

Abstract
A molecular receptor consisting of a spacer bearing two cofacially disposed terpyridyl–palladium–ligand (terpy-Pd-L) units rigidly separated by about 7 Å has been investigated for molecular recognition of planar aromatic molecules. It is found that although the receptor forms stable 1:2 host–guest association complexes with 9-methylanthracene (9-MA), the guest undergoes very rapid site exchange within the receptor and with external free 9-MA. A crystal structure of the 2:1 adduct shows one 9-MA in the molecular cleft defined by the two terpy-Pd-L units and the other resides on an outside face of one terpy-Pd-L unit. To establish the site residency time of the guests, a number of tethered molecules were prepared. These involve an anthracene molecule tethered to a pyridine ligand bound to the palladium atoms to form intramolecular host–guest adducts. Rotating-frame Overhauser effects were used to infer the site residency of the anthracene guests in the receptor. Variable-temperature 1H NMR spectroscopy of the intramolecular host–guest complexes has revealed that the site residency time of the anthracene guests is 1.6 × 10−5 sec at 20°C and 1.3 sec at −90°C in acetone solution. Whereas the guests are thermodynamically stable, they are kinetically very labile. A crystal structure of one of the tethered host–guest adducts reveals the expected structure which is the same as that determined in solution by 1H rotating-frame Overhauser enhancement spectroscopy experiments.
The current intense interest in supramolecular chemistry is the result of several parallel trends in chemistry. Synthetic chemistry has evolved to a stage where traditional methods can produce just about any molecule of modest size and complexity. These methods usually involve the formation of kinetically stable bonds in a predetermined sequence. The construction of each of these bonds allows for the deployment of a variety of synthetic methods that serve as alternatives in the synthetic orchestration. Although powerful, these methods have limitations when confronted with the task of producing the very large complex molecular structures that exist in biology and that are required for the development of material science. It is clear that a new, nontraditional, approach is required for the construction of structurally defined molecules that are in the nanoscale domain. To achieve this aim, inspiration is drawn from biology.
The overall structures of biology are usually the result of thermodynamically controlled self-assembly. The process requires first the construction of kinetically stable bonds between a sequence of molecular units. The relative geometries of these bonds and the substituents on the units possess all of the information required for the molecule to self-assemble into a defined structure, the properties of which are unique to the self-assembled molecule. Molecular recognition, a fundamental property of biological molecules, is manifested in the self-assembled structure. Synthetic supramolecular structures are generally formed by the principles just outlined.
Table 1 lists the types of bonds that are generally used for the construction of synthetic supramolecular structures (1). Included are the respective bond strength ranges and the kinetic lability and thermodynamic stability. There are exceptions to the entries but the data serve to indicate general trends. Covalent carbon bonds and
Table 1.
Supramolecular assembly bonds
 |
many third row transition metal bonds are stable and nonlabile, and such bonds are used as the permanent components of supramolecular structures. These components carry the structural information that leads to thermodynamically controlled self-assembly of the final supramolecular structure. The bonds that combine the components are required to be stable but kinetically labile in order that the most stable structure is formed rapidly. Such bonds include metal–ligand and hydrogen bonds. Thus a self-assembled supramolecular structure consists of nonlabile structurally encoded fragments that are assembled by stable but labile bonds. Once formed, the supramolecular structures can act as hosts for molecular recognition. The forces that control molecular recognition are generally weaker. Recognition depends critically on the structures of both the host and guest and on the solvent environment.
The molecular recognition interactions are listed in Table 2, where it will be noted that a range of energy interactions obtain, but in all cases the host–guest complexes are highly labile unless association and dissociation is controlled by steric impediments provided by the host (1). The interactions shown in Table 2 are well known and are illustrated in the right column. van der Waals interactions, an empirical formulation, are included in the list of interactions. The illustrations in Table 2 are largely self-explanatory, but two of these may require elaboration. Three π−π stacking interactions are shown, and on the assumption that the stability is determined by dipolar interactions, the structure drawn on the right is the most stable, followed by the slipped face-to-face structure drawn at the center. The eclipsed face-to-face structure is the least stable. The stability of host–guest associations is controlled to a significant degree by the solvation energies of the host, the guest, and the host–guest association complex. Depending on the relative contributions of these solvation energies, the association constant for host–guest formation can vary dramatically from solvent to solvent. The stability of host–guest complexes may be determined as a sum of different interactions, although usually a dominant force is ascribed to the stability.
Table 2.
Supramolecular recognition interactions
 |
The inverse distance dependence is different for a fixed ion–dipole (1/r2) compared to a freely rotating ion–dipole interaction (1/r4). For dipole–dipole interactions, fixed dipoles have a 1/r3 distance dependence, whereas freely rotating dipoles vary as 1/r6.
Noncovalent cation–π interactions are assumed to be controlled by charge–dipole interactions (1/r2) and by charge-induced dipole interactions (1/r4).
π–π interactions are assumed to be controlled by fixed dipole–dipole interactions (1/r3), by freely rotating dipole–dipole interactions (1/r6), and by dispersion interactions (1/r6).
S, solvent; G, guest; H, host.
Given the data in Table 1, it is not surprising to find that an increasing emphasis on the use of metal complexes in supramolecular assembly has emerged recently (2–6). In addition to the favorable stability–lability relationship of many metal–ligand bonds, metal-based supramolecular assemblies can be varied in overall charge and, because of the variety of bond dispositions, metal complexes can provide unique supramolecular geometries. Much of the work in this area has been directed at producing interesting large structural motifs and less attention has been devoted to molecular recognition, although the work of Fujita (5, 7–9), in particular, has demonstrated that metal-based supramolecular systems can be powerful molecular receptors.
This account describes our work on the stability and lability of guest molecules associated with a host that has unique characteristics. It will be shown that, although stable host–guest complexes can be formed, they are exceptionally labile at ambient temperatures. Few comparable studies on the dynamics of host–guest association have appeared (10–12).
The Molecular Receptor
The molecular receptors consist of several parts: a
spacer–(tridentate) chelator, 1, a spacer–chelator square
planar complex, 2, and linkers, 3, 4,
and 5. By using appropriate spacer–chelator complexes,
2, and the linkers, 3, 4, and
5, the molecular rectangle, 6, the trigonal
prism, 7, and the tetragonal prism, 8, should
form by thermodynamically controlled self-assembly. For these to form,
a number of characteristics are required to be embodied in the
spacer–chelator complex and in the linkers. The spacer–chelator
complex 2 requires rigidity to reduce the degrees of freedom
of the molecule. As the number of degrees of freedom is reduced, the
molecule becomes more disposed to self-assemble in a particular
geometry. Similarly, rigid spacers serve the same purpose. Thus,
structural characteristics of the spacer–chelator complex and the
linker carry the information that leads to thermodynamically controlled
self-assembly by a self-correcting process. For rapid self-assembly,
the ligand, L, in 2 must be a good leaving group and the
metal-linker bonds must also be kinetically labile. The lability of
metal-ligand bonds depends principally on the nature of the metal and
to some extent on the characteristics of the
ligand
 .
.
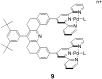
The spacer–chelator complexes discussed here are of the
type 9, the preparation of which is described elsewhere
(13). The fully oxidized spacer as well as other metal complexes have
been prepared. The molecule 9 has a number of
characteristics that were expected to lead to the formation of
supramolecular assemblies with rigid spacers. Further, 9 was
expected to act as a molecular receptor by incarcerating guests in the
molecular cleft. The receptor 9 contains a rigid spacer that
carries two cofacially disposed terpyridyl–palladium–ligand
(terpy-Pd-L) units. These two terpy-Pd-L units are separated by about 7
Å, but this separation can be contracted to some extent while
retaining a nearly eclipsed cofacial disposition by concerted rotation
of these units about the chelator–spacer single bonds. The separation
between the two terpy-Pd-L units is sufficient to incarcerate planar
aromatic molecules and square-planar complexes, the “thickness”
of which match the cavity provided by the molecular
cleft
 .
.
When L is a good leaving group, such as
CH3CN, addition of the linkers 10 or
11 to 9 leads to the molecular rectangle
6 (14) or the trigonal prism 7 (J. D.
Crowley, A.J.G., and B.B., unpublished work). The supramolecular
assemblies are formed rapidly and quantitatively in acetonitrile
solutions at 25°C. Although the molecular rectangle and trigonal
prism act as receptors for a variety of guests, the systems,
9 [L = Cl or pyridine (py)], which have a single
molecular cleft for guest incarceration are the subject of this
paper
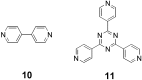 .
.
When dissolved in acetonitrile or acetone solutions, the
receptor 9 associates with a variety of planar molecules
including 12, 13, and 14. Addition of
solutions of the guest molecules to yellow solutions of 9
results in the immediate development of a deep red color, suggesting
host–guest association. In the case of 9-methylanthracene (9-MA;
12), its association with 9 (L = Cl) in
acetonitrile solutions was measured by the 1H NMR
titration method at 21°C (13, 15–17). Two equilibrium (association)
constants were obtained, K1 = 650
M−1 and K2 =
250 M−1. Whereas it is clear that one of the
association constants represents the incarceration of 9-MA in the
molecular cleft, it is not obvious how to ascribe the location of the
second 9-MA. This multiple association appears to be a general
phenomenon for 9-MA association with molecules derived from
9. Thus, for example, the molecular rectangle 6,
derived from the linker 10, associates with four 9-MA
guests, presumably by associating two 9-MA guests for each 9
fragment
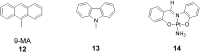 .
.
The nature of the 2:1 complex is inferred from the extended crystal structure of the adduct (Fig. 1). The solid-state structure shows that 9-MA molecules reside in the cleft as expected, but the structure also reveals a second 9-MA molecule that stacks on the outer face of the terpy-Pd-Cl units. The interplanar separation between 9-MA molecules and terpy-Pd-Cl units is 3.42 Å, which is achieved by concerted rotation of the terpy-Pd-Cl units with respect to the spacer. It is probable that in solution the receptor associates with 9-MA in a similar manner, with one 9-MA inside of the cleft and the other lying on the outside face of a terpy-Pd-Cl unit. The 1H NMR spectrum in the temperature range of −40°C to +70°C does not distinguish the site residency of the 9-MA units; an average 1H NMR spectrum is observed, indicating that the guests are undergoing rapid interchange between sites. Fast exchange also occurs between free and associated 9-MA. Thus, although the host–guest complexes are stable, they are labile with respect to site exchange and with respect to intermolecular exchange. To ascertain the site residency time of guests, a number of new compounds were prepared.
Figure 1.
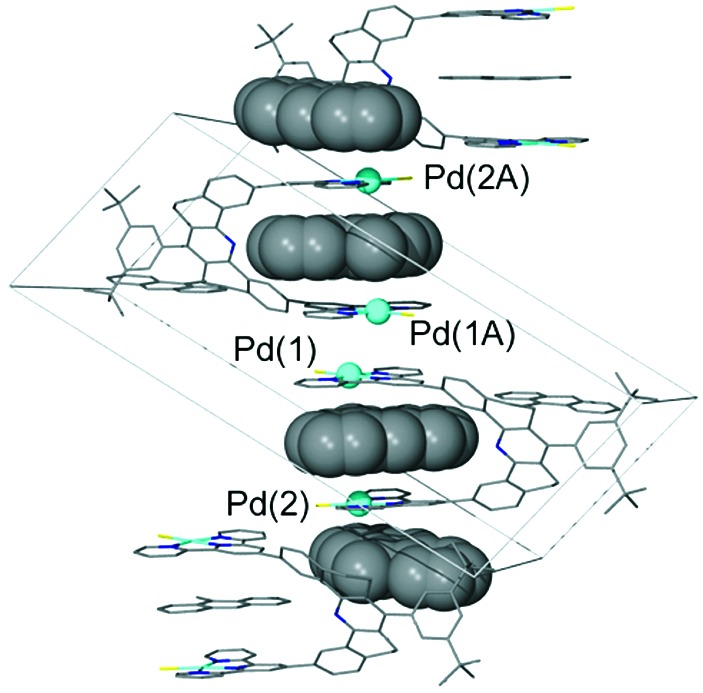
An illustration of the extended structure of [9 (L = Cl)]2+⋅2(9-MA). The box is the unit cell, 9-MA molecules of the stack are shown as space-filling models, and the terpy-Pd-Cl units are shown as stick models, as are all other molecules not involved in this stack. Palladium atoms in the stack are shown as blue spheres.
Supporting information (26 pages) contains the experimental procedures; 1H NMR, COSY, and ROESY spectra for compounds 15, 16, and 17; and the crystallographic experimental section, a table of crystal data, and views for [17](PF6)4. This material is available on the PNAS web site, www.pnas.org.
Dynamics of Host–Guest Complexes
The dynamic behavior of host–guest complexes was studied by
temperature variation of the 1H NMR spectra of
the complexes 15, 16, and 17, which,
in the cases of 16 and 17, are drawn with the
anthracene groups in their anticipated sites. The pyridine-linked
anthracene ligand required some care in design to ensure a proper
unencumbered fit of the anthracene group into the molecular cleft. As
noted previously, the anthracene associates with the receptor by
occupying the cleft and an external terpy-Pd-L face. Thus, the
anthracene groups in 16 and 17 are expected to
interchange intramolecularly between these sites and it may be possible
to measure the rate of interchange by temperature-dependent
1H NMR spectroscopy. The compound 15
was prepared as an analogue for the study of 9-MA
association
 .
.
By 1H NMR titration of 9-MA with
15 in (CD3)2CO
solution at 16°C it was determined that a 2:1 complex formed with
association constants K1 = 400 ±
50 M−1 and K2 =
100 ± 50 M−1. The two 9-MA molecules of
the complex are indistinguishable by 1H NMR
spectroscopy at temperatures between −90°C and 20°C, and the free
and bound 9-MA molecules are indistinguishable in the same temperature
range. Thus, whereas relatively stable complexes are formed, the 9-MA
guest molecules exchange rapidly between their respective sites in the
association complex and with free 9-MA. The site occupancies of the
9-MA guest associated with the receptor 15 have been
inferred from 1H rotating-frame Overhauser
enhancement spectroscopy (ROESY), a method of measuring rotating-frame
Overhauser effects (ROE) in large molecules (18–21). The ROE are
informative of the relative proximity between nuclei, in this case
proton nuclei. It was found that Overhauser effects existed between a
number of protons of the host and those of the guests. The cross-peak
Overhauser effects are consistent with the presence of a 9-MA guest in
the cleft of 15 and a 9-MA guest lying on the outer faces of
the terpy-Pd-py units. Further, the long axis of the 9-MA guests
appears to be roughly parallel to the long axis of the terpy ligands.
The cross-peak enhancements also imply that the 2:1 adducts exist as
four isomers that occur by virtue of the orientations of the 9-MA
methyl groups with respect to the spacer. These four isomers are
represented in 18, 19, 20, and
21, where proton–proton enhancements are indicated by
curved lines. Because of the rapid site exchange, the two outside faces
of the terpy-Pd-Cl units are not distinguished, giving rise to ROE
cross-peaks at both sites. For similar reasons, the four isomers are
engaged in rapid interconversions. The ROESY experiment reflects the
average population of the sites but it does provide a detailed
description of the structure of 2:1 adduct. It is gratifying that the
ROESY experiment provides a structure similar to that observed in the
solid state for the chloro receptor (Fig.
1)
 .
.
The compound 16 incorporates one tethered
anthracene guest and was prepared to determine which of the two sites
(in the cleft or above the terpy-Pd-L) was preferred in the 2:1
adducts. It may appear obvious that the 9-MA would prefer the cleft
site, but this has not been established. Because of the lower symmetry
of 16 compared with 15, a correlation
spectroscopy (COSY) experiment was performed to establish the
1H NMR proton assignments in
(CD3)2CO solution. ROESY experiments
performed on 16 in
(CD3)2CO solutions at
16°C revealed cross-peaks that indicated the anthracene guest lay
exclusively in the cleft. No Overhauser effects were detected for
protons that would give ROE cross-peaks if the anthracene occupied the
outside terpy-Pd-L face as was observed for the 2:1 adduct of the
receptor 15. The ROESY experiments indicate the structure
22 where the cross-peaks are shown as
before
 .
.
When the temperature was lowered incrementally from 20°C
to −90°C no changes in the 1H NMR spectrum of
16 were observed that would indicate that more than one site
was populated by the anthracene guest. This result, however, does not
necessarily imply that the anthracene group is not engaging in
fluxional exchange between the inside and outside sites because, if the
anthracene guest resides overwhelmingly in the cleft, its population at
the outside site will not be detected. Compound 17 is a
tethered analogue of 15 and it is expected that one
anthracene guest will lie in the cleft and the other will reside on the
outside face of the terpy-Pd-L units. This system is expected to engage
in intramolecular site exchange. Given that the anthracene guest is
tethered, it might be expected that the site exchange would be slower
than in the case of the 2:1 9-MA adduct of 15, which is
devoid of the tether steric encumbrances. The 1H
NMR spectrum of 17 at 16°C in
(CD3)2CO solutions displays
signals that indicate a symmetrical molecule, indicating that all of
the sites are equally populated by the anthracene guests. This
observation suggests that the two anthracene fragments are in rapid
exchange between the accessible sites. This process is intramolecular
because addition of the ligand to acetone solutions of 17
provides an 1H NMR spectrum at 16°C that
reveals signals for both the free and bound ligands, indicating that
the time scale for intramolecular site exchange is much faster than
intermolecular site exchange. Further, the 1H NMR
spectrum is invariant to dilution in the range of
10−3 M to 10−5 M,
indicating that the anthracene guest engages in intramolecular rather
than intermolecular association. COSY and ROESY spectra were measured
on 17 in acetone solution at 16°C to assign the signals
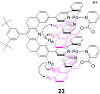
and to infer the structure of 17, respectively. The ROESY
spectrum indicates that the expected structure, 23, obtains
and, further, that ROE cross-peaks are present at all three possible
sites, which is consistent with the 1H NMR
spectrum
 .
.
The 1H NMR spectrum of 17 was measured in 10°C intervals between 20°C and −90°C in (CD3)2CO solution. Temperature-dependent chemical shifts are observed for the protons and many of the signals overlap, but there are three well separated signals that are sharp at 20°C and become broad as the temperature is lowered, and at −90°C each separates into sharp signals. This latter behavior is characteristic of fluxional site exchange. These data can be processed by conventional methods to provide the activation free energy, ΔG‡, and the corresponding enthalpy, ΔH‡, and entropy, ΔS‡, and, of course, the rate constants and site residency life-times for the fluxional site exchange (22–24).
Using any of three signals that undergo coalescence and separation, we found that ΔG‡ = 10.1 kcal/mol, ΔH‡ = 8.23 kcal/mol, and ΔS‡ = −8.47 cal/mol⋅K for the site exchange process. The average site residency time (life-time) of the guests in any site configuration is 1.6 × 10−5 sec at 20°C and 1.3 sec at −90°C.
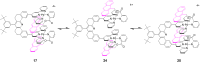
The mechanism of site exchange is consistent with the process
17 ⇌ 24 ⇌ 25, where the
intermediate 24 is produced by stepwise (intramolecular)
dissociation of the anthracene fragments from their respective sites
while maintaining Pd–N connectivity. Such a process, 17 ⇌
25, requires the rotation of the pyridine ligands, which
could conceivably be encumbered by the terpy ligands. There appears to
be a little steric hindrance to pyridine ligand rotation, because it
was found that these ligands freely rotate at −90°C in
15. That the fluxional site exchange represents the process
17 ⇌ 25 is supported by the observation that
the proton signal at δ = 9.15 ppm splits into two signals at
−90°C. These signals refer to the inner two protons of the spacer
(protons e, structure 23), and the appearance of two signals
at −90°C indicates an unsymmetrical molecule consistent with
structure 17. The molecule 17 could exist in two
interconverting forms by flipping of the spacer between the racemic and
meso forms arising from the conformations of the two reduced rings.
This interconversion in analogous molecules is a very low energy
process that would not be apparent in the temperature span of the
present study (25, 26). A possible method of interconverting from
17 to 25 is by locked rotation of terpy-Pd-L
units, but it has been demonstrated that rotation of large aromatic
molecules in place of the terpy-Pd-L substituents is a higher energy
process than the one observed here even in the absence of a guest
in the molecular cleft (26, 27). Thus, the mechanism of site
exchange is most likely that illustrated in outline by the
sequence 17 ⇌ 24 ⇌
25
 .
.
Crystal Structure
Isolation of crystals suitable for x-ray diffraction of
17 proved difficult, but eventually crystals that provided
useful data were obtained by vapor diffusion of methanol into a methyl
ethyl ketone solution of the PF salt. The crystals
formed as 17
(PF6)4⋅2Et(Me)CO. The
solid-state structure is shown in Figs. 2
and 3, where it will be seen that the
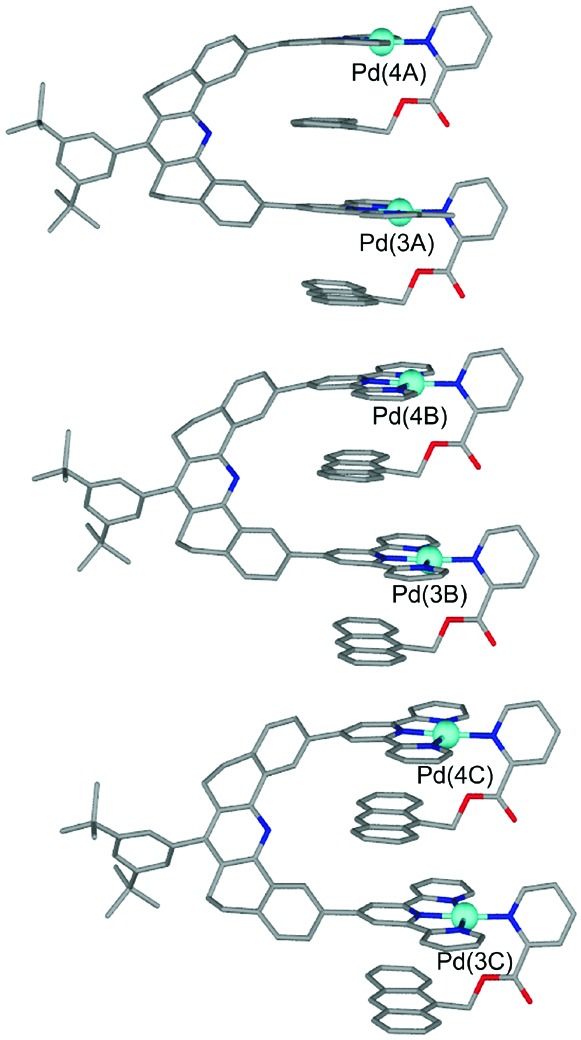
expected structure obtains. Two molecules exist in the unit cell and
each molecule is in the same absolute configuration. The bond lengths
and bond angles are unexceptional. Each of the molecules in the
unit cell has slightly different structural parameters and, in what
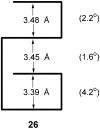
follows, average values are given. The interplanar separations are
shown in the two-dimensional illustration of the structure
26. The interplanar angles, defined by the mean plane of the
anthracene unit and the mean plane of the terpy ligand, are shown in
brackets in 26. The interplanar separations are those
expected for π-stacked aromatic molecules, and each stacking unit is
aligned roughly parallel to the other, as required. It is gratifying to
note that the crystal structure is similar to that inferred from ROESY
experiments in solution and that the structure is similar to the one in
Fig. 1, which contains untethered anthracene guests. To achieve the
interplanar separations necessary for π–π stacking, the terpy-Pd-L
units engage in concerted rotation to decrease the interplanar
separations of the two terpy-Pd-L units to 6.84 Å. Consequently, the
terpy-Pd-L planes are not perpendicular to the mean molecular plane of
the spacer, giving the molecule a twisted configuration (Fig. 3). The
ability of the receptor to adjust the interplanar separation of the
cleft is an important factor in stabilizing the
guest
salt. The crystals
formed as 17
(PF6)4⋅2Et(Me)CO. The
solid-state structure is shown in Figs. 2
and 3, where it will be seen that the
expected structure obtains. Two molecules exist in the unit cell and
each molecule is in the same absolute configuration. The bond lengths
and bond angles are unexceptional. Each of the molecules in the
unit cell has slightly different structural parameters and, in what
follows, average values are given. The interplanar separations are
shown in the two-dimensional illustration of the structure
26. The interplanar angles, defined by the mean plane of the
anthracene unit and the mean plane of the terpy ligand, are shown in
brackets in 26. The interplanar separations are those
expected for π-stacked aromatic molecules, and each stacking unit is
aligned roughly parallel to the other, as required. It is gratifying to
note that the crystal structure is similar to that inferred from ROESY
experiments in solution and that the structure is similar to the one in
Fig. 1, which contains untethered anthracene guests. To achieve the
interplanar separations necessary for π–π stacking, the terpy-Pd-L
units engage in concerted rotation to decrease the interplanar
separations of the two terpy-Pd-L units to 6.84 Å. Consequently, the
terpy-Pd-L planes are not perpendicular to the mean molecular plane of
the spacer, giving the molecule a twisted configuration (Fig. 3). The
ability of the receptor to adjust the interplanar separation of the
cleft is an important factor in stabilizing the
guest
 .
.
Figure 2.
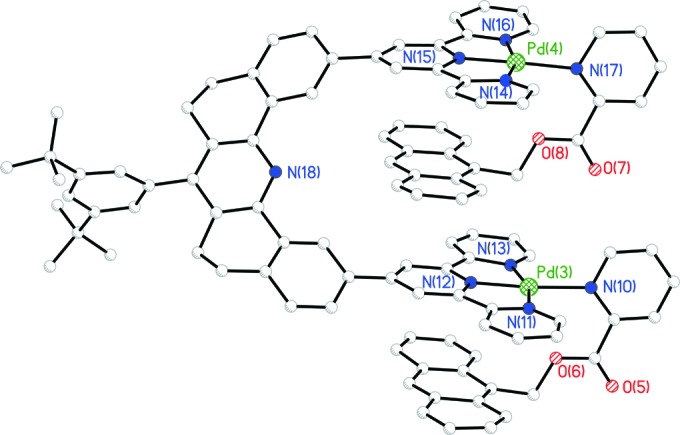
A ball-and-stick representation of a “side” view for one of the molecules in the unit cell of 17. Counter ions and solvent molecules have been removed for clarity.
Figure 3.
A ball-and-stick representation of a “front” view for one of the molecules in the unit cell of 17. Counter ions and solvent molecules have been removed for clarity.
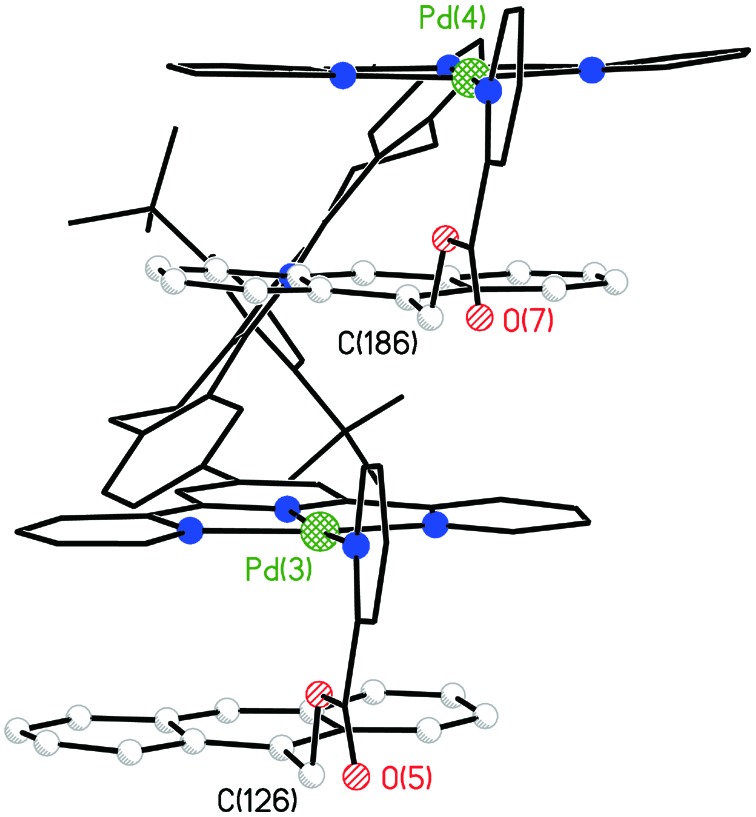
The extended solid-state structure of 17 is shown in Fig. 4, where infinite alternate stacking of anthracene and terpy-Pd-L units is observed. This structure is similar to, but different from, that observed for the structure shown in Fig. 1. The latter exists as units of eight stacks, which includes terpy-Pd-Cl–terpy-Pd-Cl stacking, whereas the former displays infinite alternate anthracene–terpy-Pd-L stacking.
Figure 4.
An illustration of a “side” view of three molecules of 17 that belong to an infinite stack of molecules in the crystal of [17]4+. Counter ions and solvent molecules have been removed for clarity.
Discussion
The work described here demonstrates a number of features of molecular recognition. Perhaps the most significant is the observation that whereas host–guest complexes can be thermodynamically stable, they, at the same time, can be exceeding labile kinetically. The forces that control host–guest formation in the present study probably involve attractive π–π interactions and may also involve charge-induced-dipole attraction between the positively charged terpy-Pd-L units and the π electrons of the anthracene guests (Table 2). Because these forces attenuate rapidly with distance, particularly if, as is probable, the dipolar interactions are not fixed, the interplanar separation of the receptor is crucial in obtaining stable host–guest association complexes. As noted, the receptor described here is able to adjust this separation with minimum energy cost.
The solution stability of host–guest complexes depends on solvation effects (Table 2), and it is interesting in this regard that one of the anthracene guests resides on the outer face of a terpy-Pd-L unit in solution. Given that the solid-state structures show extensive stacking of units, it is perhaps surprising that greater aggregation is not observed in solution. This may be because of the low concentration (≈10−3 M) at which the studies were carried out. For other guests, however, solution gels are formed, indicating extensive association, perhaps resembling that observed in the solid state.
As noted earlier, molecular recognition is a subtle phenomenon, which depends on weak interactions, and it is difficult to describe the stability of a host–guest complex in terms of a single dominant interaction. The stability usually depends on the additive effects of multiple weak interactions which, in concert, provide the molecular recognition. One of the challenges in chemistry in the near future is to understand and deploy these weak noncovalent interactions, not only for molecular recognition but also for the construction of large molecules in the nanoscale domain.
Supplementary Material
Acknowledgments
This work was supported by grants from the Basic Sciences Division of the Department of Energy.
Abbreviations
- 9-MA
9-methylanthracene
- ROE
rotating-frame Overhauser effect
- ROESY
rotating-frame Overhauser enhancement spectroscopy
- terpy
terpyridyl
- py
pyridine
- COSY
correlation spectroscopy
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 173843. These data can be obtained free of charge from www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, U.K.; E-mail: deposit@ccdc.cam.ac.uk).
References
- 1.Goshe A J, Crowley J D, Bosnich B. Helv Chim Acta. 2001;84:2971–2985. [Google Scholar]
- 2.Leininger S, Olenyuk B, Stang P J. Chem Rev. 2000;100:853–908. doi: 10.1021/cr9601324. [DOI] [PubMed] [Google Scholar]
- 3.Swiegers G F, Malefetse T J. Chem Rev. 2000;100:3483–3537. doi: 10.1021/cr000023w. [DOI] [PubMed] [Google Scholar]
- 4.Jones C J. Chem Soc Rev. 1998;27:289–299. [Google Scholar]
- 5.Biradha K, Fujita M. In: Advances in Supramolecular Chemistry. Gokel G W, editor. Stamford, CT: JAI Press; 2000. pp. 1–39. [Google Scholar]
- 6. Caulder, D. L. & Raymond, K. N. (1999) J. Chem. Soc. Dalton Trans., 1185–1200.
- 7.Fujita M. Acc Chem Res. 1999;32:53–61. [Google Scholar]
- 8.Fujita M. Chem Soc Rev. 1998;27:417–425. [Google Scholar]
- 9.Fujita M, Ogura K. Coord Chem Rev. 1996;148:249–264. [Google Scholar]
- 10.Garel L, Dutasta J-M, Collet A. Angew Chem Int Ed Eng. 1993;32:1169–1171. [Google Scholar]
- 11.Brotin T, Lesage A, Emsley L, Collet A. J Am Chem Soc. 2000;122:1171–1174. [Google Scholar]
- 12.Ballardini R, Balzani V, Credi A, Brown C L, Gillard R E, Montalti M, Philp D, Stoddart J F, Venturi M, White A J P, et al. J Am Chem Soc. 1997;119:12503–12513. [Google Scholar]
- 13.Sommer R D, Rheingold A L, Goshe A J, Bosnich B. J Am Chem Soc. 2001;123:3940–3952. doi: 10.1021/ja004279v. [DOI] [PubMed] [Google Scholar]
- 14. Goshe, A. J. & Bosnich, B. (2001) Synlett, 941–944.
- 15.Kneeland D M, Ariga K, Lynch V M, Huang C-Y, Anslyn E V. J Am Chem Soc. 1993;115:10042–10055. [Google Scholar]
- 16.Schneider H-J, Yatsimirsky A K. Principles and Methods in Supramolecular Chemistry. Chichester, U.K.: Wiley; 2000. pp. 137–190. [Google Scholar]
- 17.Tsukube H, Furuta H, Odani A, Takeda Y, Yoshihiro K, Inoue Y, Liu Y, Sakamoto H, Kimura K. In: Comprehensive Supramolecular Chemistry. Davies S E D, Ripmeester J A, editors. Vol. 8. Oxford: Pergamon; 1996. pp. 426–481. [Google Scholar]
- 18.Neuhaus D, Williamson M P. The Nuclear Overhauser Effect in Structural and Conformational Analysis. 2nd Ed. New York: Wiley; 2000. [Google Scholar]
- 19.Schneider H-J, Yatsimirsky A K. Principles and Methods in Supramolecular Chemistry. Chichester, U.K.: Wiley; 2000. pp. 230–240. [Google Scholar]
- 20.Klärner F-G, Burkert U, Kamieth M, Boese R, Benet-Buchholz J. Chem Eur J. 1999;5:1700–1707. [Google Scholar]
- 21.Mo H, Pochapsky T C. Prog Nucl Magn Reson Spectrosc. 1997;30:1–38. [Google Scholar]
- 22.Petrucci S, Eyring E M, Konya G. In: Comprehensive Supramolecular Chemistry. Davies S E D, Ripmeester J A, editors. Vol. 8. Oxford: Pergamon; 1996. pp. 483–497. [Google Scholar]
- 23.Schneider H-J, Yatsimirsky A K. Principles and Methods in Supramolecular Chemistry. Chichester, U.K.: Wiley; 2000. pp. 259–263. [Google Scholar]
- 24.Binsch G. In: Topics in Stereochemistry. Eliel E L, Allinger N L, editors. Vol. 3. New York: Interscience; 1968. pp. 97–185. [Google Scholar]
- 25.Rabideau P W. The Conformational Analysis of Cyclohexanes, Cyclohexadienes, and Related Hydroaromatic Compounds. New York: VCH; 1989. pp. 81–88. [Google Scholar]
- 26.Zimmerman S C. In: Topics in Current Chemistry. Weber E, editor. Vol. 165. Berlin: Springer; 1993. pp. 72–101. [Google Scholar]
- 27.Zimmerman S C, VanZyl C M, Hamilton G S. J Am Chem Soc. 1989;111:1373–1381. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.