

Abstract
The bridging ligands L1 and L2 contain two N,N-bidentate pyrazolyl-pyridine units linked to a central aromatic spacer unit (1,2-phenyl or 2,3-naphthyl, respectively). Reaction with Ni(II) salts and treatment with the anions tetrafluoroborate or perchlorate result in formation of dinuclear complexes having a 2:3 metal:ligand ratio, with one bridging and two terminal tetradentate ligands. In contrast, reaction of L1 and L2 with Co(II) salts, followed by treatment with tetrafluoroborate or perchlorate, results in assembly of cage complexes having a 4:6 metal:ligand ratio; these complexes have a metal ion at each corner of an approximate tetrahedron, and a bis-bidentate bridging ligand spanning each edge. The central cavity is occupied by a tetrahedral counterion that forms multiple hydrogen-bonding interactions with the methylene protons of the bridging ligands. The anionic guest fits tightly into the central cavity of the cage to which it is ideally complementary in terms of shape, size, and charge. Solution NMR experiments show that the central anion acts as a template for cage formation, with a mixture of Co(II) and the appropriate bridging ligand alone giving no assembly into a cage until the tetrahedral anion is added, at which point cage assembly is fast and quantitative. The difference between the structures of the complexes with Ni(II) and Co(II) illustrate how the uncoordinated anions can exert a profound influence on the course of the assembly process.
Metal-directed self-assembly has recently become a major tool by which coordination chemists can prepare large and elaborate complexes such as helicates, grids, boxes, rings, and cages from relatively simple components (1–11). Many examples are based on accurate control of metal-ligand coordinate bond formation, with the course of the assembly involving a labile metal ion and a multidentate ligand dictated by the metal/ligand interactions. This behavior is exemplified by the formation of helical complexes with linear oligopyridines, where the partitioning of the ligand into bidentate or terdentate binding domains is dictated by the preference of the metal ion for four-coordinate or six-coordinate geometry (3). Recently, however, it has become apparent that “innocent” anions can dictate the course of the assembly process by acting as a template around which a particular combination of metal ions and ligand can assemble in a way which would not occur in the absence of the anion. For example, Lehn and coworkers (12) showed how a trinuclear M3L3 triple helicate converted to a circular M5L5 helicate in the presence of chloride ion, which was tightly bound in the center of the resulting cationic cavity. Anions that are chosen for their innocence in terms of coordinating ability can nevertheless direct the course of an assembly process via noncovalent interactions.
Here we describe how the anions perchlorate and tetrafluoroborate act as templates for the formation of edge-bridged tetrahedral M4(μ-L)6 cages from Co(II) and bis-bidentate bridging ligands (Scheme S1). These “adamantoid” cages (Fig. 1) were first described by Saalfrank et al. (13–16) and have since been studied by several other groups (17–25). A notable feature of many such complexes is that the central cavity can accommodate guest species, with the nature of the guest depending on the charge carried by the surrounding complex cage. Thus, neutral cages tend to have either no guests (15) or solvent molecules as guests (17) in the cavity, anionic cages are found to contain tetraalkylammonium (19, 21) or ammonium (16) cations as guests, and cationic cages can contain tetrahedral anions such as [BF4]− as guests (23, 24). The twin facts that some cages can form without a guest (15, 22) and that some guests can be freely exchanged with other species (19) suggest that in these cases, the assembly of the cage arises from an ideal match between the symmetry properties of the ligands and the coordination preferences of the metal ions, and that no templating effect is necessary. In other cases, however, the guest is trapped, suggesting that the assembly of the cage is caused in these cases by a genuine template effect (16, 21, 23).
Scheme 1.

Figure 1.

Diagram of an M4(μ-L)6 tetrahedral cage based on a bis-bidentate bridging ligand and 6-coordinate metal centers.
Methods
Synthesis of L2.
A mixture of 2,3-bis(bromomethyl)naphthalene (26) (2.00 g, 6.39 mmol), 3-(2-pyridyl)pyrazole (27) (2.04 g, 14.1 mmol), aqueous NaOH (10 M, 7 cm3), and tetrahydrofuran (THF) (50 cm3) was stirred at reflux for 24 h. After cooling, the organic phase was separated, dried over MgSO4, filtered, and evaporated to dryness. The crude solid was purified by column chromatography on flash silica with ethylacetate-methanol (99:1); the desired product was the third (major) fraction. Yield: 1.42 g, 50%; electron impact mass spectrum (EIMS): m/z 442 (60%, M+), 297. 1H NMR (270 MHz, CDCl3): δ 8.63 (2 H, d; pyridyl H6), 7.96 (2 H, d; pyridyl H3), 7.78 (2 H, m; naphthyl H6/H7 or H5/H8), 7.70 (2 H, td; pyridyl H4), 7.63 (2 H, s; naphthyl H1/H4), 7.49 (2 H, m; naphthyl H5/H8 or H6/H7), 7.38 (2 H, d; pyrazolyl H4 or H5), 7.19 (2 H, ddd; pyridyl H5), 6.93 (2 H, d; pyrazolyl H5 or H4), 5.58 (4 H, s; CH2). Found: C, 76.2; H, 5.3; N, 19.3%. Required for C28H22N6: C, 76.0; H, 5.0; N, 19.0%.
Syntheses of [Co4(L2)6(ClO4)][ClO4]7, [Co4(L2)6(BF4)][BF4]7 and [Ni2(L2)3][ClO4]4.
A solution of L2 (0.049 g, 0.11 mmol) in CH2Cl2 (10 cm3) was added dropwise to a solution the appropriate metal(II) acetate hydrate (0.07 mmol) in MeOH (10 cm3); the resulting solution was stirred for 1 h at room temperature. Addition of a methanolic solution of NaBF4 or NaClO4 as appropriate resulted in precipitation of a solid, which was filtered off and dried in vacuo; yields were typically 30%. Concentrating and cooling the mother liquor could isolate further material. X-ray quality crystals were grown by diffusion of diethyl ether into solutions of the complexes in MeCN at 0°C. Electrospray mass spectroscopic data are as follows. Satisfactory elemental analytical data were obtained: [Co4(L2)6(ClO4)][ClO4]7: m/z 1129.0 (calculated 1128.3), {Co4(L2)6(ClO4)5}3+; 822.0 (calculated 821.4), {Co4(L2)6(ClO4)}4+; 600.2 (calculated 600.5), {Co(L2)(ClO4)}+. [Co4(L2)6(BF4)][BF4]7: m/z 1107.8 (calculated 1107.3), {Co4(L2)6(BF4)5}3+; 809.2 (calculated 808.7), {Co4(L2)6(BF4)}4+; 587.9 (calculated 587.8), {Co(L2)(BF4)}+. [Ni2(L2)3][ClO4]4: m/z 471.1 (calculated 471.3), {Ni(L2)2}2+.
X-Ray Crystallography.
Details of the crystal, data collection, and refinement parameters for the new structures are in Table 1. A detailed description of the methods used for unit cell determination and data collection has been published (27). Structure solutions and refinements used SHELXS-97 (28) and SHELXL-97 (29); absorption corrections were applied by using sadabs (30). Full details of the refinements are included in the supporting text, which is published on the PNAS web site, www.pnas.org, together with selected bond distances and angles.
Table 1.
Crystallographic data for the three new structures
| Compound | [Co4(L2)6(BF4)][BF4]7⋅H2O⋅7MeCN | [Co4(L2)6(ClO4)][ClO4]7⋅13MeCN | [Ni2(L2)3][ClO4]4⋅4MeCN⋅0.5(iPr2O) |
|---|---|---|---|
| Formula | C182H155B8Co4F32N43O | C194H171Cl8Co4N49O32 | C95H85Cl4N22Ni2O16.5 |
| M | 3890.7 | 4220.12 | 2058.1 |
| Crystal size/mm3 | 0.5 × 0.5 × 0.1 | 0.7 × 0.5 × 0.2 | 0.4 × 0.2 × 0.2 |
| T/K | 123 | 123 | 173 |
| System, space group | Hexagonal, R3̄ | Monoclinic, P2(1)/n | Monoclinic, P2(1)/n |
| a/Å | 18.540 (4) | 27.67 (2) | 24.7858 (10) |
| b/Å | 18.540 (4) | 18.440 (15) | 12.6972 (5) |
| c/Å | 181.09 (5) | 40.30 (3) | 30.4427 (12) |
| β/° | 90 | 109.424 (15) | 95.8960 (10) |
| U/Å3 | 53909 (22) | 19391 (27) | 9529.9 (7) |
| Z | 12 | 4 | 4 |
| ρcalc/g cm−3 | 1.438 | 1.446 | 1.434 |
| μ/mm−1 | 0.463 | 0.532 | 0.586 |
| Data/restraints/parameters | 15680, 4, 1615 | 17981, 228, 1339 | 11264, 88, 1311 |
| Final R1, wR2* | 0.0721, 0.2142 | 0.2161, 0.5475 | 0.0563, 0.1685 |
Details in common: Siemens SMART diffractometer; Mo-Kα radiation (0.71073 Å°).
The value of R1 is for selected data with F ≥ 4σ(F); the value of wR2 is for all data.
Results and Discussion
Complexes with L1: Studying the Template Effect by 1H NMR Spectroscopy.
In a recent communication we described briefly the structures of the complexes [Co4(L1)6(BF4)][BF4]7 and [Ni2(L1)3][BF4]4 (24). These are recalled in Figs. 2 and 3, and the contrast between them clearly illustrates how participation of the anion can influence the course of the assembly. In [Ni2(L1)3][BF4]4, one ligand L1 is acting as a bridge, but the other two are acting as tetradentate chelates, such that the structure of the cation is [(L1)Ni(μ-L1)Ni(L1)]4+. This is an unusual arrangement of ligands in M2L3 complexes, and is similar to the structure of the Fe(III) complex of the tetradentate siderophore alcilagin (31). In contrast, [Co4(L1)6(BF4)]7+ is a tetrahedral cage, with the [BF4]− anion being complementary in both charge and shape to the cavity; with respect to the Co4 tetrahedron, the [BF4]− tetrahedron is inverted such that each F atom is directed to the space at the center of a Co3 triangular face. The Co–N bond distances lie in the range 2.11–2.21 Å (average 2.151 Å), typical for high-spin Co(II). All four metal ions have the same optical configuration, such that each complex cage has approximate T symmetry (although the crystal is a racemate). The Co⋅⋅⋅Co separations (all different) are between 8.98 and 10.07 Å; the Co⋅⋅⋅B distances are all comparable (5.61–5.98 Å), indicating that the anion is located approximately centrally in the cavity.
Figure 2.

Crystal structure of the complex cation of [Co4(L1)6(BF4)][BF4]7, showing (a) one bridging ligand and the encapsulated anion and (b) a space-filling view with the ligands colored differently.
Figure 3.

Crystal structure of the complex cation of [Ni2(L1)3][BF4]4. The terminal and bridging ligands are shaded differently.
Although the [BF4]− anion can act as a weak ligand (32), it is clear that the encapsulation cannot be ascribed to Co⋅⋅⋅F interactions in this case, because each fluorine atom of the trapped anion is directed toward the center of a triangular face of the Co4 tetrahedron, away from the Co apices that are in any case coordinately saturated (Co⋅⋅⋅F distances, 5–6 Å). This behavior contrasts with that found by Huttner and coworkers (23), who found that incorporation of [BF4]− into a tetrahedral cage was helped by weak M⋅⋅⋅F interactions, with the F atoms directed exactly toward the metal vertices rather than between them. In [Co4(L1)6(BF4)]7+, however, there are close contacts between the fluorine atoms and the CH2 spacers of the ligands, with nonbonded C⋅⋅⋅F separations in the range 3.01–3.21 Å (and F⋅⋅⋅H contacts in the range 2.3–2.6 Å), indicative of C–H⋅⋅⋅F hydrogen-bonding interactions (33, 34), which no doubt help to stabilize the assembly. In addition, multiple stacking interactions between overlapping aromatic fragments of adjacent ligands are evident. Variable-temperature 11B and 19F NMR spectra showed that the trapped [BF4]− anion did not exchange with the remaining seven free [BF4]− anions on the NMR time scale (24). The reason for the quite different structure of this compared with the complex with Ni(II) is unclear, but may be related to the slightly different ionic radii of Co(II) and Ni(II).
The formation of [Co4(L1)6(BF4)][BF4]7 prompted us to carry out solution 1H NMR studies to see whether a genuine anion-based template effect occurs. The 1H NMR spectrum of the preformed complex [Co4(L1)6(BF4)][BF4]7 in CD3CN/CD3OD is in Fig. 4a. The paramagnetism of the high-spin Co(II) centers means that the signals from the ligand protons are in the 15–85 ppm range. Assuming that the cage adopts a T-symmetric structure in solution we expect 10 resonances—i.e., for each equivalent half of L1—and this is what occurs (although two of the signals, D and E, are very weak). Assignment of individual signals is not obvious, but on the basis that the degree of shift depends on distance from the paramagnetic centers, we assign the least-shifted peaks I and J to the phenyl spacer.
Figure 4.

1H NMR spectra (300 MHz) of (a) [Co4(L1)6(BF4)][BF4]7 (redissolved crystals) in CD3OD/CD3CN; (b) a mixture of Co(MeCO2)2· 2H2O and L1 in a 4:6 ratio in CD3OD/D2O; and (c) the solution in b after addition of aqueous NaBF4 (similar results were obtained with NaClO4).
When Co(II) acetate and L1 are mixed in a 2:3 molar ratio in CD3OD/D2O (necessary to dissolve the metal salt), the 1H NMR spectrum shown in Fig. 4b results. It lies in the normal chemical shift range, but is broad and poorly resolved because of some degree of interaction with the paramagnetic metal centers. There are no signals at shifts more positive than 10 ppm, indicating that no significant amount of the cage complex exists under these conditions. On addition of one equivalent of [BF4]− to the sample, the spectrum changes dramatically (Fig. 4c) and becomes essentially identical to that in Fig. 3a measured for the preformed complex. This spectrum is rather noisy because of the poor solubility of the cage in this solvent mixture, but all of the principal features of the spectrum of the cage complex in Fig. 4a are present at the correct positions, and no other peaks are apparent. Addition of [BF4]−, therefore, results in quantitative assembly of the [Co4(L1)6(BF4)]7+ cage in as much time as it took to make the addition and record a new spectrum (about a minute), and a genuine anion-templated assembly is occurring.
This simple 1H NMR experiment allows us to screen for their templating ability in this system in solution. We found that [PF6]− does not act as a template, but that [ClO4]− does, with addition of [ClO4]− to a mixture of Co(II) and L1 resulting in a spectrum identical to that obtained by addition of [BF4]− (Fig. 4c). These findings are in agreement with expectations based on the basis of the size and shape of these anions: hexafluorophosphate is clearly too large to fit in the cavity, whereas perchlorate—which is the same size and shape as tetrafluoroborate—is an effective template. We accordingly undertook further syntheses and structural studies with the related ligand L2 by using both [BF4]− and [ClO4]− as anions.
Syntheses of L2 and Formation of Cage Complexes with Co(II).
We prepared L2 by using the same general method as used for L1 (24, 35), expecting that it will coordinate in the same manner as L1 but with additional inter-ligand aromatic stacking interactions in the complexes provided by the naphthyl units. Reaction of Ni(II) acetate with L2 in a 2:3 molar ratio in CH2Cl2/MeOH afforded a blue solution from which a precipitate appeared on addition of methanolic NaClO4. On the basis of the electrospray mass spectrum [which showed only the fragment {Ni(L2)2}2+] and by analogy with the structure of the Ni(II) complex of L1, we tentatively identified this complex as [Ni2(L2)3][ClO4]4; this was confirmed by x-ray crystallography (Fig. 5). The dinuclear complex contains one tetradentate chelating ligand attached to each Ni(II) center, with the third ligand acting as a bis-bidentate bridge spanning the two metals, which are separated by 9.16 Å. The bridging ligand adopts a helical twist that is emphasized in the space-filling view shown in Fig. 5b; this view also emphasizes the inter-ligand aromatic stacking. The pseudooctahedral metal ions both have a meridional tris-chelate geometry, with unremarkable bond distances and angles (see supporting text). This structure illustrates the ability of L2 to act as either a chelating or bridging ligand as circumstances dictate because of the flexibility provided by the methylene units.
Figure 5.

Crystal structure of the complex cation of [Ni2(L2)3][ClO4]4 showing (a) a conventional view with the terminal and bridging ligands shaded differently and (b) a space-filling view emphasizing the aromatic stacking with the bridging ligand in blue and the terminal ligands purple.
Reaction of Co(II) acetate with L2 in a 2:3 molar ratio in CH2Cl2/MeOH afforded an orange solution from which a precipitate appeared on addition of methanolic NaBF4 or NaClO4. From electrospray mass spectra, and by analogy with the behavior of L1, we tentatively identified these as the cage complexes [Co4(L2)6(BF4)][BF4]7 and [Co4(L2)6(ClO4)][ClO4]7, respectively. In particular, the peak at highest m/z value in each case in the mass spectrum corresponds to the species {Co4(L2)6(X)5}3+ (X = BF4 or ClO4 respectively), confirming the presence of the intact cage.
We followed the anion-templated assembly process in the manner described above (Fig. 6). The 1H NMR spectrum of the preformed complex gave a characteristic highly shifted pattern of peaks, similar to that seen for the cage complex with L1 but with three signals (I, J, and K) in the 10–20 ppm region rather than two. We assign these three peaks as arising from the 2,3-disubstituted naphthyl unit (with twofold symmetry), in agreement with their distance from the paramagnetic centers. In all other respects there is a clear correspondence between the signals of the cage complex with L2 (Fig. 6a) and with L1 (Fig. 4a). When Co(II) acetate and L2 were mixed in a 2:3 ratio in MeOD/D2O in an NMR tube, however, only a broad poorly resolved spectrum was seen in the normal region (5.5–9 ppm) with no sign of the highly shifted pattern of signals from the cage (Fig. 6b). Addition of one equivalent of either [BF4]− or [ClO4]− immediately resulted in appearance of the characteristic spectrum of the cage (Fig. 6c). This spectrum is noisy because the cage is poorly soluble, and the two weakest signals (D and E) are not resolved; but the remaining nine signals correspond exactly with those in Fig. 6a, confirming the templating action of the anions in these complexes also. The absence of any other signals means that assembly of the cage is quantitative in the presence of the template.
Figure 6.

1H NMR spectra (300 MHz) of (a) [Co4(L2)6(ClO4)][ClO4]7 (redissolved crystals) in CD3OD/CD3CN; (b) a mixture of Co(MeCO2)2⋅2H2O and L2 in a 4:6 ratio in CD3OD/D2O; and (c) the solution in b after addition of aqueous NaClO4 (similar results were obtained with NaBF4).
[Co4(L2)6(BF4)][BF4]7 gave x-ray-quality crystals from MeCN/ether (Figs. 7 and 8). The figures illustrate only one of the crystallographically independent cage cations; the alternate one is similar, and both contain [BF4]− ions in their central cavity. The Co–N separations are in the range 2.09–2.17 Å, and the coordination environment about each metal center is approximately octahedral with a facial tris-chelate geometry (see supporting text). The gross geometry is similar to that of the complex with L1 described above: the metal ions form an approximately tetrahedral cage, with the Co⋅⋅⋅Co separations lying between 9.3 and 10 Å. The cage has approximate T symmetry with all four metal centers having the same configuration, such that the racemic crystals contain equal amounts of ΔΔΔΔ and ΛΛΛΛ forms (in solution the symmetry is exactly T on the basis of the NMR spectrum). The fluoroborate anion within each cavity is inverted with respect to the Co4 tetrahedron such that each F atom is directed toward the space at the center of a Co3 triangular face; the anion is approximately central in the cavity, with all Co⋅⋅⋅B distances being in the range 5.68–6.16 Å. The F atoms make close contacts with the CH2 spacer units in some of the ligands, with nonbonded C⋅⋅⋅F distances lying in the range 3.07–3 38 Å and the associated H⋅⋅⋅F distances varying from 2.33 to 2.53 Å (Fig. 7b). These distances are characteristic of C–H⋅⋅⋅F hydrogen bonds (33, 34), and the number of such interactions (three per F atom, or 12 per cage) presumably contributes significantly to stabilization of the cage structure. From the space-filling view in Fig. 8, the intertwining of the six ligands, resulting in substantial aromatic stacking interactions between them, is clear. It is also apparent from this view that the [BF4]− anion in the cavity is almost completely encapsulated.
Figure 7.

Crystal structure of the complex cation of [Co4(L2)6(BF4)][BF4]7. (a) A view showing two of the bridging ligands and the encapsulated anion. (b) The array of CH⋅⋅⋅F hydrogen-bonding interactions involving the CH2 units of the ligands and the encapsulated anion.
Figure 8.
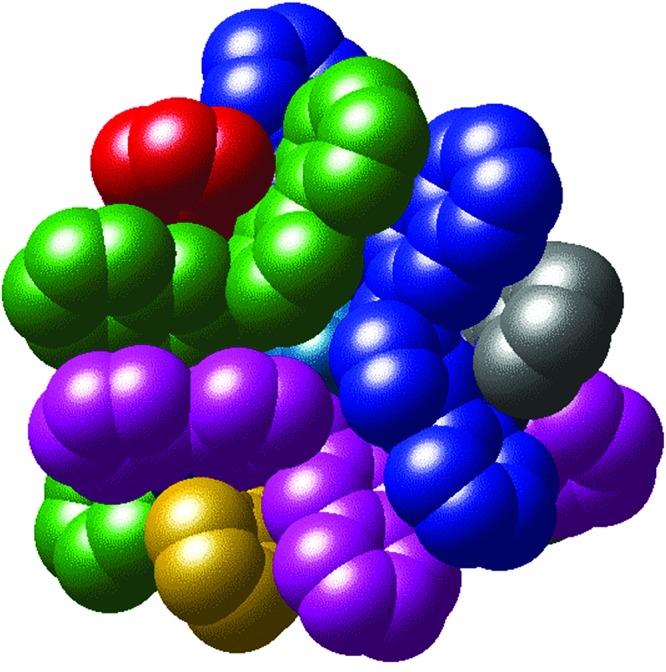
Space-filling view of the complex cation of [Co4(L2)6(BF4)][BF4]7 with each ligand colored differently. Note that the fluoroborate ion (pale blue) is barely visible through the gap in the center of the face.
[Co4(L2)6(ClO4)][ClO4]7 also afforded crystals, which decomposed very fast because of the presence of 13 MeCN molecules per complex. The structural determination is accordingly of lower precision than that of the [BF4]− complex (R1 = 21.6%), and detailed discussion of structural parameters is not warranted. The gross structure is, as expected from the 1H NMR data, essentially identical to that of [Co4(L1)6(BF4)][BF4]7, with a perchlorate anion in the central cavity. The Co⋅⋅⋅Co separations lie in the range 9.2–10.3 Å, and the anion is centrally located with the Co⋅⋅⋅Cl distances lying between 5.7 and 6.1 Å. The perchlorate oxygen atoms are involved in short contacts with the CH2 spacers of the ligands L2 (nonbonded C⋅⋅⋅O separations in the range 3.0–3.3 Å), corresponding to C–H⋅⋅⋅O hydrogen-bonding interactions (34).
The behavior of these cage complexes contrasts with some recent examples described by Raymond and coworkers (10, 17), based on bridging ligands whose two binding sites are carefully arranged to optimize cage formation. These ligands cannot act as tetradentate chelates to a single metal, because the binding sites are divergent and give M4L6 cages quantitatively without the assistance of a template effect. As described previously, the cavity may contain a counterion or solvent molecules as guest species, but this is not a prerequisite for cage formation and the guests can be freely exchangeable with other species. With L1 and L2, however, the flexibility imparted by the methylene spacers means that both tetradentate chelating and bis-bidentate bridging coordination modes are available, giving either M4L6 or M2L3 species. The balance between these seems to be in favor of the M2L3 complex in the absence of a template effect, with a templating anion altering the balance in favor of cage formation when M = Co(II) but not when M = Ni(II). It follows that larger tetrahedral anions (e.g., pertechnetate, tetrahalometallates) may act as templates for assembly of larger M4L6 cage complexes based on longer bridging ligands.
Conclusions
The tetrahedral cage complexes [Co4(μ-L)6X][X]7, where X = [BF4]− or [ClO4]− and L = L1 or L2, form by an anion-directed templating effect in solution. NMR spectra show that the cages form quantitatively only in the presence of a suitable templating anion (perchlorate or tetrafluoroborate), which is a good fit for the central cavity and which participates in hydrogen-bonding interactions with the ligand CH2 groups. The anion is completely enclosed, is centrally located in the cavity, and is inverted with respect to the Co4 tetrahedron. These templating anions seem to be ideally complementary in terms of size, shape, and charge for the cavities in which they are located. In contrast, with Ni(II), open-chain complexes [Ni2L3]X4 form in which the same anions do not act as templates for cage assembly.
Supplementary Material
Acknowledgments
We thank the Leverhulme Trust and the Engineering and Physical Sciences Research Council for financial support.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: Atomic coordinates have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, Cambridge CB2 1EZ, United Kingdom, www.ccdc.cam.ac.uk (CSD reference nos. 172967, 172968, and 173833).
References
- 1.Baxter P N W. In: Comprehensive Supramolecular Chemistry. Atwood J L, Davies J E D, MacNicol D D, Vögtle F, Lehn J-M, editors. Vol. 9. Oxford: Pergamon; 1996. , 165–211. [Google Scholar]
- 2.Constable E C. In: Comprehensive Supramolecular Chemistry. Atwood J L, Davies J E D, MacNicol D D, Vögtle F, Lehn J-M, editors. Vol. 9. Oxford: Pergamon; 1996. , 213–252. [Google Scholar]
- 3.Constable E C. Prog Inorg Chem. 1994;42:67–138. [Google Scholar]
- 4.Fujita M. In: Comprehensive Supramolecular Chemistry. Atwood J L, Davies J E D, MacNicol D D, Vögtle F, Lehn J-M, editors. Vol. 9. Oxford: Pergamon; 1996. , 253–282. [Google Scholar]
- 5.Philp D, Stoddart J F. Angew Chem Int Ed Engl. 1996;35:1155–1196. [Google Scholar]
- 6.Piguet C, Barnardinelli G, Hopfgartner G. Chem Rev. 1997;97:2005–2062. doi: 10.1021/cr960053s. [DOI] [PubMed] [Google Scholar]
- 7.Leininger S, Olenyuk B, Stang P J. Chem Rev. 2000;100:853–908. doi: 10.1021/cr9601324. [DOI] [PubMed] [Google Scholar]
- 8.Sweigers G F, Malefetse T J. Chem Rev. 2000;100:3483–3537. [Google Scholar]
- 9.Fujita M. Chem Soc Rev. 1998;27:417–425. [Google Scholar]
- 10. Caulder, D. L. & Raymond, K. N. (1999) J. Chem. Soc. Dalton Trans., 1185–1200.
- 11.Caulder D L, Raymond K N. Acc Chem Res. 1999;32:975–982. [Google Scholar]
- 12.Hasenknopf B, Lehn J-M, Kneisel B O, Baum G, Fenske D. Angew Chem Int Ed Engl. 1996;35:1838–1840. [Google Scholar]
- 13.Saalfrank R W, Stark A, Peters K, von Schnering H G. Angew Chem Int Ed Engl. 1988;27:851–853. [Google Scholar]
- 14.Saalfrank R W, Stark A, Bremer M, Hummel H-U. Angew Chem Int Ed Engl. 1990;29:311–314. [Google Scholar]
- 15.Saalfrank R W, Hörner B, Stalke D, Salbeck J. Angew Chem Int Ed Engl. 1993;32:1179–1182. [Google Scholar]
- 16.Saalfrank R W, Burak R, Breit A, Stalke D, Herbst-Irmer R, Daub J, Porsch M, Bill E, Müther M, Trautwein A X. Angew Chem Int Ed Engl. 1994;33:1621–1623. [Google Scholar]
- 17.Beissel T, Powers R E, Raymond K N. Angew Chem Int Ed Engl. 1996;35:1084–1086. [Google Scholar]
- 18.Beissel T, Powers R E, Parac T N, Raymond K N. J Am Chem Soc. 1999;121:4200–4206. [Google Scholar]
- 19.Caulder D L, Powers R E, Parac T N, Raymond K N. Angew Chem Int Ed Engl. 1998;37:1840–1843. [Google Scholar]
- 20.Parac T N, Caulder D L, Raymond K N. J Am Chem Soc. 1998;120:8003–8004. [Google Scholar]
- 21.Scherer M, Caulder D L, Johnson D W, Raymond K N. Angew Chem Int Ed Engl. 1999;38:1588–1592. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1587::AID-ANIE1587>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 22.Enemark E J, Stack T D P. Angew Chem Int Ed Engl. 1998;37:932–935. doi: 10.1002/(SICI)1521-3773(19980420)37:7<932::AID-ANIE932>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 23.Mann S, Huttner G, Zsolnai L, Heinze K. Angew Chem Int Ed Engl. 1996;35:2808–2809. [Google Scholar]
- 24.Fleming J S, Mann K L V, Carraz C-A, Psillakis E, Jeffery J C, McCleverty J A, Ward M D. Angew Chem Int Ed Engl. 1998;37:1279–1281. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1279::AID-ANIE1279>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 25. Paul, R. L., Couchman, S. M., Jeffery, J. C., McCleverty, J. A., Reeves, Z. R. & Ward, M. D. (2000) J. Chem. Soc. Dalton Trans., 845–851.
- 26.Wang C, Mosberg H I. Tetrahedron Lett. 1995;36:3623–3626. [Google Scholar]
- 27.Jones P L, Amoroso A J, Jeffery J C, McCleverty J A, Psillakis E, Rees L H, Ward M D. Inorg Chem. 1997;36:10–18. [Google Scholar]
- 28.Sheldrick G M. SHELXS-97: A Program for Automatic Solution of Crystal Structures. Göttingen, Germany: Univ. of Göttingen; 1997. [Google Scholar]
- 29.Sheldrick G M. SHELXL-97: A Program for Crystal Structure Refinement. Göttingen, Germany: Univ. of Göttingen; 1997. [Google Scholar]
- 30.Sheldrick G M. SADABS: A Program for Absorption Correction of Crystallographic Data. Göttingen, Germany: Univ. of Göttingen; 1996. [Google Scholar]
- 31.Hou Z, Sunderland C J, Nishio T, Raymond K N. J Am Chem Soc. 1996;118:5148–5149. [Google Scholar]
- 32. Mann, K. L. V., Jeffery, J. C., McCleverty, J. A. & Ward, M. D. (1998) J. Chem. Soc. Dalton Trans., 3029–3035.
- 33.Grepioni F, Cojazzi G, Draper S M, Scully N, Braga D. Organometallics. 1998;17:296–307. [Google Scholar]
- 34.Desiraju D, Steiner T. The Weak Hydrogen Bond in Structural Chemistry and Biology. Oxford: OUP; 1999. [Google Scholar]
- 35. Fleming, J. S., Mann, K. L. V., Couchman, S. M., Jeffery, J. C., McCleverty, J. A. & Ward, M. D. (1998) J. Chem. Soc. Dalton Trans., 2047–2052.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.