

Abstract
The phenylalanine-4-hydroxylase (PAH) gene encodes the PAH enzyme, which is necessary for the conversion of l-phenylalanine (l-Phe) to l-tyrosine (l-Tyr). Deleterious mutations in PAH can disrupt its function, leading to the toxic buildup of phenylalanine in the brain and causing the inherited genetic disorder phenylketonuria (PKU). This condition results in behavioral issues, epilepsy, and intellectual disability. This in silico study has been conducted to investigate the structure and dynamics of both wild-type PAH and its variants, including I65T and R408W, which are prevalent in U.S. patients, as well as D282G and A202T, commonly observed in PKU patients from China and Korea. SIFT, PolyPhen-2, PhD-SNP, and MutPred2 methods, which utilize either sequence-based or machine learning algorithms, predicted the four mutations to be disease-causing and deleterious with the potential to disrupt the PAH structure and impair its function, thereby confirming their association with PKU. Four replicates of 500 ns molecular dynamics (MD) simulations resulting in a cumulative simulation time of 2 μs for all variants demonstrated that all these variants adversely affect the PAH structure and dynamics. The MM/GBSA binding free energy of I65T with BH4, a crucial cofactor in the hydroxylation of l-Phe to l-Tyr, is found to be −12.8 kcal/mol compared to −16.5 kcal/mol for the wild type. Similarly, the R408W variant decreased BH4 binding with a calculated free energy of −11.4 kcal/mol. Additionally, the binding affinity of the tetramerization domains in R408W significantly reduced by at least 26.7 kcal/mol compared to the wild type. This study highlights how different pathogenic mutations in PAH impact the protein’s structure, dynamics, and binding affinity, possibly leading to advancements in targeted drug development for PKU.

1. Introduction
Phenylketonuria (PKU) is an inherited genetic disorder caused by a pathogenic change in the phenylalanine hydroxylase (PAH) gene, a gene responsible for producing the enzyme necessary for phenylalanine metabolism in human. , Phenylalanine hydroxylase (PAH) enzyme converts l-phenylalanine to l-tyrosine by hydroxylating the aromatic side chain. Failure or any compromised enzymatic activity of PAH can lead to the bioaccumulation of phenylalanine in the blood, urine, and brain, which ultimately causes phenylketonuria (PKU). The most severe forms of PKU can result in permanent neurological disabilities, including mental retardation, behavioral issues, epilepsy, microcephaly, and stunted growth. , This disease affects approximately 1 in 15,000 births in the United States and can persist through adolescence into adulthood. −
The human PAH protein structure adopts a tetrameric conformation, formed through a 2-fold symmetry of two dimers (Figure ) with each subunit consisting of an N-terminal regulatory domain (residues 1–142), a catalytic domain (residues 143−410), and a C-terminal oligomerization domain (residues 411–452). The enzyme is a nonheme iron-dependent enzyme that requires (6R)-l-erythro-5, 6, 7, 8-tetrahydrobiopterin (BH4) as an essential cofactor in the hydroxylation of l-Phe. The regulatory mechanisms of the protein require reversible conformational changes that are transmitted throughout the enzyme upon the binding of BH4 and l-Phe. The cofactor BH4 plays an important role to prevent wild-type and variant PAH from degradation by the ubiquitin-proteasome-dependent pathway, whereas its absence causes folding defects and increases the rates of degradation. , Analysis of the crystal structure of the BH4 bound and unbound states of PAH protein as well as modeling studies points to the interaction of BH4 with serine-23 through hydrogen bonds with both oxygen atoms in the dihydroxy propyl side chain. Serine-251 and glycine-247 also form hydrogen bonds with BH4, whereas leucine-249, leucine-248, and phenylalanine-254 help to stabilize BH4 at the protein’s center through van der Waals, pi–sigma, and pi–pi stacking interactions.
1.

(A) Structure of the wild-type full-length human PAH protein complexed with BH4 (red). (B) Ligand BH4 (pink) complexed with a single PAH chain and labeled with the studied residues in the β4 mutation sites: I65T (blue), A202T (red), D282G (green), and R408W (orange). (C) Bonding interactions between BH4 and residues within the catalytic domain of PAH.
Several studies have reported that most patients with elevated phenylalanine levels are attributed to missense mutations. ,, Approximately 548 mutations are identified in the PAH gene, of which 50% are missense mutations resulting from nonsynonymous single nucleotide polymorphisms (nsSNPs) leading to the incorporation of a different amino acid in the encoded protein sequence. , Furthermore, PAH mutations have been observed to vary among patients from different demographic regions. Patients in the United States exhibit the greatest mutational heterogeneity in PAH, with prominent mutations occurring in amino acids R408W, Y414C, F39L, I65T, L348 V, E280 K, and P281L. In a study on 102 independent Korean PKU patients, nine novel missense mutations (N207D, K95del, A447P, G344D, P69S, S391I, A202T, G103S, and I306L) were identified. It was suggested that the A202T mutation might disrupt hydrophobic interactions among the ALA202, TYR198, TYR206, and LEU348 residues in the PAH protein while the G344D mutation could alter intermolecular interactions with a specific β4 strand and α10 helical domain. Song et al. discovered Y154H, R157K, S349A, A395D, E280G, and D282G mutations in PAH by examining 185 patients in northern China. These investigations also revealed significant differences in some mutations compared to those found in Europe.
A low-phenylalanine diet and sapropterin dihydrochloride are currently available treatment options for managing PKU. , Sapropterin dihydrochloride is a synthetic version of naturally occurring BH4. , Currently, this treatment represents the only effective approach which functions by saturating the mutant PAH structure with excessive BH4, which helps counteract their reduced binding affinity and restores the protein activity. , But only 20–56% of PKU patients are reported to respond to this treatment. Numerous studies have demonstrated that nsSNPs can induce structural changes in proteins, thereby impacting their function. − Several mutations in the PAH gene have been associated with misfolding and instability of the PAH protein. , A previous in silico study identified two variants, I65N and L311P, as potentially linked to severe PKU [31]; however, these variants have not been observed in patients. In this study, we provide a comprehensive analysis of various PAH mutations found in patients to elucidate their structural and functional consequences in PKU. Our findings offer valuable insights into the underlying mechanisms of the disease and may contribute to the development of novel, effective treatment strategies.
The nonsynonymous single nucleotide polymorphisms I65T, A202T, D282G, and R408W of PAH protein associated with PKU are thoroughly investigated in the present study. Variants I65T and R408W are common in PKU patients in the United States with a frequency of 4.1% and 18.7%, respectively, whereas A202T is a novel variant of Korean PKU patients and D282G in China. ,, To confirm the deleterious effects of these mutations on the PAH protein structure and to explore possible pathogenic mechanisms, advanced bioinformatics tools such as SIFT, PolyPhen-2, PhD-SNP, and MutPred2 − and long molecular dynamics simulations, using the crystallographic structure of human phenylalanine hydroxylase (PDB ID: 6N1K), were conducted for both wild-type and mutant systems. To investigate the mutational effect on PAH protein’s binding with BH4, MM/GBSA binding free energies of the wild type as well as variants of PAH were conducted using the crystallographic structure of the protein–BH4 complex (PDB ID: 6HYC). Additionally, MM/GBSA binding affinity calculations for the regulatory domains (PDB ID: 5FII) and tetrameric domains (PDB ID: 6HYC) of both wild-type and mutant PAH structures were carried out to unveil the impact of mutations on dimerization and tetramerization domains of PAH.
2. Methods
2.1. Predicting Deleterious Impact, Pathogenicity, and Protein Structure Disruption Mechanism
The Universal Protein Resource (UniProt) and Protein Data Bank (PDB) are used to identify pathogenic mutations in the PAH gene and to acquire the wild-type human PAH protein sequence in FASTA format. , The structural stability and pathogenicity of mutant proteins were reassessed using SIFT, PolyPhen-2, PhD-SNP, and MutPred2. − Sorting Intolerance From Tolerance (SIFT) (https://sift.bii.a-star.edu.sg/) predicts the effect of amino acid substitutions on protein function based on sequence homology. ,, SIFT has an accuracy of 70% and provides a numerical score indicating the likelihood of protein function disruption. , The scores fall between 0 and 1 where a score less than 0.05 is considered to affect the protein function. SIFT prediction is based upon the degree of conservation of amino acid residues in sequence alignments that are derived from related structures and are collected using Position-Specific Iterated BLAST (PSI-BLAST). Then the PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) was employed which predicts the impact of missense mutations on proteins by analyzing the sequence, structural, and phylogenetic data of proteins. , PolyPhen-2 is a trained machine learning model for probabilistic classifiers that has shown an accuracy of 68% in predicting disease-related nsSNPs. , PolyPhen-2 predicts with confidence score ranges of 0 to 1, where scores less than 0.15 are predicted to be benign SNPs and scores ranging from 0.15 to 1 are possibly damaging SNPs. PolyPhen-2 is not dependent on protein sequence alignment like SIFT. Therefore, these two algorithms can be used in conjunction to best predict whether and to what degree an amino acid substitution will affect protein function.
Then, PhD-SNP (https://snps.biofold.org/phd-snp/phd-snp.html), a support vector machine (SVM)-based predictor of human deleterious single nucleotide polymorphisms, was used to predict variant pathogenicity. , This SVM classifier is trained with a data set comprising more than one million amino acid polymorphisms through supervised machine learning techniques to predict variant pathogenicity. PhD-SNP predicts the likelihood of an nsSNP resulting in a new protein structure and relating it to a genetic disease in humans, with an accuracy of 74%.
Finally, MutPred2 (http://mutpred.mutdb.org/), a machine learning-based software, was used to further predict the disease-causing potential of amino acid substitutions. MutPred2 assists in interpreting pathogenicity with molecular alteration scores and provides a comprehensive framework for systematically ranking the underlying mechanisms. It analyzes both wild-type and mutated sequences using multiple sequence-based protein property predictors. The tool extracts six categories of features from the provided amino acid sequence and substitution, which are processed by an ensemble of neural networks designed to classify variants as pathogenic or not. MutPred2 provides two key outputs, the general score (g score), which estimates the likelihood of disease association for the nsSNP, and the property score (p score), which assesses the impact on specific structural or functional properties. , Final predictions hypotheses are categorized based on these two scores: actionable hypotheses for g > 0.5 and p < 0.05, confident hypotheses for g > 0.75 and p < 0.05, and very confident hypotheses for g > 0.75 and p < 0.01.
2.2. Molecular Dynamics Simulations
Molecular dynamics (MD) simulations were carried out utilizing the AMBER20 software suite using the full-length crystal structure of human phenylalanine hydroxylase from the protein data bank (PDB ID: 6N1K). , The systems force fields were prepared using the CHARMM-GUI Web server utilizing the CHARMM36m additive force field for proteins, CGenFF for BH4, and TIP3P was used to model water. The wild-type and I65T, A202T, D282G, and R408W mutant systems were prepared using the CHARMM-GUI solution builder. , A rectangular water box was fitted around the protein with a radius of 10 Å from the surface of the complex, and a pH of 7.0 was ensured for each of the systems. Using the Monte Carlo ion placing method, 0.15 M KCl ions were added to mirror physiological conditions. Each of the systems underwent the steepest descent energy minimization of 2500 cycles with an additional 5000 cycles following a conjugate gradient algorithm. Then the systems were equilibrated for 2 ns under NVT conditions, applying a positional restraint of 1 kcal/mol to keep the complex in place. Upon completion of equilibration, both the wild-type and mutant protein systems were subjected to 500 ns production molecular dynamics (MD) simulations under the NPT conditions. Four independent replicate simulations, each lasting 500 ns, were conducted for each PAH system, resulting in a cumulative simulation time of 2000 ns (2 μs) for the wild-type and mutant systems individually. Langevin dynamics was used to ensure a constant temperature at 303.15 K, and a friction coefficient (γ) of 1.0 ps–1 was used for the Langevin thermostat. A Monte Carlo barostat was used to ensure a constant pressure of 1.0 bar under the NPT condition. The cutoff for nonbonded atom interactions was set to 12 Å. All bonds involving hydrogen were constrained using the SHAKE algorithm and water was restrained using the SETTLE algorithm. , The integration of the dynamics was performed using a leapfrog algorithm in every 2 fs time step. To calculate the binding free energy between BH4 and the PAH protein, additional sets of molecular dynamics simulations were performed using the crystal structure of human phenylalanine hydroxylase in complex with the cofactor BH4 (PDB ID: 6HYC). For each of the wild-type and mutant systems (I65T, A202T, D282G, and R408W), 100 ns simulations were conducted following minimization and equilibration using the same procedure outlined here. For each system, at least three replicate 100 ns MD simulations were conducted, and their average values were reported. Similarly, MD simulations were also conducted for each of the wild-type and mutant system to understand the tetramerization domain (TD) and the regulatory domain (RD) by building simulation systems using the PDB 6HYC and PDB 5FII, respectively. Three 400 ns MD simulations were also conducted with the tetramerization domain at the C-terminus of PAH to understand how the dimer–dimer interaction is affected by the R408W mutation, and their average MM/GBSA binding free energy values are reported.
2.3. Binding Free Energy Calculations
The binding free energies between BH4 and the wild type and four mutant systems were performed using the Molecular Mechanics-Generalized Born Surface Area (MM/GBSA) method with the MMPBSA Python script inside of Ambertools20. The Ambertools20 ante-MMPBSA Python script was used to create the complex, receptor, and ligand parameter files from the solvated parameter files used in the MD simulations. The generalized Born method was set to igb = 5, along with a salt concentration set to 0.15 M. The dielectric constant of the solvent was set to 78.5, and the dielectric constant of the solute to 1.0. Both dielectric constants are Amber default and recommended values. Wang et al. investigated 21 protein–protein complexes to gain the performance of MM/GBSA in different conditions and suggested using 1.0 as a solute dielectric constant.
In MM/GBSA calculations, the binding free energy (ΔG bind) between the ligand and PAH was calculated as
| 1 |
where ΔE MM,gas, ΔG sol, and TΔS are the changes of the gas phase molecular mechanics energy, the solvation free energy, and the conformational entropy upon binding, respectively. The ΔE MM,gas is the sum of the internal energy (ΔE internal) arising from bond, angle, and dihedral interactions, nonbonded electrostatic energy (ΔE ELE), and van der Waals energy (ΔE VDW).
| 2 |
In MM/GBSA calculations, the ΔE int is canceled since the complex, receptor, and ligand parameter files are created from the same trajectory. The solvation free energy (ΔG sol) is calculated from the sum of the polar solvation energy (ΔG GB) using the generalized Born model, and the nonpolar energy is calculated based on the solvent accessible surface area (ΔG surf) according to the LCPO algorithm.
| 3 |
The conformational entropy change TΔS is usually computed by normal-mode analysis on a set of conformational snapshots taken from MD simulations. In this case, the contribution from the conformational entropy is neglected, as the inclusion of the entropy term may not improve the ranking of the binding affinities of BH4 with the wild type and the four mutant systems. The estimation of the binding free energy between two regulatory domains and between two tetramerization domains was carried out similarly, treating one domain as the ligand and the other as the receptor.
3. Results and Discussion
3.1. Impact of Pathogenic Mutations on the Protein Structure from SIFT, PolyPhen-2, PhD-SNP, and MutPred2
While I65T, A202T, D282G, and R408W variants of PAH are identified in PKU cases, their exact role in the disease remains unclear. ,, We aimed to reassess their disease association and to explore the reason for their association by analyzing their effects on protein structure and function with advanced machine learning and protein sequence-based computational tools. First, the protein sequences with the mutations were analyzed by the Sorting Intolerant From Tolerant (SIFT) program, which uses the physical properties of amino acids to classify amino acid substitutions affecting protein function. , SIFT performs PSI-BLAST to assess the degree of conservation of amino acid residues in the given sequences and returns scores between 0 and 1. SIFT scores less than 0.05 represent the mutations intolerable for the protein and can affect its function. Among the four mutations, the SIFT algorithm found I65T, D282G, and R408W to affect the structure of the protein with a tolerance score below 0.05 (Table ), indicating these mutations most likely affect the function of the protein.
1. Scores from SIFT and PolyPhen-2 with the Prediction of How the Variant Will Affect Protein Structure and Function.
| missense mutation | SIFT tolerance score | SIFT prediction | PolyPhen-2 score | PolyPhen-2 prediction |
|---|---|---|---|---|
| I65T | 0.01 | affect protein function | 1.00 | probably damaging |
| A202T | 0.08 | tolerated | 1.00 | probably damaging |
| D282G | 0.01 | affect protein function | 1.00 | probably damaging |
| R408W | 0.00 | affect protein function | 1.00 | probably damaging |
Next, PolyPhen-2 was used to corroborate the results generated by SIFT. PolyPhen-2 is a machine learning-based tool that makes predictions based on structural and phylogenic information about the protein with a score between 0 and 1. , The PolyPhen-2 score (PSIC) < 0.15 indicates the mutation is benign, between 0.15 and 1 is possibly damaging, and between 0.85 and 1 represents probably damaging to the protein structure and function. The four mutations I65T, A202T, D282G, and R408W obtained a PolyPhen score 1.00, indicating all these mutations are strongly associated to affect the structure and function of PAH protein (Table ).
Both SIFT and PolyPhen-2 indicate that these mutations are deleterious and damaging for PAH, but their role in causing the disease in humans needs further validation. To establish their disease association, we used PhD-SNP, an SVM-based classifier, to predict if these specific nsSNPs are neutral or disease-causing. , Our analysis with PhD-SNP predicted all the mutations to be disease-causing (Table ), suggesting their connection to PKU. To understand the possible underlying mechanism for the loss of protein function and disease cause, we employed MutPred2, a machine learning software tool that integrates genetic and molecular data to predict the pathogenicity with the underlying mechanism after the substitutions of a residue within a protein sequence. MutPred2 provides two outputs: the general score (g score) indicating the likelihood that the nsSNP is disease-related and the property score (p score) indicating whether specific structural or functional properties are disrupted by the mutation. Based on the g and p scores, the prediction hypotheses by Mutpred2 are considered actionable (g > 0.5 and p < 0.05), confident (g > 0.75 and p < 0.05), and very confident (g > 0.75 and p < 0.01). , In our analysis by MutPred2, all four variants were found to have confident reliability of structural disruption (Table ). Mutant I65T has g and p scores of 0.859 and 0.0095, respectively, which suggests a gain of intrinsic disorder for this mutation with a very confident reliability. In addition, MutPred2 elicited altered stability with a confident hypothesis (g score = 0.859 and p score = 0.006), a loss of strand (g score = 0.859 and p score = 0.04), and a loss of N-linked glycosylation at N61 (g score = 0.859 and p score = 0.04) for the same mutation (I65T). The R408W mutation is predicted to disrupt the protein with gain of a strand mechanism with a very confident hypothesis (g score = 0.792 and p score = 0.0033). The R408W is also predicted to disrupt the protein structure with several other mechanisms such as altered transmembrane protein (P = 0.0015), altered DNA binding (P = 0.02), and loss of relative solvent accessibility (P = 0.01).
2. PhD-SNP and MutPred2 Prediction on the Disease Association of the Studied Mutations in PAH and the Protein Structure Disruption Mechanism with the Confidence Scores.
| missense mutation | PhD-SNP | g score | p score | mechanism disrupted | probability | confidence |
|---|---|---|---|---|---|---|
| I65T | disease | 0.859 | 0.0095 | gain of intrinsic disorder | 0.39 | very confident |
| I65T | disease | 0.859 | 0.006 | altered stability | 0.29 | very confident |
| I65T | disease | 0.859 | 0.04 | loss of strand | 0.26 | confident |
| I65T | disease | 0.859 | 0.04 | loss of N-linked glycosylation at N61 | 0.01 | confident |
| A202T | disease | 0.834 | 0.04 | altered metal binding | 0.25 | confident |
| A202T | disease | 0.834 | 0.04 | gain of relative solvent accessibility | 0.25 | confident |
| D282G | disease | 0.867 | 0.03 | loss of helix | 0.28 | confident |
| R408W | disease | 0.792 | 0.02 | altered ordered interface | 0.30 | confident |
| R408W | disease | 0.792 | 0.01 | loss of relative solvent accessibility | 0.29 | confident |
| R408W | disease | 0.792 | 0.0033 | gain of strand | 0.29 | very confident |
| R408W | disease | 0.792 | 0.0015 | altered transmembrane protein | 0.24 | very confident |
| R408W | disease | 0.792 | 0.02 | altered DNA binding | 0.20 | confident |
MutPred2 predicts the I65T mutation to gain intrinsic disorder, while R408W results in the gain of a strand. Both of these mechanistic disruptions are supported with high confidence g and p scores. Likewise, the A202T mutation is predicted to affect metal binding and increase relative solvent accessibility, whereas the D282G mutation may disrupt the protein structure by causing the loss of a helical region. The American College of Medical Genetics and Genomics recommends using multiple computational tools to assess the impact of mutations on protein function. Employing various tools allows for a broader range of features to be considered and helps cross-validate predictions, enhancing their accuracy and reliability. Using four different tools enabled us to thoroughly evaluate the detrimental effects of the mutations on protein structure, function, and disease development and to understand the mechanisms by which these mutations disrupt protein function.
3.2. Molecular Dynamics Simulation
3.2.1. All Mutations Impact Protein Dynamics
The root-mean-square deviation (RMSD), root-mean-square fluctuations (RMSF), and radius of gyration (R g) values obtained from all four replicate molecular dynamics (MD) simulations for wild-type and I65T, A202T, D282G, and R408W mutant proteins are shown in the Supporting Information, Figures S1–S3. The average RMSD values obtained from all four MD simulations are shown in Table S1. The RMSD for the wild-type protein fluctuates between 4.20 and 5.41 Å, with an average of 4.94 Å. On the I65T, A202T, D282G, and R408W mutant structures, the average RMSD values were 6.94, 6.45, 7.96, and 6.87 Å, respectively. Among the four simulations, the least RMSD values were 4.20, 6.03, 6.04, 7.07, and 6.58 Å for the wild type, I65T, A202T, D282G, and R408W, respectively. Figure presents the data obtained from MD simulation with the lowest RMSD over time among the four independent simulations for the wild type, I65T, A202T, D282G, and R408W. All the four studied mutant structures showed greater average RMSD values than the wild-type structure. The wild-type protein stays relatively stable throughout the simulation and all the mutant structures showed higher deviation (more than 2 to 3 Å) compared to the wild type. The D282G mutation exhibits the greatest deviation (7.07 Å) among the four mutant structures (Figure A). The elevated RMSD observed for the PAH variants is consistent with findings from MutPred2 and PolyPhen-2, which also identified these mutations as significantly impacting the structural integrity of PAH in comparison with the wild-type protein.
2.
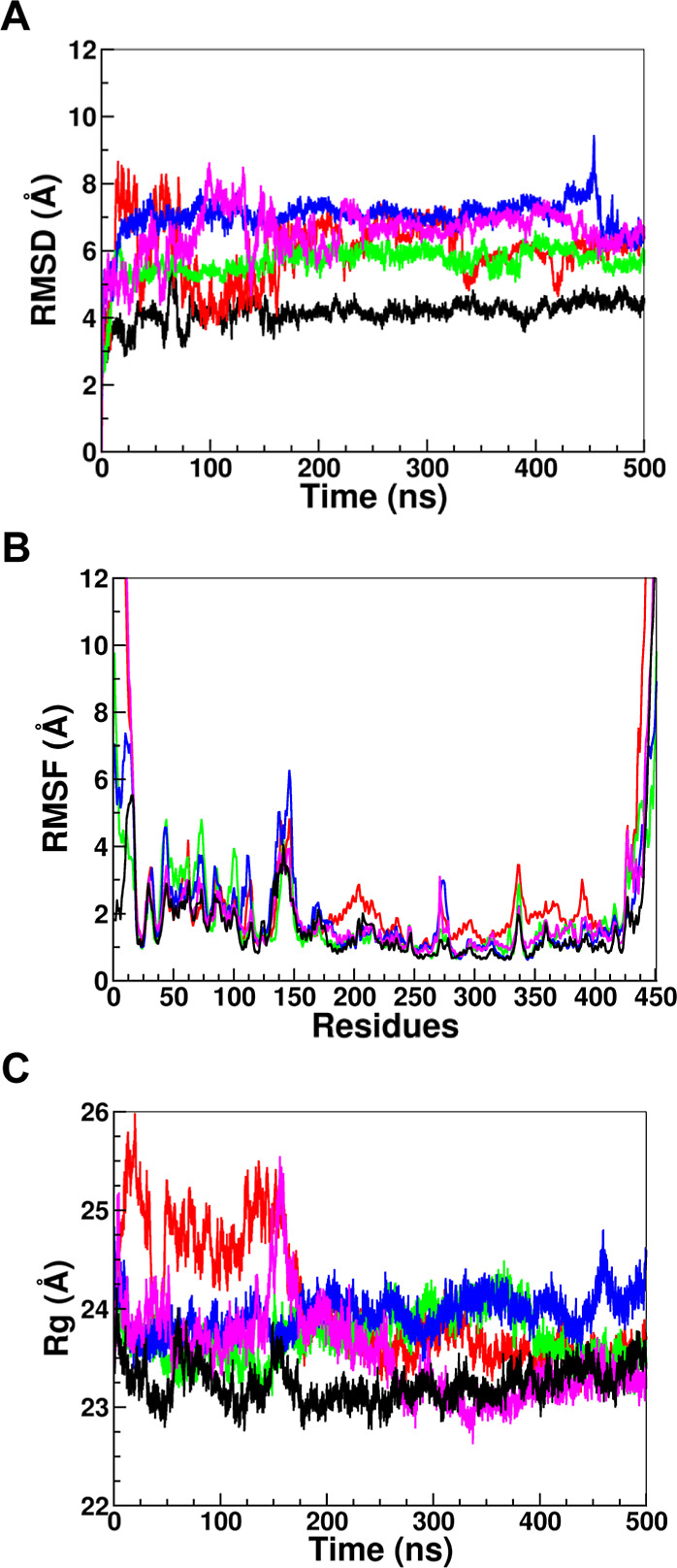
(A) Root-mean-square deviation (RMSD), (B) root-mean-square fluctuation (RMSF), and (C) radius of gyration (R g) values for the wild type and PAH variants I65T, A202T, D282G, and R408W, represented in black, red, green, blue, and magenta, respectively.
In addition, root-mean-square fluctuation (RMSF) was calculated to analyze the dynamic behavior of each residue of the wild-type PAH as well as of all of the mutant protein structures. Comparison of the RMSF plots of the wild-type and mutant proteins allows the identification of residues and regions that are more significantly impacted by the mutations. Overall, increased residue fluctuations have been observed in the mutant protein structures compared to the wild type, indicating a reduction in stability for the mutant configurations. Several residues from the N-terminal and C-terminal regions displayed higher fluctuations in all variants, including the wild type, suggesting that these regions are highly flexible. This is consistent with the fact that these sites are involved in dimerization and tetramerization, with other PAH chains that form the tetramer. Since the catalytic domain (residues 143–410) plays a key role in the conversion of l-Phe to l-Tyr, attention was directed toward this region. Three primary fluctuation peaks were observed: between residues 140 and 150, 265 and 275, and 320 and 340. The D282G and R408W variants exhibited higher fluctuations in residues 140 to 150 and 265 to 275, while the I65T and A202T variants showed increased fluctuations in residues 320 to 340. The D282G mutation impacts residues 140–150 and 265–275, which lie within loop regions adjacent to residue 282. The substitution of aspartic acid with glycine at position 282 disrupts a key hydrogen bond with arginine at position 270 (Supporting Information Table S2). In the R408W mutation, hydrogen bonds originally formed between R408 and residues L308 and L311 are lost upon substitution with tryptophan, resulting in increased residue fluctuations in the 320–340 region. Similarly, the A202T mutation introduces a bulkier threonine at position 202, which destabilizes nearby residues and contributes to heightened fluctuations within the same 320–340 region.
Furthermore, the radius of gyration (R g) was calculated for each of the wild-type and mutant protein structures. The radius of gyration is the mass weighted root-mean-square distance of each of the atoms from the center of the mass, reflecting its degree of folding and overall structural stability. The wild-type proteins showed stable R g throughout the whole simulation period with an average R g of approximately 23 Å, while all four mutant proteins had an average R g exceeding 24 Å.
Hydrogen bonds between different protein residues, known as intramolecular hydrogen bonds, and those formed between protein residues and surrounding water molecules, termed intermolecular hydrogen bonds, play a crucial role in maintaining the structural stability of proteins. The total number of intramolecular hydrogen bonds within the residues of wild-type and PAH variants ranges from 180 to 229, suggesting that the overall hydrogen bond count may not be a critical factor in the onset of PKU (see Supporting Information Figure S4). However, notable alterations were found in specific intramolecular hydrogen bonds involving various residues before and after mutation as well as in the intermolecular hydrogen bonds formed between the mutated sites and the surrounding water molecules (see Sections and 3.2.5).
3.2.2. Principal Component Analysis Reveals Significant Correlated Movements for I65T and R408W
We further incorporated principal component analysis (PCA) to calculate eigenvectors and the respective eigenvalues along the two principal components (Figure ). In PCA, the number of dimensions needed to describe protein dynamics is reduced by using a covariance matrix constructed from Cartesian coordinates that define atomic displacements in each conformation comprising the trajectories. In this calculation, the root-mean-square atomic displacement of the coordinates was fit to the average structure obtained from the trajectory to generate the covariance matrix, which was then diagonalized, and the fit coordinates were saved along PC-1 and PC-2. The displacement of each of the Cα atoms along the eigenvectors helps distinguish the concerted motions of the protein in each direction. For the wild-type protein, movements were confined between −50 and 100 along PC-1 and between −60 and 110 along PC-2 (Figure A). In contrast, the I65T mutant exhibited a broader range of movement, from −90 to 200 along PC-1 and from −80 to 100 along PC-2, indicating greater flexibility in both directions (Figure B). The R408W mutant also demonstrated a wider range of movement along PC-1 and PC-2 compared to the wild-type protein structure, from −80 to 120 along PC-1 and from −120 to 100 along PC-2 (Figure E). These wider ranges of movement due to I65T and R408W mutations suggest potential destabilization of the protein structure due to these mutations. The A202T and D282G variant structures showed less mobility compared with the wild-type structure. For A202T, the movement was confined between −60 and 70 along PC-1 and between −50 and 50 along PC-2. The D282G mutant protein structure showed movement between −90 and 40 along PC-1 and between −40 and 90 along PC-2. These relatively moderate conformational shifts indicate that the A202T and D282G mutations are unlikely to cause major disruptions in the overall protein folding.
3.

Projection of motion along the first two principal component (PC) eigenvectors for Cα atoms of the wild type and variants of PAH, (A) wild-type PAH shown in black, (B) I65T variant in red, (C) A202T in green, (D) D282G in blue, (E) R408W in magenta, and (F) wild type and all variants overlaid.
3.2.3. Regions Exhibiting Extensive Changes in the Protein Structure
Some regions of the protein structure may experience structural changes greater than others, potentially affecting the enzymatic activity of PAH and impairing its ability to convert phenylalanine to tyrosine. Five representative frames were extracted at 125 ns intervals throughout the 500 ns molecular dynamics (MD) simulation (Figure ), beginning at 0 ns and continuing through 125, 250, 375, and 500 ns. The red structures indicate the initial stage, white structures represent intermediate conformations, and blue structures correspond to the final phase of the simulation. Over the course of the simulation, the wild-type protein structure exhibited lower fluctuations (Figure A), indicative of its good structural stability. In contrast, the mutant protein structures displayed greater flexibility throughout the simulation with destabilization varying across different regions of the protein. The tetramerization and dimerization and regulatory domains exhibited higher fluctuations in all four mutant proteins. Given the catalytic domain’s essential role in converting l-Phe to l-Tyr, focus is placed on residues near the BH4 and l-Phe binding sites, particularly those spanning residues 265 to 275 and 320 to 340. Notably, residues 265 to 275 exhibited greater fluctuations in the D282G and R408W variants (Figure D,E), whereas residues 320 to 340 showed increased fluctuations in the I65T and A202T variants (Figure B,C). These findings suggest that different mutations impact the catalytic mechanism of PAH in distinct ways, potentially altering the efficiency of the conversion of l-Phe to l-Tyr.
4.

Five superimposed structures selected from the 500 ns MD simulation of each system, (A) wild type, (B) I65T, (C) A202T, (D) D282G, and (E) R408W, with red, white, and blue colors representing the initial, middle, and final structures, respectively. The boxes represent the highest fluctuating residues, 265 to 275 (yellow box) in D282G and R408W, and 320 to 340 (orange box) in I65T and A202T, of the catalytic domain of PAH.
3.2.4. Analysis of Intramolecular Hydrogen Bonding to Investigate the Impact of Mutations
Intramolecular hydrogen bonds (H bonds) involving different residues before and after mutation were analyzed to better understand how mutations influence the structure and dynamics of PAH. H bonds are vital for maintaining a protein’s structural and functional integrity, as well as ensuring proper folding. The intramolecular hydrogen bonds were calculated between ILE65, ALA202, ASP282, and ARG408 and all residues within the wild-type protein, as well as between the mutant residues THR65, THR202, GLY282, and TRP408 and all residues within the mutant protein structures (Figure and Supporting Information Table S2). In the wild-type protein, the ILE65 residue forms an H bond with LEU62 at a frequency of 12%. For the I65T variant, T65 establishes strong H bonds with LEU62 and THR81, with significantly higher frequencies of 62.1% and 47.4%, respectively. For the wild type, ALA202 forms a H bond with TYR198, but when it was mutated to threonine, it reduced H bonding with surrounding residues in addition to hydrophobic interaction among ALA202, TYR198, TYR206, and LEU348. Losses of H bond after mutation can increase structural flexibility and hamper the appropriate folding of PAH protein. In the case of wild-type PAH, aspartic acid in position 282 (D282) forms a complex hydrogen bond network with multiple surrounding residues such as ARG270, GLU286, and HIS285. The ARG270NH2/ASP282OD1, ARG270HE1/ASP282OD2, GLU286HN/ASP282°, and HIS285N/ASP282OD1 pairs were found to form H bonds with 100.0%, 96.4%, 68.1%, and 66.5% occupancy, respectively. In the D282G variant, the mutation of ASP282 to GLY282 weakens the hydrogen bond network, resulting in hydrogen bond occupancies of 57.3% with HIS285HN and 19.1% with GLU286HN. The reduction in hydrogen bonding may be due to glycine’s smaller size relative to aspartic acid, as well as the absence of aspartic acid’s additional carboxyl group, which facilitates strong hydrogen bonding with neighboring residues. Next, the hydrogen bonding frequency for wild-type arginine at position 408 was calculated and compared to the H-bond frequencies after mutation to tryptophan. ARG408 was observed to form very strong H bonds with LEU311°, LEU308°, and PHE410HN, with occupancy values of 98.8%, 97.9%, and 19.7%, respectively. In this position, the mutated tryptophan formed H bonds with ALA403°, ASN426ND2, and ILE406° with 91.7%, 46.6%, and 14.9%, respectively, which suggests an alteration and shifting of the H-bond network toward the N-terminus of the PAH protein that can lead to conformational changes of the protein structure. These findings support the predictions made by SIFT, PolyPhen2, PhD-SNP, and MutPred2 that these mutations are highly deleterious to the PAH protein structure and may impair or inactivate its function.
5.

Intramolecular hydrogen bond of the wild type and mutant residues with the neighboring amino acids; (A), (C), (E), and (G) represent the H bond of wild-type residues, I65, A202, D282, and R408, with the surrounding residues, while (B), (D), (F), and (H) show the H bonds of mutant residues, T65, T202, G282, and W408, with the surrounding residues.
3.2.5. Analysis of Intermolecular Hydrogen Bonding with the Solvent Molecules to Investigate the Impact of Mutations
The intermolecular H bonds between the residues in the selected mutation position and the water molecules before and after mutations were calculated to explore the impact of solvent on the PAH structure and dynamics. The intermolecular hydrogen bonds were analyzed between wild-type residues ILE65, ALA202, ASP282, and ARG408, and water molecules, as well as between mutations THR65, THR202, GLY282, and TRP408, and water molecules (Supporting Information Table S3). The wild-type ILE65 formed practically no hydrogen bonds with water as the highest H bonds are with ILE65N/WAT17586O and ILE65N/WAT22659O with H-bond occupancies of only 0.2% and 0.1% as well as the H-bond distance and angle for ILE65N/WAT17586O are 3.0 Å and 160.6°, and those for ILE65N/WAT22659O are 3.0 Å and 153.8°. On the contrary, THR65 mutation increases H-bond occupancies to 17.9%, 8.4%, 8.3%, and 8.2% for THR65OG1/WAT15473O, THR65OG1/WAT15473H2, THR65OG1/WAT15473H1, and THR65OG1/WAT20996O, respectively. This change in H bonding significantly impacts the structure and dynamics in the I65T variant resulting in higher RMSD and R g values compared to the wild-type protein (Figure A,C). For both wild-type as well as A202T and D282G variants, intermolecular hydrogen bonding of water with residues 202 and 282 was very low with maximum H-bond occupancies ranging from 1% to 3.7%. Substitution of ALA202 with TYR202 and ASP282 with GLY282 was not found to alter the hydrogen bond formation with water molecules significantly. However, for R408W, the ARG408 forms six H bonds with occupancies ranging from 5.0% to 11.6%, and substitution of ARG408 to TRP408 decreased the intermolecular H bonding with water significantly, with the highest H-bond occupancy of only 0.5%. Since tryptophan (TRP) is a nonpolar amino acid, its hydrophobic nature influences the structural changes in the R408W variant due to hydrophobic interactions.
3.2.6. Binding Free Energy Confirms Decreased Binding of I65T and R408W Variants with BH4
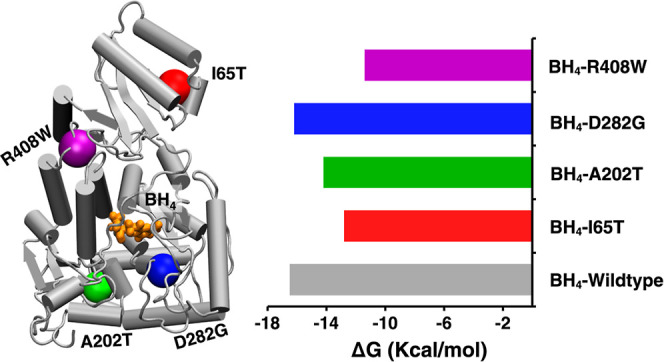
Tetrahydrobiopterin (BH4) is a natural cofactor for PAH protein, which is necessary for stabilization of the protein structure by ensuring appropriate folding. To calculate the binding free energy of the wild-type and mutant PAH protein–BH4 complexes, the MM/GBSA (Molecular Mechanics-Generalized Born Surface Area) method was used. The calculated binding free energy for the wild type, I65T, A202T, D282G, and R408W with BH4 was found to be −16.5 kcal/mol, −12.8 kcal/mol, −14.2 kcal/mol, −16.2 kcal/mol, and −11.4 kcal/mol, respectively (Table and Figure A). The I65T and R408W variants showed a significantly reduced binding affinity to BH4. The reason for the reduction in binding free energy could be explained by the contribution of van der Waals (ΔE VDW) and electrostatic (ΔE ELE) interaction energy terms. ΔE VDW decreased from −27.9 kcal/mol in the wild type to −25.2 kcal/mol in I65T and −26.8 kcal/mol in R408W. Similarly, ΔE ELE is reduced by about 50% for R408W compared to the wild-type protein with −16.4 kcal/mol in the wild type vs −8.4 kcal/mol in R408W. The ΔG GB values for R408W are also decreased by about 4.1 kcal/mol. Additionally, the ΔG surf values did not show a significant change between the wild type and mutant protein variants.
3. Binding Free Energies of BH4 Bound to the Wild-Type Protein and I65T, A202T, D282G, and R408W Variants Obtained from the MM/GBSA Calculation .
| system | ΔE VDW | ΔE ELE | ΔG GB | ΔG surf | ΔG bind (kcal/mol) |
|---|---|---|---|---|---|
| BH4–wild type | –27.9 | –16.4 | 32.1 | –4.3 | –16.5 ± 0.5 |
| BH4–I65T | –25.2 | –16.0 | 32.4 | –4.0 | –12.8 ± 0.4 |
| BH4–A202T | –25.7 | –9.3 | 24.8 | –4.0 | –14.2 ± 0.5 |
| BH4–D282G | –26.8 | –14.4 | 29.1 | –4.1 | –16.2 ± 0.4 |
| BH4–R408W | –26.8 | –8.4 | 28.0 | –4.2 | –11.4 ± 0.8 |
Binding free energies are presented as the mean ± standard error of mean. All units are in kcal/mol.
6.

(A) Binding free energy comparison of BH4 with the PAH wild type and its variants (I65T, A202T, D282G, and R408W); (B) pairwise energy decomposition analysis illustrating the contribution of individual residues to BH4 binding in wild-type PAH and its variants, I65T, A202T, D282G, and R408W; black, red, green, blue, and magenta energy bars are for the wild type, I65T, A202T, D282G, and R408W, respectively; (C) hydrogen bonding of BH4 with selected residues in wild-type PAH.
Currently, Kuvan (sapropterin dihydrochloride) is the only FDA-approved medication for treating PKU. Kuvan is a synthetic form of BH4 that binds to PAH and in many cases restores the ability to metabolize phenylalanine (Phe) and reduce Phe accumulation in the body. However, not all PKU patients respond to Kuvan treatment. The I65T and R408W mutations in PAH that have shown reduced binding with BH4 might adversely impact the treatment outcomes with synthetic BH4 supplements like Kuvan.
3.2.7. Pairwise Decomposition Analysis Reveals Key Residues of PAH Involved in BH4 Binding
Superimposed structures of the active site residues at the BH4 binding site for the wild type and I65T reveal minimal conformational changes due to the mutation, except in the loop regions (Supporting Information, Figure S5). Very little conformational changes in the active site were also observed in the A202T, D282G, and R408W variants. To further understand the effect of mutation, a pairwise energy decomposition analysis using MM/GBSA was conducted to evaluate the contribution of individual residues to the binding affinity of BH4 with both the wild type and PAH variants. This analysis revealed that residues SER23, GLY247, LEU248, LEU249, SER251, PHE254, LEU255, and TYR323 (see Figure C) contribute more significantly to the binding energy compared to other residues. Among these residues, each of SER23, LEU248, LEU249, SER251, and PHE254 was found to contribute more than 2 kcal/mol of free energy (see Supporting Information in Table S4).
PHE254 in both wild-type and mutant proteins exhibits strong binding to BH4, with binding affinity values ranging from −4.6 to −4.7 kcal/mol (Figure B). This strong interaction is attributed to the formation of a stable intermolecular hydrogen bond between PHE254 and BH4 (Figure C). All variants also show the same interaction of PHE254 with BH4 (see Supporting Information, Figure S6). SER251 contributes to −4.1 kcal/mol binding affinity with the wild-type protein, but the interaction energy decreased to −3.8 kcal/mol for A202T, I65T, and R408W. SER251 also makes a hydrogen bond with BH4, but the H-bond occupancy slightly decreased for A202T, I65T, and R408W. For LEU249, the binding interaction energy decreased in the I65T, D282G, and R408W variants (−3.0, −3.1, and −3.2 kcal/mol, respectively) compared to the wild-type protein (−3.3 kcal/mol). Similarly, for GLY247, the binding interaction energy decreased for all variants compared to that of the wild-type protein (Figure B). Certain residues, including SER23, LEU248, LEU255, and TYR325, contribute to BH4 binding with PAH. However, their binding affinity remains largely unchanged or exhibits only minimal variation upon mutation in the I65T, A202T, D282G, and R408W variants.
3.2.8. The R408W Can Disrupt PAH Tetramerization
Human PAH is a tetrameric protein with each subunit comprising a regulatory domain (residues 1–110), a catalytic domain (residues 111–410), and a tetramerization domain (residues 411–452). ARG408 is located at the end of the catalytic domain within the hinge loop, which connects the tetramerization domain (TD) to the core of the PAH monomer (Figure A). The side chain of ARG408 is buried and forms a hydrogen bond with the main chain of the PAH monomer, likely facilitating oligomerization by anchoring the tetramerization arm to the catalytic domain. To investigate the impact of the mutation on PAH tetramerization, a complex consisting of two PAH subunits, including a portion of the catalytic domain and the tetramerization domain (residues 390–452), was constructed for both the wild type and R408W variant, as illustrated in Figure A.
7.
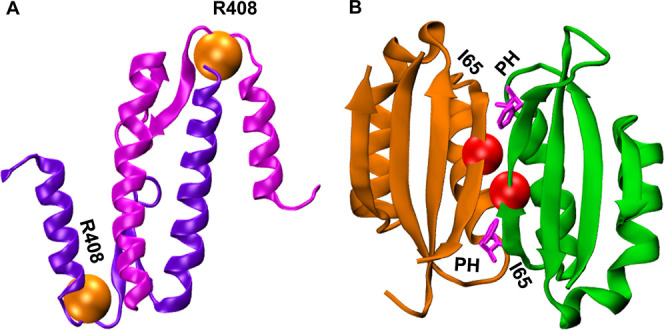
(A) Phenylalanine hydroxylase (PAH) N-terminal regulatory domain homodimer showing the ligand PHE (magenta) binding and the site for I65T mutation (red). (B) Displaying the R408W mutation site (orange) close to the tetramerization domain of two subunits of PAH protein.
The binding free energy between the two subunits was found to be −90.7 kcal/mol for the wild-type protein, which decreased significantly to −62.2 kcal/mol for the R408W variant when only one subunit is mutated from ARG408 to TRP408. When ARG408 in both subunits were mutated to TRP408, the binding free energy between the subunits significantly decreased to −64.0 kcal/mol (Table ). This finding indicates that the R408W mutation weakens interactions among the tetramerization domains, potentially disrupting the PAH tetramerization. These findings align with a previous enzyme kinetic study, which also demonstrated that the R408W mutation can significantly disrupt the protein’s oligomeric state and potentially cause inactivation.
4. Binding Free Energies between two Subunits of PAH Involved in Tetramerization for the Wild Type and R408W .
| system | ΔE VDW | ΔE ELE | ΔG GB | ΔG surf | ΔG bind (kcal/mol) |
|---|---|---|---|---|---|
| wild type–TD | –110.7 | –386.1 | 424.4 | –18.4 | –90.7 ± 2.1 |
| R408W–TD (single-chain mutant) | –83.9 | –198.6 | 234.0 | –13.8 | –62.2 ± 1.7 |
| R408W–TD (double-chain mutant) | –105.6 | –192.5 | 250.9 | –16.8 | –64.0 ± 4.1 |
| wild type–TD (tetramer) | –224.6 | –457.0 | 575.0 | –33.5 | –140.1 ± 1.6 |
| R408W–TD (tetramer with mutation) | –191.6 | –447.8 | 538.3 | –29.3 | –130.3 ± 1.6 |
Binding free energies are presented as mean ± standard error of mean. All units are in kcal/mol.
The reason for the reduction in binding free energy for R408W could be explained by the contribution of E VDW and ΔE ELE interaction energy terms. ΔE VDW decreased from −110.7 kcal/mol in the wild type to −83.9 kcal/mol when only one subunit is mutated from ARG408 and to −105.6 kcal/mol when both the subunits are mutated from ARG408 to TRP408. Similarly, ΔE ELE decreased from −386.1 kcal/mol to −198.6 and −192.5 kcal/mol when ARG408 is mutated to TRP408 in one and two subunits, respectively. ΔG GB is also found to change significantly for the wild type and R408W variant. The ΔG surf values did not show a significant change between the wild-type and mutant protein variants.
The tetramerization domain at the C-terminus of PAH facilitates the initial dimerization of monomers followed by the association of two dimers to form a tetramer. The R408W mutation in this region may disrupt tetramer formation by altering the dimer–dimer interactions. To investigate this, we modeled two dimers (a tetramer) of the C-terminal tetramerization domain using the PAH crystal structure (PDB ID: 6HYC, Figure A). Three replicate molecular dynamics simulations (each 400 ns) were performed for both the wild-type and R408W mutant systems, where the mutation was introduced into each chain. The calculated average MM/GBSA binding free energies were found to be −140.06 ± 1.6 kcal/mol for the wild-type and −130.3 ± 1.6 kcal/mol for the mutant system (see Supporting Information, Table S5). Notably, the van der Waals interaction energy (ΔE VDW) was significantly weakened in the mutant (−191.6 kcal/mol) compared to the wild type (−224.6 kcal/mol), and the polar solvation energy (ΔG GB) was substantially reduced (538.3 kcal/mol vs. 575.0 kcal/mol). These results suggest that the R408W mutation impairs both dimerization and tetramerization of the PAH protein, potentially disrupting the protein structure and reducing binding affinity among its functional subunits, thereby impairing overall protein function.
PAH undergoes ligand-induced homodimerization by binding to Phe in the regulatory domains (RD), a process crucial for enzyme activation through an allosteric mechanism. The ILE65 residue is located within this RD, prompting an investigation into whether the I65T mutation destabilizes dimerization and affects binding of Phe with PAH (Figure B). A previous study demonstrated that a different mutation at the same position, the I65N variant, reduced the binding affinity between the two regulatory domains, impairing homodimerization. To assess this, we performed molecular dynamics simulations using a complex of two regulatory domains from wild-type PAH and the I65T variant, based on PDB ID: 5FII. The calculated binding free energies for the two regulatory domains in the wild type and I65T remained comparable (Table ). While ΔE VDW and ΔG surf showed minimal variation, ΔE ELE and ΔG GB differed between the wild type and I65T; however, the overall binding free energies remained within 3 kcal/mol.
5. Binding Free Energies between two Subunits of PAH Involved in Dimerization for the Wild Type and I65T .
| binding interaction between regulatory domains | |||||
|---|---|---|---|---|---|
| system | ΔE VDW | ΔE ELE | ΔG GB | ΔG surf | ΔG bind (kcal/mol) |
| wild type–RD | –81.9 | –192.7 | 223.3 | –12.8 | –64.1 ± 1.1 |
| I65T–RD (single-chain mutant) | –85.3 | –176.8 | 208.1 | –13.0 | –67.1 ± 1.1 |
| I65T–RD (double-chain mutant) | –82.0 | –169.0 | 198.8 | –13.1 | –65.4 ± 1.0 |
Binding free energies are presented as mean ± standard error of mean. All units are in kcal/mol.
The MM/GBSA binding free energies of phenylalanine (Phe) with the RD domains for both the wild type and I65T variants remained comparable (Table ). While the interaction energy slightly increased following the mutation, the difference was not significant, with values of −43.5 kcal/mol for the wild type and −48.6 kcal/mol for I65T. Additionally, ΔE VDW, ΔG surf, ΔE ELE, and ΔG GB remained similar between the wild type and I65T variants. This suggests that the I65T mutation does not substantially affect the ligand-induced dimerization of the PAH subunits.
6. MM/GBSA Binding Free Energies of Phenylalanine (PHE) with the Regulatory Domain of the Wild Type and I65T .
| PHE binding in regulatory domain | |||||
|---|---|---|---|---|---|
| system | ΔE VDW | ΔE ELE | ΔG GB | ΔG surf | ΔG bind (kcal/mol) |
| wild type–RD | –37.4 | –157.3 | 158.5 | –7.3 | –43.5 ± 0.8 |
| I65T–RD | –38.1 | –162.2 | 159.0 | –7.4 | –48.6 ± 1.0 |
Binding free energies are presented in kcal/mol as mean ± standard error of mean.
4. Conclusions
Computational analyses are instrumental in identifying structural features and elucidating the molecular mechanisms underlying protein destabilization caused by mutations. This study focuses on human phenylalanine hydroxylase (PAH), an essential enzyme responsible for converting l-phenylalanine to l-tyrosine. Impaired PAH function leads to the accumulation of phenylalanine, resulting in phenylketonuria (PKU), a metabolic disorder characterized by cognitive impairment, behavioral abnormalities, epilepsy, microcephaly, and stunted growth. Investigating the pathogenicity of nonsynonymous single nucleotide polymorphisms (nsSNPs) and their effects on protein structure and function is crucial for identifying therapeutic targets and developing effective treatments. To assess the impact of pathogenic mutations, we employed advanced computational approaches to analyze the structural and functional consequences of four PAH variants: I65T, A202T, D282G, and R408W. Pathogenicity predictions from multiple in silico methods consistently indicated a strong association between these variants and structural alterations in PAH. Molecular dynamics simulations were conducted to further investigate the effects of these mutations on protein stability and BH4 binding affinity. Structural analyses, including root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), radius of gyration (R g), and principal component analysis (PCA), revealed that these mutations contribute to destabilization of the protein. Furthermore, MM/GBSA binding free energy calculations demonstrated a reduction in PAH–BH4 binding affinity for the I65T and R408W variants, which may affect the efficacy of PKU treatment with sapropterin dihydrochloride. Additionally, analysis of the R408W mutation indicated a significant disruption of tetramerization domain interactions, suggesting an adverse effect on PAH’s quaternary structure. Our findings are in alignment with an experimental kinetic study which also showed R408W to significantly disrupt the protein’s oligomeric state and potentially cause PAH inactivation. Overall, this computational study provides mechanistic insights into the structural and functional consequences of PKU-associated mutations in PAH. These findings contribute to a better understanding of disease pathology and may aid in the development of targeted therapeutic strategies.
Supplementary Material
Acknowledgments
This work used Anvil supercomputers at Purdue University through allocation CHE210078 to S.M.I. from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296. This research has been funded by COBRE (P20GM145765), RCMI (U54MD015959), DE-INBRE (P20GM103446) funded by NIH/NIGMS, and CREST (2431276) funded by NSF Division of Chemistry (MPS/CHE).
All data is available upon reasonable request. MM/GBSA and pairwise energy decomposition analyses were performed using AmberTools23 with academic license. MD simulations were performed using Amber20 on the Anvil supercomputer at Purdue University through XSEDE support. Xmgrace (v 5.1.25) was used to generate data plots. VMD (v 1.9.4a57) was used to generate molecular graphics and is available at https://www.ks.uiuc.edu/Research/vmd/.
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.5c01993.
RMSD plots and average RMSD values for the wild-type and mutant protein systems across four replicate simulations; comparative analyses of RMSF, radius of gyration (R g), and total hydrogen bond formation for the wild-type and mutant systems, from four replicate simulations; detailed information on intramolecular and intermolecular hydrogen bond formation; binding energy values from pairwise decomposition analysis highlighting key residues involved in BH4 binding; and visual representations of the key residues and their interaction modes with BH4 (PDF)
S.M.I. designed the study and wrote the manuscript with input from all authors. J.S. and M.M.H. also wrote and edited the paper and K.S. edited the paper. J.S., M.M.H., and K.S. conducted analyses. S.M.I. secured funding and supervised the work.
The authors declare no competing financial interest.
References
- Blau N., Van Spronsen F. J., Levy H. L.. Phenylketonuria. Lancet. 2010;376(9750):1417–1427. doi: 10.1016/S0140-6736(10)60961-0. [DOI] [PubMed] [Google Scholar]
- Mitchell J. J., Trakadis Y. J., Scriver C. R.. Phenylalanine Hydroxylase Deficiency. Genet Med. 2011;13(8):607–617. doi: 10.1097/GIM.0b013e3182141b48. [DOI] [PubMed] [Google Scholar]
- Strisciuglio P., Concolino D.. New Strategies for the Treatment of Phenylketonuria (PKU) Metabolites. 2014;4:1007–1017. doi: 10.3390/metabo4041007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumaily K. M., Mujamammi A. H. P.. A New Look at an Old Topic, Advances in Laboratory Diagnosis, and Therapeutic Strategies. Int. J. Health Sci. (Qassim) 2017;11(5):63. [PMC free article] [PubMed] [Google Scholar]
- Hata, I. ; Yuasa, M. ; Isozaki, Y. . In Phenylketonuria BT - Human Pathobiochemistry: From Clinical Studies to Molecular Mechanisms; Oohashi, T. , Tsukahara, H. , Ramirez, F. , Barber, C. L. , Otsuka, F. , Eds.; Springer Singapore: Singapore, 2019; pp 101–110. 10.1007/978-981-13-2977-7_10. [DOI] [Google Scholar]
- Flydal M. I., Alcorlo-Pagés M., Johannessen F. G., Martínez-Caballero S., Skjærven L., Fernandez-Leiro R., Martinez A., Hermoso J. A.. Structure of Full-Length Human Phenylalanine Hydroxylase in Complex with Tetrahydrobiopterin. Proc. Natl. Acad. Sci. U.S.A. 2019;116:11229–11234. doi: 10.1073/pnas.1902639116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlandsen H., Pey A. L., Gámez A., Pérez B., Desviat L. R., Aguado C., Koch R., Surendran S., Tyring S., Matalon R., Scriver C. R., Ugarte M., Martínez A., Stevens R. C.. Correction of Kinetic and Stability Defects by Tetrahydrobiopterin in Phenylketonuria Patients with Certain Phenylalanine Hydroxylase Mutations. Proc. Natl. Acad. Sci. U.S.A. 2004;101(48):16903–16908. doi: 10.1073/pnas.0407256101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Døskeland A. P., Martinez A., Knappskog P. M., Flatmark T.. Phosphorylation of Recombinant Human Phenylalanine Hydroxylase: Effect on Catalytic Activity, Substrate Activation and Protection against Non-Specific Cleavage of the Fusion Protein by Restriction Protease. Biochem. J. 1996;313(2):409–414. doi: 10.1042/BJ3130409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters P. J., Parniak M. A., Akerman B. R., Jones A. O., Scriver C. R.. Missense Mutations in the Phenylalanine Hydroxylase Gene (PAH) Can Cause Accelerated Proteolytic Turnover of PAH Enzyme: A Mechanism Underlying Phenylketonuria. J. Inherit Metab Dis. 1999;22(3):208–212. doi: 10.1023/A:1005533825980. [DOI] [PubMed] [Google Scholar]
- Seeliger D., de Groot B. L.. Conformational Transitions upon Ligand Binding: Holo-Structure Prediction from Apo Conformations. PLoS Comput. Biol. 2010;6(1):e1000634. doi: 10.1371/journal.pcbi.1000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee M. M., Greengard O., Knox W. E.. Liver Phenylalanine Hydroxylase Activity in Relation to Blood Concentrations of Tyrosine and Phenylalanine in the Rat. Biochem. J. 1972;127(4):675–680. doi: 10.1042/bj1270675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. W., Lee D. H., Kim N. D., Lee S. T., Ahn J. Y., Choi T. Y., Lee Y. K., Kim S. H., Kim J. W., Ki C. S.. Mutation Analysis of PAH Gene and Characterization of a Recurrent Deletion Mutation in Korean Patients with Phenylketonuria. Exp. Mol. Med. 2008;40(5):533. doi: 10.3858/EMM.2008.40.5.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates C. M., Sternberg M. J. E.. The Effects of Non-Synonymous Single Nucleotide Polymorphisms (NsSNPs) on Protein–Protein Interactions. J. Mol. Biol. 2013;425(21):3949–3963. doi: 10.1016/j.jmb.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Guldberg P., Levy H. L., Hanley W. B., Koch R., Matalon R., Rouse B. M., Trefz F., De la Cruz F., Henriksen K. F., Guttler F.. Phenylalanine Hydroxylase Gene Mutations in the United States: Report from the Maternal PKU Collaborative Study. Am. J. Hum. Genet. 1996;59(1):84. [PMC free article] [PubMed] [Google Scholar]
- Yoo S. J., Hong Y. H., Lee Y. W., Jung S. C., Ki C. S., Lee D. H.. The Study of DNA Mutations of Phenylketonuria in Koreans. Journal of Genetic Medicine. 2008;5(1):26–33. [Google Scholar]
- Song F., Qu Y., Zhang T., Jin Y., Wang H., Zheng X.. Phenylketonuria Mutations in Northern China. Mol. Genet. Metab. 2005;86:107–118. doi: 10.1016/j.ymgme.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Van Wegberg A. M. J., MacDonald A., Ahring K., Bélanger-Quintana A., Blau N., Bosch A. M., Burlina A., Campistol J., Feillet F., Giżewska M., Huijbregts S. C., Kearney S., Leuzzi V., Maillot F., Muntau A. C., Van Rijn M., Trefz F., Walter J. H., Van Spronsen F. J.. The Complete European Guidelines on Phenylketonuria: Diagnosis and Treatment. Orphanet J. Rare Dis. 2017;12(1):162. doi: 10.1186/s13023-017-0685-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett J. R.. Sapropterin Dihydrochloride (Kuvan/Phenoptin), an Orally Active Synthetic Form of BH4 for the Treatment of Phenylketonuria. IDrugs. 2007;10(11):805–813. [PubMed] [Google Scholar]
- Gordon P., Thomas J. A., Suter R., Jurecki E.. Evolving Patient Selection and Clinical Benefit Criteria for Sapropterin Dihydrochloride (Kuvan) Treatment of PKU Patients. Mol. Genet. Metab. 2012;105:672–676. doi: 10.1016/j.ymgme.2011.12.023. [DOI] [PubMed] [Google Scholar]
- Hofto M. E., Cross J. N., Cafiero M.. Interaction Energies between Tetrahydrobiopterin Analogues and Aromatic Residues in Tyrosine Hydroxylase and Phenylalanine Hydroxylase. J. Phys. Chem. B. 2007;111(32):9651–9654. doi: 10.1021/jp072518w. [DOI] [PubMed] [Google Scholar]
- Erlandsen H., Stevens R. C.. A Structural Hypothesis for BH4 Responsiveness in Patients with Mild Forms of Hyperphenylalaninaemia and Phenylketonuria. J. Inherit Metab Dis. 2001;24(2):213–230. doi: 10.1023/A:1010371002631. [DOI] [PubMed] [Google Scholar]
- Goldenberg M. M.. Pharmaceutical Approval Update. Pharm. Ther. 2008;33(2):114. [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Purohit R.. Computational Investigation of Pathogenic NsSNPs in CEP63 Protein. Gene. 2012;503(1):75–82. doi: 10.1016/j.gene.2012.04.032. [DOI] [PubMed] [Google Scholar]
- Singh P. K., Mistry K. N.. A Computational Approach to Determine Susceptibility to Cancer by Evaluating the Deleterious Effect of NsSNP in XRCC1 Gene on Binding Interaction of XRCC1 Protein with Ligase III. Gene. 2016;576(1):141–149. doi: 10.1016/j.gene.2015.09.084. [DOI] [PubMed] [Google Scholar]
- Al Mehdi K., Fouad B., Zouhair E., Boutaina B., Yassine N., Chaimaa A. E. C., Najat S., Hassan R., Rachida R., Abdelhamid B., Halima N.. Molecular Modelling and Dynamics Study of NsSNP in STXBP1 Gene in Early Infantile Epileptic Encephalopathy Disease. Biomed Res. Int. 2019;2019:1–14. doi: 10.1155/2019/4872101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loong C. K. P., Zhou H.-X., Bryant Chase P.. Familial Hypertrophic Cardiomyopathy Related E180G Mutation Increases Flexibility of Human Cardiac Α-tropomyosin. FEBS Lett. 2012;586(19):3503–3507. doi: 10.1016/j.febslet.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma D., Jacobs D. J., Livesay D. R.. Changes in Lysozyme Flexibility upon Mutation Are Frequent, Large and Long-Ranged. PLoS Comput. Biol. 2012;8(3):e1002409. doi: 10.1371/journal.pcbi.1002409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M., Kumar R., Singh R., Kaur J.. Thirty-Degree Shift in Optimum Temperature of a Thermophilic Lipase by a Single-Point Mutation: Effect of Serine to Threonine Mutation on Structural Flexibility. Mol. Cell. Biochem. 2017;430(1–2):21–30. doi: 10.1007/s11010-017-2950-z. [DOI] [PubMed] [Google Scholar]
- Flydal M. I., Martinez A.. Phenylalanine Hydroxylase: Function, Structure, and Regulation. IUBMB Life. 2013;65(4):341–349. doi: 10.1002/iub.1150. [DOI] [PubMed] [Google Scholar]
- Waters P. J.. HowPAH Gene Mutations Cause Hyper-Phenylalaninemia and Why Mechanism Matters: Insights from in Vitro Expression. Hum Mutat. 2003;21(4):357–369. doi: 10.1002/humu.10197. [DOI] [PubMed] [Google Scholar]
- Lopez A., Havranek B., Papadantonakis G. A., Islam S. M.. In Silico Screening and Molecular Dynamics Simulation of Deleterious PAH Mutations Responsible for Phenylketonuria Genetic Disorder. Proteins: Struct., Funct., Bioinf. 2021;89(6):683–696. doi: 10.1002/prot.26051. [DOI] [PubMed] [Google Scholar]
- Ng P. C.. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003;31(13):3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaser R., Adusumalli S., Leng S. N., Sikic M., Ng P. C.. SIFT Missense Predictions for Genomes. Nat. Protoc. 2016;11(1):1–9. doi: 10.1038/nprot.2015.123. [DOI] [PubMed] [Google Scholar]
- Adzhubei I. A., Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., Kondrashov A. S., Sunyaev S. R.. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods. 2010;7(4):248. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei, I. ; Jordan, D. M. ; Sunyaev, S. R. . Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc Hum Genet 2013, 76(1). 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E., Calabrese R., Casadio R.. Predicting the Insurgence of Human Genetic Diseases Associated to Single Point Protein Mutations with Support Vector Machines and Evolutionary Information. Bioinformatics. 2006;22(22):2729–2734. doi: 10.1093/bioinformatics/btl423. [DOI] [PubMed] [Google Scholar]
- Capriotti E., Fariselli P.. PhD-SNPg: A Webserver and Lightweight Tool for Scoring Single Nucleotide Variants. Nucleic Acids Res. 2017;45(1):W247–W252. doi: 10.1093/nar/gkx369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Krishnan V. G., Mort M. E., Xin F., Kamati K. K., Cooper D. N., Mooney S. D., Radivojac P.. Automated Inference of Molecular Mechanisms of Disease from Amino Acid Substitutions. Bioinformatics. 2009;25(21):2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Full-Length Human Phenylalanine Hydroxylase (PAH) in the Resting State. 2019. DOI: 10.2210/PDB6N1K/PDB. [DOI] [Google Scholar]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2017;45(D1):D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim N. L., Kumar P., Hu J., Henikoff S., Schneider G., Ng P. C.. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012;40(W1):W452–W457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F., Koonin E. V.. Iterated Profile Searches with PSI-BLASTa Tool for Discovery in Protein Databases. Trends Biochem. Sci. 1998;23(11):444–447. doi: 10.1016/S0968-0004(98)01298-5. [DOI] [PubMed] [Google Scholar]
- Galehdari H., Saki N., Mohammadi-Asl J., Rahim F.. Meta-Analysis Diagnostic Accuracy of SNP-Based Pathogenicity Detection Tools: A Case of UTG1A1 Gene Mutations. Int. J. Mol. Epidemiol Genet. 2013;4(2):77–85. [PMC free article] [PubMed] [Google Scholar]
- Pejaver V., Urresti J., Lugo-Martinez J., Pagel K. A., Lin G. N., Nam H. J., Mort M., Cooper D. N., Sebat J., Iakoucheva L. M., Mooney S. D., Radivojac P.. Inferring the Molecular and Phenotypic Impact of Amino Acid Variants with MutPred2. Nat. Commun. 2020;11(1):5918. doi: 10.1038/s41467-020-19669-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, D. A. ; Belfon, K. ; Ben-Shalom, I. Y. ; Brozell, S. R. ; Cerutti, D. S. ; T.E. Cheatham, V. W. D. C., III ; Darden, T. A. ; Duke, R. E. ; Giambasu, G. ; Gilson, M. K. ; Gohlke, H. ; Goetz, A. W. ; Harris, R. ; Izadi, S. ; Izmailov, S. A. ; Kasavajhala, K. ; Kovalenko, A. ; Krasny, R. ; Kurtzman, T. ; Lee, T. S. ; LeGrand, S. ; Li, P. ; Lin, C. ; L, J. ; Luchko, T. ; Luo, R. ; Man, V. ; Merz, K. M. ; Miao, Y. ; Mikhailovskii, O. ; Monard, G. ; Nguyen, H. ; Onufriev, A. ; Pan, F. ; Pantano, S. ; Qi, R. ; Roe, D. R. ; Roitberg, A. ; Sagui, C. ; Schott-Verdugo, S. ; Shen, J. ; Simmerling, C. L. ; Skrynnikov, N. R. ; Smith, J. ; Swails, J. ; Walker, R. C. ; Wang, J. ; Wilson, L. ; Wolf, R. M. ; Wu, X. ; Xiong, Y. ; Y, X. ; Kollman, D. M. Y. . P. A. AMBER 2020; University of California: San Francisco, 2020. [Google Scholar]
- Jo S., Kim T., Iyer V. G., Im W. C. H. A. R. M. M.-G. U. I.. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29(11):1859–1865. doi: 10.1002/JCC.20945. [DOI] [PubMed] [Google Scholar]
- Lee J., Cheng X., Swails J. M., Yeom M. S., Eastman P. K., Lemkul J. A., Wei S., Buckner J., Jeong J. C., Qi Y., Jo S., Pande V. S., Case D. A., Brooks C. L., MacKerell A. D., Klauda J. B., Im W.. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016;12(1):405–413. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., Mackerell A. D.. CHARMM General Force Field (CGenFF): A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010;31(4):671. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L.. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983;79(2):926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- Jo S., Kim T., Iyer V. G., Im W.. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J. Comput. Chem. 2008;29(11):1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Qi Y., Lee J., Cheng X., Shen R., Islam S. M., Roux B., Im W.. CHARMM-GUI DEER Facilitator for Spin-pair Distance Distribution Calculations and Preparation of Restrained-ensemble Molecular Dynamics Simulations. J. Comput. Chem. 2020;41(5):415–420. doi: 10.1002/jcc.26032. [DOI] [PubMed] [Google Scholar]
- Bosco P., Calì F., Meli C., Mollica F., Zammarchi E., Cerone R., Vanni C., Palillo L., Greco D., Romano V.. Eight New Mutations of the Phenylalanine Hydroxylase Gene in Italian Patients with Hyperphenylalaninemia. Hum Mutat. 1998;11(3):240–243. doi: 10.1002/(SICI)1098-1004(1998)11:3<240::AID-HUMU9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Kollman P. A.. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992;13(8):952–962. doi: 10.1002/jcc.540130805. [DOI] [Google Scholar]
- Van Gunsteren W. F., Berendsen H. J. C.. A Leap-Frog Algorithm for Stochastic Dynamics. Mol. Simul. 1988;1(3):173–185. doi: 10.1080/08927028808080941. [DOI] [Google Scholar]
- Miller B. R., McGee T. D., Swails J. M., Homeyer N., Gohlke H., Roitberg A. E.. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012;8(9):3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- Wang E., Weng G., Sun H., Du H., Zhu F., Chen F., Wang Z., Hou T.. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 10. Impacts of Enhanced Sampling and Variable Dielectric Model on Protein–Protein Interactions. Phys. Chem. Chem. Phys. 2019;21(35):18958–18969. doi: 10.1039/C9CP04096J. [DOI] [PubMed] [Google Scholar]
- Weiser J., Shenkin P. S., Still W. C.. Approximate Atomic Surfaces from Linear Combinations of Pairwise Overlaps (LCPO) J. Comput. Chem. 1999;20(2):217–230. doi: 10.1002/(SICI)1096-987X(19990130)20:2<217::AID-JCC4>3.0.CO;2-A. [DOI] [Google Scholar]
- Fusetti F., Erlandsen H., Flatmark T., Stevens R. C.. Structure of Tetrameric Human Phenylalanine Hydroxylase and Its Implications for Phenylketonuria. J. Biol. Chem. 1998;273(27):16962–16967. doi: 10.1074/jbc.273.27.16962. [DOI] [PubMed] [Google Scholar]
- Gersting S. W., Kemter K. F., Staudigl M., Messing D. D., Danecka M. K., Lagler F. B., Sommerhoff C. P., Roscher A. A., Muntau A. C.. Loss of Function in Phenylketonuria Is Caused by Impaired Molecular Motions and Conformational Instability. Am. J. Hum. Genet. 2008;83(1):5. doi: 10.1016/j.ajhg.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D., Kopec J., Fitzpatrick F., Mccorvie T. J., Yue W. W.. Structural Basis for Ligand-Dependent Dimerization of Phenylalanine Hydroxylase Regulatory Domain. Sci. Rep. 2016;6(1):1–10. doi: 10.1038/srep23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available upon reasonable request. MM/GBSA and pairwise energy decomposition analyses were performed using AmberTools23 with academic license. MD simulations were performed using Amber20 on the Anvil supercomputer at Purdue University through XSEDE support. Xmgrace (v 5.1.25) was used to generate data plots. VMD (v 1.9.4a57) was used to generate molecular graphics and is available at https://www.ks.uiuc.edu/Research/vmd/.