing an intricate host molecule while wondering whether, in the end, it will disappoint owing to uncertainties or disorder in the geometry of the host–guest complex? I refer to this problem because it is a general one. As supramolecular chemistry addresses ever larger and more complicated assemblies, so will the necessary synthetic effort expand. Time and money will become increasingly important considerations, especially for systems in which a commercial potential is envisioned. The task is facilitated, of course, by the ability of chemists to roam far afield from biological mechanisms that have had billions of years to evolve and will likely never be truly emulated.
There is a strategy for sidestepping our difficulty in predicting host–guest architectures. One can construct a particular host and then combinatorially screen a population of potential guests or substrates (a “catalyst in search of a substrate”). Alternatively, one can combinatorially screen a library of easily accessible hosts for a given guest or substrate. Catalytic antibody research employs the latter approach (3). I realize that there are those who regard such combinatorial searches as “acerebral,” to cite a term I once heard used with regard to combinatorial catalysis (4), but such an attitude shows little understanding or sympathy for the severe problems with complexity often facing the host–guest chemist.
Over time, the definition of supramolecular chemistry expanded to encompass any organized entity in which two or more chemical species are held together by intermolecular forces. Supramolecular chemistry laid claim to films, gels, liquid crystals, nanostructures, polymers—all systems that self-assemble. The field, being at the triple meeting point of chemistry, biology, and physics, happily embodied all of these as well. Supramolecularity became a sort of hodgepodge of general science excepting that at the nuclear, single-molecule, and celestial levels. For me to say that I am doing supramolecular chemistry would now seem to have no more information-content than, for example, the statement that I am a biologist. The latter may inform the listener that I am not engaged in the history of the Reformation, or the construction of a bridge, but diverse interests (the cause of cancer, the search for new species of earthworms in Australia, the physiology of the octopus brain, etc.) remain as viable possibilities. The point here is that supramolecular chemistry, in taking under its wing all organized molecular systems, has become fuzzy and difficult to define. The association of a peptide, the density of ice, the viscosity of an oil, the conductance of an alloy, the reflection from a film, the hardness of a ceramic—all these fall under the province of supramolecular chemistry as it is presently being defined in the literature. Because the noncovalent bond is ubiquitous, supramolecule enthusiasts can stake a claim to a large fraction of Nature.
The term “self-assembly” has been applied to multimolecular systems to differentiate them from simpler host–guest complexes. It is a historical fact that colloid chemistry had been involved with self-assembly for decades before the coining of the word “supramolecular.” Yet the word “colloid” seldom if ever appears even in the most prominent reviews of supramolecularity and self-assembly (5). Poor colloid chemistry! For decades, colloid chemistry was a stepchild of physical chemistry as the attention and glory focused on quantum mechanics. When (finally!) self-assembly was accepted into the forefront of science, supramolecular chemistry immediately embodied it. The work of Ostwald, Svedberg, Langmuir, Debye, and other colloid stalwarts, plus that of their scientific descendants, was largely sidestepped. Sooner or later, I predict, supramolecular chemistry will exploit more fully reflectometry, small-angle neutron scattering, small-angle x-ray scattering, ellipsometry, tensiometry, pulse-gradient spin-echo NMR, light scattering, phase diagrams, and the many other tools of the colloid chemist. Supramolecular chemistry and colloid chemistry will, at that point, join hands and the rather artificial distinction will disappear.
There is, admittedly, a perceived difference between colloids and supramolecular self-assemblies, and this may be, in part, a source of the dichotomy. Colloids are regarded by some as mere collections of molecules, often of commercial origin, that impose an organizational mode upon the chemist. Supramolecular self-assemblies, on the other hand, are ostensibly composed of molecules that have been “designed” to organize in a certain manner. This distinction is decidedly unfair. Colloid chemists often design, synthesize, and examine new self-assembling molecules. To illustrate the point, I will cite below several works with new compounds, including two of our own, whose properties can be classified as either supramolecular or colloidal. The purpose here is not to quibble over semantics but to address a needless distinction. In an ideal world, chemists would not be forced to select a journal or symposium, and hence a readership or listenership, that artificially excludes a group of scientists with virtually identical interests. In any case, and more importantly, the examples given below were selected to demonstrate the potential of supramolecular or colloidal assemblies—whatever term one prefers.
Before getting into the specifics, let me say something about the “potential” to which I have just alluded. In a fit of exuberance, I recently wrote that it is only a matter of time before colloid chemistry intellectually dominates biology (6). Here is the reasoning: molecular biology focuses on the behavior of DNA. We are now close to elaborating the entire human genome, which will facilitate the primary sequencing of many new human proteins. This elaboration is a marvelous accomplishment with far-reaching consequences. But collections of protein structures, important though they may be, tell us no more about the nature of life than a collection of bricks tells us about the architecture of a building. If anyone has any doubt over this assertion, imagine that all human proteins and other cell components are mixed in a flask. A living system will obviously not be created. The point is that identification of the building blocks is only the very beginning. We must now learn how the building blocks are organized through self-assembly. And it is here where colloid chemistry or supramolecular chemistry will begin to dominate biology.
And now on to some examples. In the past few years we, and many others worldwide, have synthesized a family of amphiphilic compounds designated as “gemini” surfactants (7). Their potential use in DNA transfection, synthesis of macroporous materials, detergency, etc., has already been reviewed (8). Geminis have (in sequence) a long hydrocarbon chain, an ionic group, a spacer, an ionic group, and a long hydrocarbon chain. In the surfactant of Scheme S2, the two ionic groups are of opposite charge, creating a zwitterionic gemini. It turns out that these compounds are relatively easy to synthesize and, as such, we have assembled a library of about 100 of them (with the chains being, among other variations, short–short, long–long, short–long, and long–short). The first question we addressed is how the morphology of self-assembly varies with the gemini structure. In answering this question, we constructed a “structural phase diagram” (Fig. 1). The y axis represents the number of carbons on the phosphate-bound chain, whereas the x axis represents the number of carbons on the ammonium-bound chain. Each of the 42 points on the phase diagram expresses the phase-state of one particular gemini.
Scheme 2.
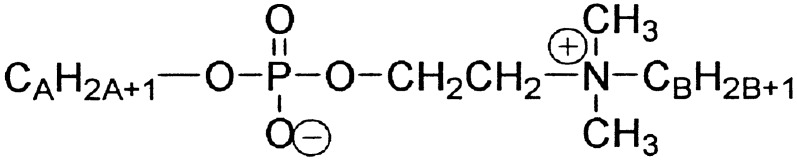
Figure 1.

A structural phase diagram for 42 geminis.
Because the structural phase diagram can be fully described only by means of a lengthy story, I will confine the discussion to a single region: the coacervate phase symbolized by the crosses. When compounds in this region are dissolved in water, oily droplets soon appear. These droplets are about 85% water, and yet they are immiscible with water. How can this be? With the aid of cryo–high-resolution scanning electron microscopy, one can detect a morphology resembling a sponge. Apparently, the geminis self-assemble into a porous network composed of highly interconnected planar bilayers. Although the network entrains a huge amount of water, the bilayer walls prevent this water from comixing with the bulk water in which the coacervate droplets are suspended.
It might be claimed (correctly) that our colloidal phase was discovered by screening, not design. Yet as a result of this screening, we can now confidently predict that certain unknown compounds (A = 9/B = 10, for example) will self-assemble into a sponge morphology. More importantly, one wants to know why such simple compounds self-assemble into coacervates. Some understanding along these lines, obtained by molecular modeling, is available in the primary literature (7). There is, however, a more general lesson for supramolecular chemistry that should be mentioned here. Self-assembly by design is both a tribute to the human imagination and a limitation. The latter is illustrated by the fact that current principles of supramolecular chemistry could not have easily predicted our sponge-like self-assembly. Thus, progress in the field of self-assembly requires, in addition to clever a priori design, the investigation of thoughtfully selected synthetic compounds whose mode of self-assembly may, at first, be unsuspected.
Block copolymers dissolved in block-selective solvents self-assemble into a variety of morphologies including spheres, cylinders, and vesicles. Winnik, Manners and their coworkers (9) synthesized a polyferrocene block attached to a polysiloxane block in a monomer ratio of 1:13, respectively. The authors pointed out that organometallic block copolymers offer the possibility of self-assembled materials with redox-active, semiconducting, optical, and “nanowire” capabilities. When the block copolymer was placed in hexane (a good solvent for the polysiloxane block but a precipitant for the polyferrocene block), a remarkable self-assembly into a network of hollow tubules was achieved. According to transmission electron micrographs, the tubules formed at l mg/ml are 29 nm wide (11 nm of which is interior cavity) and 360–930 nm long. Assembly into single-walled nanotubes is believed to arise from fusion of smaller subunits such as vesicular aggregates and shorter tubules. If the tubules are formed in the presence of an inert guest (n-butylferrocene), the guest becomes encapsulated within the cavity. A proposed schematic of a tubule cross-section (in which the polyferrocene block is represented by dark rods) is given in Scheme S5.
Scheme 5.

I now turn to a system composed of eight different components. Each component has its own location within the system as well as its own particular role in doing something useful, namely the destruction of environmentally dangerous compounds. Molecular organization is key here. In its absence, the eight-component system would be merely an intractable and useless mess. The system is called a microemulsion—a self-organizing assembly composed of a surfactant, cosurfactant (usually a short-chain alcohol), water, and hydrocarbon (“oil”) (10). “Oil-in-water” microemulsions consist of an optically clear and thermodynamically stable suspension of hydrocarbon droplets, 5–50 nm in diameter, in a continuous water phase. It is an amazing thing to watch an aqueous surfactant/cosurfactant solution rapidly dissolve 10–20% of hexane. A schematic diagram of a microemulsion is shown in Scheme S6. The hexane and water form a single bulk phase because the surfactant and cosurfactant molecules gather at the droplet/water interface and reduce the oil/water surface tension to near zero.
Scheme 6.
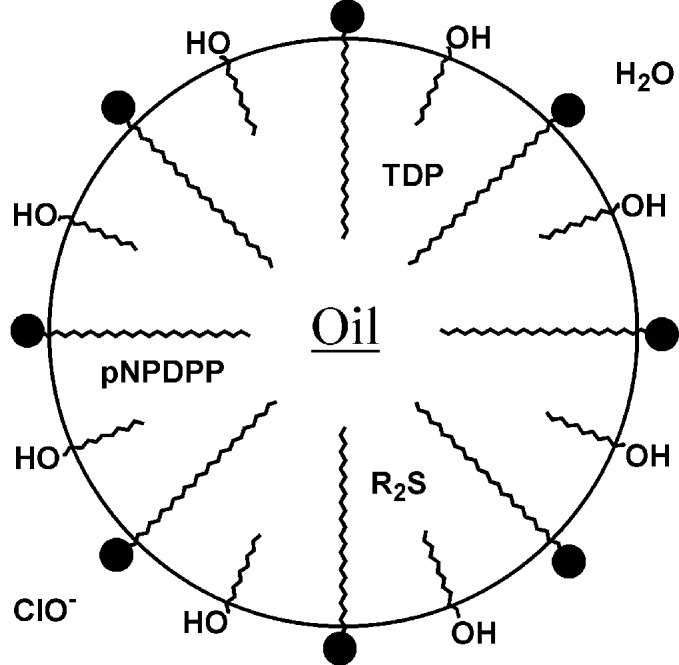
The microemulsion schematic shows that the oil droplet has dissolved three water-insoluble chemical warfare simulants: TDP and pNPDPP (nerve agents) and R2S (mustard), the exact structures of which are not important here. We have shown that when one adds commercial bleach (hypochlorite) to the microemulsion, all three chemical warfare simulants are destroyed by combined oxidative/hydrolytic pathways (11). The method is rapid, cheap, and mild. No special equipment (stirrer, heater, photolyzer, etc.) is required. Self-organization has converted the complicated eight-component mixture (water, hydrocarbon, surfactant, cosurfactant, three simulants, and hypochlorite) into a useful system. Living cells, of course, discovered the value of self-organization long ago.
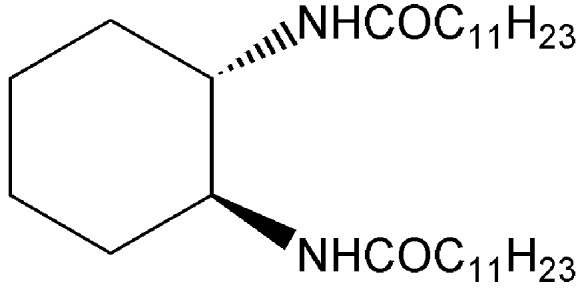
The next example illustrates how even a simple organic building block can assemble into a supramolecule of nanoscale dimensions. Hanabusa et al. (12) found that the dilauroyl amide of trans-(1R,2R)-l,2-diaminocyclohexane is a “universal” gelling agents for a host of organic solvents including hexane, methanol, acetone, benzene, and dimethyl sulfoxide. Cooling a heated solution of 6 g of diamide in 1 liter of hexane, for instance, creates a gel that is stable for months. The cis isomer of the diamide is not a gelator, whereas a racemic mixture of the 1R,2R compound with its 1S,2S stereoisomer exits only as an unstable gel. Gels are among the most useful supramolecular (colloidal) systems, with wide applications in photography, drug delivery, cosmetics, sensors, and food processing, to name a few.
Most organic gels are formed from fibrous aggregates that become entangled into three-dimensional networks. Solvents, entrained within the interstices of the network, suffer impaired flow. Strong circular dichroism peaks of the diamide gels suggest that the fibers are composed of helical stacks. Transmission electron microscopy images confirm the presence of right-handed helical fibers (40–70 nm in width). On the basis of molecular modeling, the authors suggest that the two equatorial amides in the primary fiber are antiparallel to each other, and perpendicular to the cyclohexane ring (Fig. 2). Intermolecular hydrogen-bonding create tape-like molecular aggregates which, in turn, interlock into chiral helices by means of van der Waals interactions among the long chains.
Figure 2.
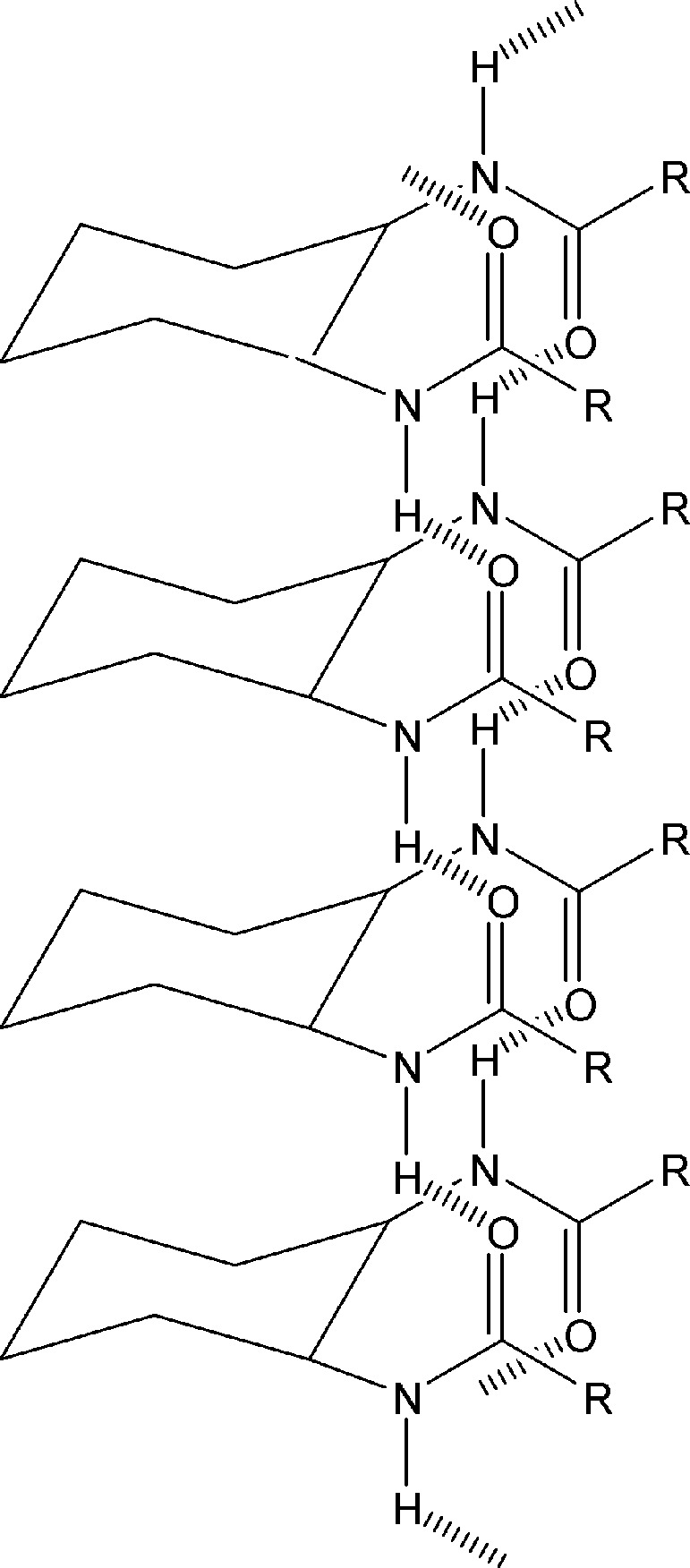
Proposed structure for fiber of trans-(1R,2R)-l,2-diaminocyclohexane dilauroyl amide.
Self-assemblies are not merely beautiful structures (although this is certainly a source of their appeal), they allow alternative solutions to problems that have hitherto been addressed only at the single-molecule level. Consider the following objective: one wants to selectively target cancer cells that, owing to a malfunctioning biochemistry, exude into their immediate environment an excessive amount of a given enzyme (as occurs, for example, with bone cancer). How might this goal be achieved? In the single-molecule mode, a prodrug is a likely possibility. The prodrug must, accordingly, split into its parent drug on exposure to the cancer cell's enzyme. Self-assembly, however, provides a different strategy. It should be possible to encase a cytotoxic drug in a self-assembled system, such as a vesicle. If a component of the vesicle walls is constructed such that it is also a substrate of the enzyme in question, then the enzyme will selectively cleave the vesicle and release the drug in the vicinity of the cancer cells (Fig. 3). The beauty of this approach is that any cytotoxic drug (or mixture of drugs) can be used, and it does not have to first be converted into a prodrug. I now cite a system of ours that demonstrates the feasibility of such chemistry (13).
Figure 3.

Cancer treatment using a self-assembled system.
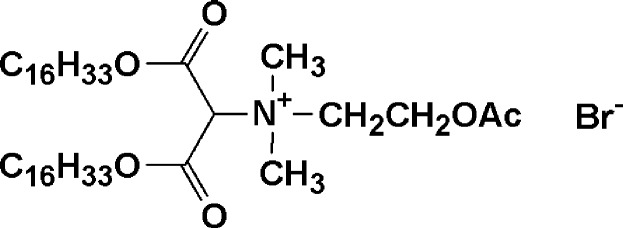
The compound
has two notable features: (i) it has two hydrocarbon tails and an ionic headgroup (the elements necessary for spontaneous formation into a vesicular bilayer). (ii) It has an acetylcholine-like moiety whose —OAc group is amenable to hydrolysis by acetylcholinesterase (an enzyme secreted in excess by neuroblastomas). When the latter occurs, the —OAc is converted into an —OH. Subsequently, the —OH attacks an ester group in the same molecule to form a lactone, thereby ejecting one of the long chains. Because two chains are needed to form a stable vesicular bilayer, the vesicles disintegrate and release their contents.
Proof that the above mechanism does indeed take place involved encapsulating an acetylcholinesterase substrate within the vesicles composed of our compound. The substrate produces a fluorescent product on hydrolysis. When enzyme was added to the vesicle system, there was an immediate burst of fluorescence, proving that the enzyme is able to rapidly chew through the vesicle walls; substrate is thus released into the bulk water where it is enzymatically hydrolyzed. If the acetylcholinesterase was heat-denatured, or if a different enzyme was used, no fluorescence was produced, demonstrating that this is an enzyme-specific release system.
I now end this Perspective by reporting on the recent work of Mrksich et al. (14) that demonstrates how the clever manipulation of a self-assembled monolayer creates a biologically useful device. The device allows two different cell populations to be attached to a solid surface in a predetermined pattern. Here how this was accomplished: “microcontact printing” on a gold layer on glass coverslips gave a surface coated with a series of alternating stripe-shaped regions (Fig. 4). One region was covered with a protein layer (fibronectin), whereas contiguous regions had, fixed to the gold by means of sulfurs, a mixture of two thiols (Fig. 5 Left). Note in particular that one of the thiol chains terminates with a hydroquinone moiety.
Figure 4.

Controlled attachment of two different cell types to a solid surface.
Figure 5.

Reactions used to attach a peptide to a gold surface by means of a hydroquinone thiol.
When the above system was exposed to a population of fibroblasts, the cells attached to only to the fibronectin-coated regions. There was no affinity for the region occupied by the two thiols. In the next step, the hydroquinone moiety of the thiol region was electrochemically oxidized to a quinone moiety (Fig. 5 Center). This oxidation was a key step in the process because the quinone provided a handle with which to attach a pentapeptide that had, bonded to it, a cyclopentadiene ring. The quinone and the cyclopentadiene ring combined in a Diels–Alder reaction, thereby fixing the peptide to the thiol-bearing regions of the assembly (Fig. 5 Right).
A second population of cells with an affinity for the peptide was then adsorbed onto the thiol-bearing regions. In this manner, side-by-side attachment of two different cell types was accomplished in a predetermined pattern (Fig. 4). It was pointed out that the ability to control the locations of different cell types, and to vary the distances between cell types in a systematic manner offers, among other things, new opportunities for mechanistic studies of heterotypic cell–cell signaling. Clearly, a useful task has been accomplished that would be difficult to achieve in the absence of self-assembly.
The approach—the philosophy—of supramolecular/colloidal chemistry will, in my opinion, be a dominant force in science for a long time. It is not one of those fields that, like a comet, appears with much fanfare only to fade quickly into obscurity. A prolonged life seems likely for several reasons. (i) After a century or more of intense interest in “compounds” (i.e., the covalent bond), it seems natural to expect that the noncovalent bond will take a turn at the head of the table. (ii) Supramolecular/colloidal systems tend to be complex, and dealing properly with complexity is a task that generally requires long periods of time. (iii) Because fields of science endure only if they ultimately have commercial applications, and because supramolecular/colloidal systems play a role in almost all major industries, there is further reason for optimism here. (iv) Finally, as mentioned previously, once the components of a cell are fully identified, biology will progress only if we understand how those components are assembled. And that will happen only when we have mastered that all-important phenomenon—the noncovalent bond.
Scheme 1.
Scheme 3.

Scheme 4.

Scheme 7.
Scheme 8.
Acknowledgments
I am grateful to the National Institutes of Health and the Army Research Office for making possible our own research into supramolecular and colloidal systems.
References
- 1. Trend, J. E., Kipke, C. A., Rossmann, M., Yafuso, M. & Patil, S. L (1993) U.S. Patent 5,474,743.
- 2.Khanjin N A, Snyder J P, Menger F M. J Am Chem Soc. 1999;121:11831–11846. [Google Scholar]
- 3.Janjic N, Tramontano A. J Am Chem Soc. 1989;111:9109–9110. [Google Scholar]
- 4.Menger F M, Eliseev A V, Migulin V A. J Org Chem. 1995;60:6666–6667. [Google Scholar]
- 5.Lehn J-M. Supramolecular Chemistry. Weinheim, Germany: VCH; 1995. [Google Scholar]
- 6.Menger F M, Caran K L, Seredyuk V A. Angew Chem Int Ed Engl. 2001;40:3905–3907. [PubMed] [Google Scholar]
- 7.Menger F M, Peresypkin A V. J Am Chem Soc. 2001;123:5614–5615. doi: 10.1021/ja003779l. [DOI] [PubMed] [Google Scholar]
- 8.Menger F M, Keiper J S. Angew Chem Int Ed Engl. 2000;39:1906–1920. doi: 10.1002/1521-3773(20000602)39:11<1906::aid-anie1906>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 9.Raez J, Barjovanu R, Massey J A, Winnik M A, Manners I. Angew Chem Int Ed Engl. 2000;39:3862–3865. doi: 10.1002/1521-3773(20001103)39:21<3862::AID-ANIE3862>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 10.Hager M, Holmberg K. Tetraherdon Lett. 2000;41:1245–1248. [Google Scholar]
- 11.Menger F M, Park H. Recl Trav Chim Pays-Bas. 1994;113:176–180. [Google Scholar]
- 12.Hanabusa K, Yamada M, Kimura M, Hirofusa S. Angew Chem Int Ed Engl. 1996;35:1949–1951. [Google Scholar]
- 13.Menger F M, Johnston D. J Am Chem Soc. 1991;113:5467–5468. [Google Scholar]
- 14.Yousaf M N, Houseman B T, Mrksich M. Proc Natl Acad Sci USA. 2001;98:5992–5996. doi: 10.1073/pnas.101112898. [DOI] [PMC free article] [PubMed] [Google Scholar]