

Abstract
A family of nanoscale-sized supramolecular cage compounds with a trigonal prismatic framework was prepared by means of spontaneous self-assembly from the combination of a predesigned molecular “clip” with tritopic pyridyl subunits. As confirmed by x-ray crystallography, the smallest structure of the reported series is ≈1 × 2 nm and possesses a nitrate anion incarcerated inside its molecular cavity. The largest structure has dimensions of ≈ 1 × 4 nm.
The formation of discrete supramolecular entities driven and held together by metal coordination is an intense new area of investigation at the forefront of supramolecular chemistry (1–10). Because self-assembly is guided by the chemical information encoded into the molecular subunits, diverse structures with predetermined shape, size, and functionality can readily be designed. Indeed, a wide variety of aesthetic structures have been realized, such as molecular grids, helicates, rings, catenanes, tetrahedra, cubes, cuboctahedra, etc. Once assembled, many of the hollow structures have been shown to be capable of encapsulating molecules through electrostatic and/or dispersion forces. Often times, ions will template the formation of an assembly (11–21). When considering that metal-containing assemblies often possess magnetic, photophysical, and/or redox properties not accessible from purely organic systems, studies in basic host–guest chemistry hold new promise for technologies in molecular sensing (22–28), separations, and catalysis (29, 30).
Because lower-symmetry hosts can ultimately be expected to show enhanced guest selectivity, especially toward planar aromatic guests, prismatic cages represent a natural progression in the development of this area. Although M3L2-type cages are relatively simple three-dimensional constructs, they remain uncommon. Of those that have been reported (31–40), most usually either require the use of templates to assemble in solution, or assemble only in the solid state. Part of the reason for this limitation is possibly the fact that, in most cases, flexible ligands were used. By contrast, structures derived from rigid tritopic linkers with cis-metal ions are either: (i) tetrahedral M6L4 cages (41) where L is a planar ligand, or (ii) double-square M6L4 cages (42) where L is a 109° linker ligand. Construction of the M3L2, D3h species is complicated by the fact that rigid tritopic linkers with ideal mutual angles of 60° are not easily accessible. A noteworthy trigonal bipyramidal structure (35), made from Pd(II) ions and a calix[3]arene subunit, was shown to be able to reversibly bind a molecule of C60.
By exploiting incommensurate symmetry requirements for differing metallic subunits, an alternative approach to structures of this general topology was recently reported. Raymond and Wong (43–45) successfully prepared a series of M2M′3L6 supramolecular clusters where a multifunctionalized ligand (L) was cleverly designed to selectively interact with two types of metal ions (one hard and one soft).
Double oxidative addition to rigid aromatic ligands offers a unique way to direct the open coordination sites of metals (46), because nearly any desired angle can be fashioned provided the corresponding aryl halide is accessible. Presented here is a ligand-directed approach to molecular prisms, thus providing a versatile method for the construction of supramolecular structures of variable size and functionality, in high yield, and without the use of templates. Specifically, the modular self-assembly of a new family of metalla-bicyclic structures (3a–3c), directed by a molecular “clip” (1) as a preconstructed shape-defining unit, is described. The structure and properties of these nanoscopic, macrocyclic species have been studied by electrospray ionization (ESI)-mass spectrometry, multinuclear NMR, and x-ray crystallography.
Experimental Section
General Methods.
Building unit 2a (47) was synthesized by a published method and 2b and 2c were prepared in an analogous manner. NMR spectra were recorded on a Varian XL-300 or a Unity 300 spectrometer. Deuterated solvents were used as received. Proton chemical shifts are reported relative to residual protons of the deuterated acetone (δ 2.05). 31P {1H} chemical shifts (δ) are reported relative to an unlocked, external sample of H3PO4 (0.0 ppm). Elemental analyses were performed by Atlantic Microlab, Norcross, GA.
General Procedure for the Preparation of Compounds 3a–3c.
In a 2-dram (8 g) vial equipped with a magnetic stir bar were placed solid 1,8-bis(trans-Pt(PEt3)2(NO3))anthracene (20.0 mg, 0.0172 mmol) with a 0.67 molar ratio of the appropriate tritopic bridging ligand. Next, 1 ml of an acetone-d6/D2O mixture was added to the vial (1:1 for 3a, 1:2 for 3b and 3c), which was then sealed with Teflon tape and heated in an oil bath at 50–55°C, with stirring. After the specified length of time (3–5 h for 3a, 20–24 h for 3b and 3c), the initial pale yellow suspension gradually became homogeneous. The solution was then transferred to an NMR tube for analysis. The product was precipitated with KPF6, collected on a frit, washed with excess water, and dried in vacuo.
Bicyclo[Tris[1,8-bis(trans-Pt(PEt3)2(NO3))anthracene)]bis[Tris(4-pyridyl)methanol]] (3a).
1H NMR (acetone-d6/D2O, 300 MHz): δ 9.18 (d, 6H, 3JHH = 6.3 Hz, Hα-Py), 8.98 (m, 9H, Hα-Py, H9), 8.37 (s, 3H, H10), 8.31 (d, 6H, 3JHH = 6.0 Hz, Hβ-Py), 7.75 (m, 12H, Hβ-Py, H4, 5), 7.69 (d, 6H, 3JHH = 7.8 Hz, H2,7), 7.14 (m, 6H, H3,6), 1.40 (m, 72H, PCH2CH3), 0.89 (m, 108H, PCH2CH3). 31P{1H} NMR (acetone-d6/D2O, 121.4 MHz): δ 9.5 (s, 1JPPt = 2,652 Hz). ESI-MS: 2,069 [calculated for (M − 2PF6)2+: 2,069], 1,331 [calculated for (M − 3PF6)3+: 1,331]. Analysis calculated for C196H230F30N7O5P17Pt6: C, 39.58; H, 5.23; N, 2.21. Found: C, 39.64; H, 5.24; N, 2.21.
Bicyclo[Tris[1,8-bis(trans-Pt(PEt3)2(NO3))anthracene]bis[1,3,5-Tris(4- ethynylpyridyl)adamantane]] (3b).
1H NMR (acetone-d6/D2O, 300 MHz): δ 9.17 (s, 3H, H9), 8.95 (d, 6H, 3JHH = 5.7 Hz, Hα-Py), 8.76 (d, 6H, 3JHH = 5.7 Hz, Hα-Py), 8.32 (s, 3H, H10), 7.85 (m, 12H, Hβ-Py), 7.66 (m, 12H, H2,4,5,7), 7.13 (m, 6H, H3,6), 1.37 (m, 72H, PCH2CH3), 0.82 (m, 108H, PCH2CH3). 31P{1H} NMR (acetone-d6/D2O, 121.4 MHz): δ 8.9 (s, 1JPPt = 2,656 Hz). ESI-MS: 2287 [calculated for (M − 2PF6)2+: 2,287], 1,476 [calculated for (M − 3PF6)3+: 1,476), 1,071 [calculated for (M − 4PF6)4+: 1,071). Analysis calculated for C204H272F36N6P18Pt6: C, 46.94; H, 5.25; N, 1.61. Found: C, 47.07; H, 5.27; N, 1.71.
Bicyclo[Tris[1,8-bis(trans-Pt(PEt3)2(NO3))anthracene]bis[1,1,1-Tris(4- phenyl(4′-ethynylpyridyl)ethane]] (3c).
1H NMR (acetone-d6/D2O, 300 MHz): δ 9.33 (s, 3H, H9), 9.06 (d, 6H, 3JHH = 5.4 Hz, Hα-Py), 8.91 (d, 6H, 3JHH = 5.7 Hz, Hα-Py), 8.40 (s, 3H, H10), 8.03 (m, 12H, Hβ-Py), 7.73 (d, 6H, 3JHH = 8.7 Hz, H4,5), 7.71 (d, 6H, 3JHH = 6.0 Hz, H2,7), 7.62 (d, 12H, 3JHH = 8.4 Hz, H3,5-phenylene), 7.24 (d, 12H, 3JHH = 8.7 Hz, H2,6-phenylene), 7.17 (m, 6H, H3,6), 1.46 (m, 72H, PCH2CH3), 0.87 (m, 108H, PCH2CH3). 31P{1H} NMR (acetone-d6/D2O, 121.4 MHz): δ 8.8 (s, 1JPPt = 2,641 Hz). ESI-MS: 2409 [calculated for (M − 2PF6)2+: 2,409], 1,557 [calculated for (M − 3PF6)3+: 1557), 1,132 [calculated for (M − 4PF6)4+: 1,132]. Analysis calculated for C196H258F36N6P18Pt6: C, 46.07; H, 5.09; N, 1.64. Found: C, 45.26; H, 5.03; N, 1.74.
X-Ray Data Collection, Structure Solution, and Refinement.
A single crystal of either 3a or 3c was selected from the reaction product and mounted on a thin glass fiber by using silicone grease. The data were collected at −100°C, using a narrow frame method with scan widths of 0.3° in ω and exposure times of 20 s. A hemisphere of intensity data were collected in 1,081 frames with the crystal-to-detector distance of 50.4 mm, which corresponds to a maximum 2θ value of 54.1°. Frames were integrated with the Bruker SAINT program (Billerica, MA). A semiempirical absorption correction based on simulated ψ-scans was applied to the data set. Both structures were solved by a combination of direct methods and difference Fourier methods, and refined with full-matrix least squares techniques. Details of the data collection, structure solution, and refinement are given in Table 1. Tables of the atomic coordinates, thermal displacement parameters, and selected bond distances and angles complete with ORTEP representations of 3a and 3c are provided in the supporting information, which is published on the PNAS web site, www.pnas.org.
Table 1.
Crystal data and structure refinement results for 3a and 3c
| Empirical formula | C146H230F36N6O2P18Pt6 (3a) | C194H258F36N6P18Pt6 (3c) |
| Formula weight | 4,513.36 | 5,086.06 |
| Color/shape | Yellow/trigonal prism | |
| Crystal system | Triclinic | Rhombohedral |
| Space group | P1̄ (#2) | R3̄c (#167) |
| Unit cell dimensions | a = 20.6055(5) Å | a = 30.6988(2) Å |
| b = 20.8610(5) Å | b = 30.6988(2) Å | |
| c = 29.2406(5) Å | c = 52.3624(8) Å | |
| α = 79.88(1)° | α = 90.00° | |
| β = 69.55(1)° | β = 90.00° | |
| γ = 61.39(1)° | γ = 120.00° | |
| Volume | 10,337.9(4) Å3 | 42,735.9(8) |
| Z | 2 | 6 |
| Density (calculated) | 1.450 mg/m3 | 1.186 mg/m3 |
| Absorption coefficient | 4.257 mm−1 | 3.096 mm−1 |
| F(000) | 4,452 | 15,156 |
| Crystal size | 0.31 × 0.19 × 0.17 mm | 0.24 × 0.17 × 0.15 |
| θ Range for data collection | 1.49 to 28.40 | 1.72 to 28.21 |
| Reflections collected | 123,286 | 11,538 |
| Independent/observed reflections | 48,757 (Rint = 0.088)/23700 ([I > 2σ(I)]) | 88,416 (Rint = 0.1265)/4256 ([I > 2σ(I)]) |
| Absorption correction | Semiempirical from simulated Ψ-scans | Semiempirical from simulated Ψ-scans |
| Range of relative transmission factors | 1.00, 0.841 | 1.00, 0.883 |
| Final R indices [I > 2σ(I)] | R = 0.0634, Rw = 0.1688 | R = 0.1492, Rw = 0.3827 |
| Goodness-of-fit on F | 0.965 | 1.300 |
| Weighting scheme | 1/[σ2(F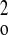 ) +
(0.1064P)2 + 0.0000P] where P = (F ) +
(0.1064P)2 + 0.0000P] where P = (F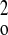 +
2F +
2F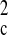 )/3w )/3w |
1/[σ2(F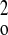 )
+ (0.2000P)2 + 0.0000P], where P =
(F )
+ (0.2000P)2 + 0.0000P], where P =
(F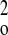 + 2F + 2F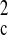 )/3w )/3w |
| Largest difference peak and hole | 3.47 and −2.33 eÅ−3 | 3.77 and −1.08 eÅ−3 |
Results and Discussion
The preparation of several discrete nanoscopic cage complexes can be achieved with simplicity and, given the proper conditions, in essentially quantitative yield. The assembly of these cages is most easily monitored by their 1H and 31P{1H} NMR spectra. For example, on gentle heating, a suspension of a 3:2 stoichiometric combination of 1 and 2a in a 1:1 (vol:vol) acetone-d6/D2O mixture (Scheme S1) gradually dissolves (3 h) to give a bright yellow homogeneous solution of the smallest molecular cage assembly described in this paper, structural motif 3a. 31P{1H} NMR (121.4 MHz) analysis of the reaction solution is consistent with the formation of a highly symmetrical species by the appearance of a sharp singlet with concomitant 195Pt satellites, shifted 5.0 ppm upfield (−Δδ) relative to 1 (Δ1JPPt = −203 ppm). Likewise, reaction of component 1 with tritopic bridging ligands 2b and 2c yielded the analogous molecular cages 3b and 3c, respectively, of differing dimensions and topology (Scheme S1).
Scheme 1.

Self-assembly of coordination cages 3a–3c.
Examination of the 1H NMR (300 MHz) spectra of cages 3a–3c was also indicative of highly symmetrical structures and displayed significant spectroscopic differences from their monomeric subunits. Particularly diagnostic were the significant downfield shifts of the pyridyl signals (Δδ ≈ 0.5 ppm), associated with the loss in electron density on coordination by the nitrogen lone pair to the platinum metal center. In accordance with previously reported molecular rectangles (48, 49), the inner and outer pyridyl protons were found to be inequivalent because of restricted rotation around the platinum–pyridine bond (50–54). After the reactions were judged to be complete by NMR, each product was isolated as its hexafluorophosphate salt by precipitation with excess KPF6.
Additional support for these structures was provided by ESI-MS.
Analysis of the hexafluorophosphate salts of
3a–3c all showed prominent M −
2PF6 and M − 3PF6
peaks (isotopically unresolved). Interestingly, the respective
2+ and 3+ peaks for
3a at m/z 2,069 and 1,331 were found
to be consistent with only five of the six nitrate counterions
exchanged for PF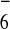 . This formulation was also
supported by elemental analysis. The nature of this distinction was
revealed by single crystal x-ray analysis of 3a.
. This formulation was also
supported by elemental analysis. The nature of this distinction was
revealed by single crystal x-ray analysis of 3a.
Quality single crystals of 3a grew after 2 days at ambient
temperature by vapor diffusion of diethyl ether into a concentrated
acetonitrile solution of the complex. It crystallizes in the triclinic
system (space group P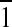 ) with an asymmetric unit cell that
contains a whole molecular prism, an NO
) with an asymmetric unit cell that
contains a whole molecular prism, an NO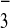 and five
PF
and five
PF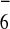 anions. The entire molecular prism is situated so
as to void any crystallographically imposed symmetry on the molecule.
However, a pseudo-3-fold rotation axis can still be identified to run
through the two OH groups. The most significant feature is that a
nitrate anion was found incarcerated inside the cavity of the cationic
complex (Fig. 1). It appears to be
strongly bound because it was not exchanged, even in the presence of a
large excess of KPF6. It is interesting to note
that the principal molecular axis of the NO
anions. The entire molecular prism is situated so
as to void any crystallographically imposed symmetry on the molecule.
However, a pseudo-3-fold rotation axis can still be identified to run
through the two OH groups. The most significant feature is that a
nitrate anion was found incarcerated inside the cavity of the cationic
complex (Fig. 1). It appears to be
strongly bound because it was not exchanged, even in the presence of a
large excess of KPF6. It is interesting to note
that the principal molecular axis of the NO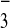 is
approximately in alignment with the pseudo-C3
axis of the prism; the three oxygen atoms of the nitrate anion extrude
into the space created between the molecular “clips.” It is
tempting to conjecture that the size and symmetry match between the
supermolecular cation and the nitrate anion may contribute a great deal
to this cation–anion affinity. As a point of note, the assembly of
3a was observed to be about an order of magnitude faster
than the formation of 3b and 3c. This
finding suggests that the nitrate anion is possibly providing a
templating effect, facilitating the formation of 3a.
Computer-generated van der Waals surfaces based on the x-ray data show
that the NO
is
approximately in alignment with the pseudo-C3
axis of the prism; the three oxygen atoms of the nitrate anion extrude
into the space created between the molecular “clips.” It is
tempting to conjecture that the size and symmetry match between the
supermolecular cation and the nitrate anion may contribute a great deal
to this cation–anion affinity. As a point of note, the assembly of
3a was observed to be about an order of magnitude faster
than the formation of 3b and 3c. This
finding suggests that the nitrate anion is possibly providing a
templating effect, facilitating the formation of 3a.
Computer-generated van der Waals surfaces based on the x-ray data show
that the NO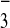 fits securely inside the host framework.
The solid-state asymmetry of the molecule is probably caused by an
outward distortion of the Pt–N bonds that appear to be a result of
slightly mismatched angles between the rigid molecular subunits; the
average trans-(C—Pt—N) bond angle is about 174°. The
dimensions of 3a offer a more detailed description of the
overall topology. The diameter is ≈19.1 Å, as defined by a circle
that the arms of the propeller sweep out when rotated on its 3-fold
axis (measured to the centroids of the anthracenes). The height, as
defined by the distance between the central carbon atoms of ligand
3a, is 9.34 Å. As expected, the pyridine groups were found
perpendicular to the coordination planes of the platinum metal centers.
fits securely inside the host framework.
The solid-state asymmetry of the molecule is probably caused by an
outward distortion of the Pt–N bonds that appear to be a result of
slightly mismatched angles between the rigid molecular subunits; the
average trans-(C—Pt—N) bond angle is about 174°. The
dimensions of 3a offer a more detailed description of the
overall topology. The diameter is ≈19.1 Å, as defined by a circle
that the arms of the propeller sweep out when rotated on its 3-fold
axis (measured to the centroids of the anthracenes). The height, as
defined by the distance between the central carbon atoms of ligand
3a, is 9.34 Å. As expected, the pyridine groups were found
perpendicular to the coordination planes of the platinum metal centers.
Figure 1.

Two perspectives of the x-ray structure of 3a. (Upper) The framework is shown as a stick model, and the encapsulated nitrate is represented with a space-filling model. All other counterions and the methyl groups from the triethylphosphines are omitted for clarity. (Lower) Space-filling model viewed down the C2 axis. Colors: C, blue; H, white; N, green; O, red; P, orange; Pt, gray.
Single crystals of the BF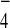 salt of 3b were
grown from a DMF/Et2O solvent system.
Examination and geometric data collection of three single crystals from
different batches of samples consistently revealed that 3b
crystallizes in the C-centered orthorhombic system with a =
54.868(4) Å, b = 76.086(4) Å, c = 27.958(2) Å, and V
= 116714(1) Å3. Intensity data collection on a yellow-orange single
crystal with dimensions of 0.33 × 0.25 × 0.21 mm at
−100°C afforded a total of 483,454 reflections. The systemic absence
of the reflections suggested the possible space group to be
C2221. Unfortunately, neither Patterson
approaches nor direct methods have yielded any reasonable structure
solution thus far. However, given that the NMR spectra and the ESI-MS
data for 3b are consistent with the smaller prism
(3a, see above), as well as the larger prism (3c,
see below), the structures of which have both been unambiguously
determined by x-ray crystallography, the structure of 3b can
be assigned as a trigonal prism with a high degree of confidence.
salt of 3b were
grown from a DMF/Et2O solvent system.
Examination and geometric data collection of three single crystals from
different batches of samples consistently revealed that 3b
crystallizes in the C-centered orthorhombic system with a =
54.868(4) Å, b = 76.086(4) Å, c = 27.958(2) Å, and V
= 116714(1) Å3. Intensity data collection on a yellow-orange single
crystal with dimensions of 0.33 × 0.25 × 0.21 mm at
−100°C afforded a total of 483,454 reflections. The systemic absence
of the reflections suggested the possible space group to be
C2221. Unfortunately, neither Patterson
approaches nor direct methods have yielded any reasonable structure
solution thus far. However, given that the NMR spectra and the ESI-MS
data for 3b are consistent with the smaller prism
(3a, see above), as well as the larger prism (3c,
see below), the structures of which have both been unambiguously
determined by x-ray crystallography, the structure of 3b can
be assigned as a trigonal prism with a high degree of confidence.
Quality single crystals of the largest assembly in this series,
metalla-prismatic cage 3c, grew over the course of 1 week at
ambient temperature by vapor diffusion of diethyl ether into a
concentrated DMF solution of the complex. The asymmetric unit cell of
3c (space group R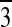 c) contains half of a
molecular “clip” (1) and a third of ligand
2c. The entire molecular trigonal prism is generated by
means of a 3-fold rotary inversion axis that runs through the central
carbon atoms of the ligands (Fig. 2).
Therefore, the molecule possesses crystallographically imposed
D3h symmetry. Because of the domination by
the heavy Pt atoms on the electron density maps, three terminal carbon
atoms on the triethylphosphine ligands could not be located from the
difference Fourier maps. On the other hand, the PF
c) contains half of a
molecular “clip” (1) and a third of ligand
2c. The entire molecular trigonal prism is generated by
means of a 3-fold rotary inversion axis that runs through the central
carbon atoms of the ligands (Fig. 2).
Therefore, the molecule possesses crystallographically imposed
D3h symmetry. Because of the domination by
the heavy Pt atoms on the electron density maps, three terminal carbon
atoms on the triethylphosphine ligands could not be located from the
difference Fourier maps. On the other hand, the PF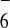 ions are located completely outside the prism, and situated near a
2-fold rotation axis, making it crystallographically disordered. The
structure refinement of 3c was based on a model reflecting
such disorders and gave r = 0.149. The packing diagram
shows that the molecular prisms stack on top of each other along the
crystallographic {001} direction (Fig.
3). Because the molecular axes of the
prisms coincide with each other and the molecular clips from the
neighboring prisms are in a staggered conformation, propeller-like
hexagonal patterns are generated by the cations packed throughout the
crystal lattice. The greater flexibility of the arms of the ligand
could account for the higher symmetry in the solid-state structure of
3c vs. 3a. This bending is accommodated primarily
by the acetylenic moieties. The overall dimensions of this structure
are about 38 Å in diameter, with a distance of about 11.5 Å
separating the central carbon atoms of ligand 2c. These
dimensions place 3c among the largest discrete
metallacyclic assemblies characterized by x-ray crystallography
to date.
ions are located completely outside the prism, and situated near a
2-fold rotation axis, making it crystallographically disordered. The
structure refinement of 3c was based on a model reflecting
such disorders and gave r = 0.149. The packing diagram
shows that the molecular prisms stack on top of each other along the
crystallographic {001} direction (Fig.
3). Because the molecular axes of the
prisms coincide with each other and the molecular clips from the
neighboring prisms are in a staggered conformation, propeller-like
hexagonal patterns are generated by the cations packed throughout the
crystal lattice. The greater flexibility of the arms of the ligand
could account for the higher symmetry in the solid-state structure of
3c vs. 3a. This bending is accommodated primarily
by the acetylenic moieties. The overall dimensions of this structure
are about 38 Å in diameter, with a distance of about 11.5 Å
separating the central carbon atoms of ligand 2c. These
dimensions place 3c among the largest discrete
metallacyclic assemblies characterized by x-ray crystallography
to date.
Figure 2.

Two perspectives of the x-ray structure of 3c. (Upper) Stick model as viewed down the C3 axis. Counterions and the methyl groups from the triethylphosphines are omitted for clarity. (Lower) Space-filling model viewed down the C2 axis. Colors: C, blue; H, white; N, green; P, orange; Pt, gray.
Figure 3.
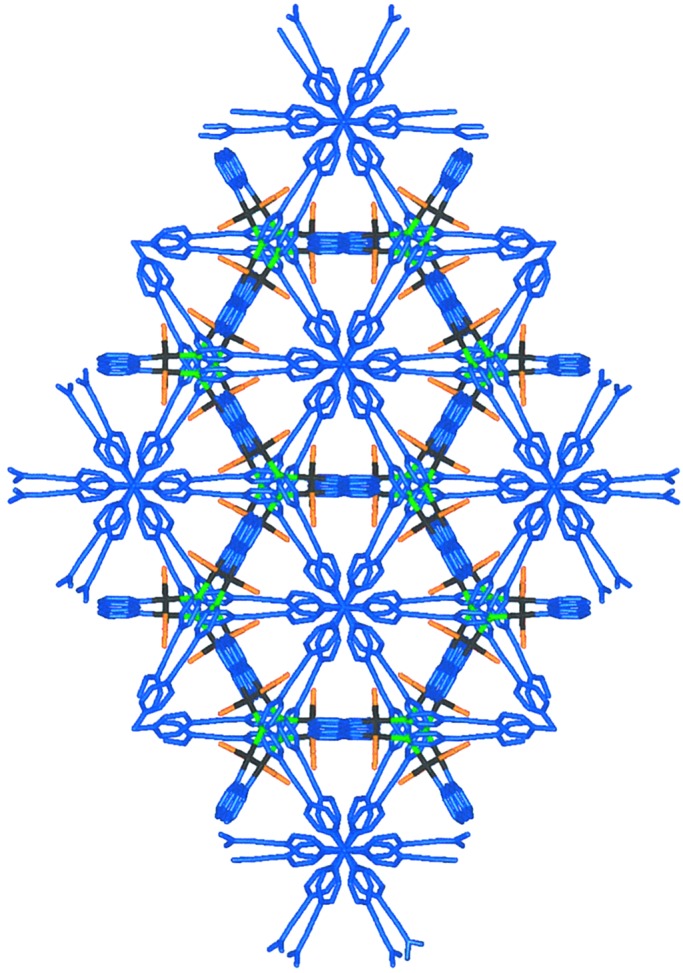
Crystal packing diagram of 3c as viewed down the {001} direction.
Conclusions
In summary, metalla-bicyclic supramolecular cages 3a–3c were prepared by means of spontaneous self-assembly based on a molecular “clip” (1) as a rigid modular subunit. Multinuclear NMR and ESI mass spectral data, in conjunction with x-ray crystallography, unambiguously established their structure, which showed that the sizes of the propeller-type three-dimensional structures range from 1 × 2 nm to 1 × 4 nm. The formation of the smallest of these structures, 3a, was accompanied by the entrapment of a nitrate ion inside its molecular cavity. Although the chemistry of metal-assembled molecular containers is steadily expanding, many of the desired features of that comprise guest selectivity are far from fully developed. Anionic guests appear to be the simplest and easiest starting points for controlled studies of inclusion phenomenon because of the inherent electrostatic affinity for the host framework. Ongoing investigations for this and related series of structures are being undertaken to assess the desired features of guest selectivity, reversibility, and dynamics.
Supplementary Material
Acknowledgments
We thank the National Science Foundation (Grant CHE-9818472) and the National Institute of Health (Grant 5R01GM57052) for support.
Abbreviation
- ESI
electrospray ionization
Footnotes
Data deposition: The atomic coordinates have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, Cambridge CB2 1EZ, United Kingdom (CSD reference nos. 172584 and 172585).
References
- 1.Holliday B J, Mirkin C A. Angew Chem Int Ed. 2001;40:2022–2043. [PubMed] [Google Scholar]
- 2. Fujita, M., Umemoto, K., Yoshizawa, M., Fujita, N., Kusukawa, T. & Biradha, K. (2001) Chem. Commun. 509–518.
- 3.Uller E, Demleitner I, Bernt I, Saalfrank R W, editors. Structure and Bonding. Berlin: Springer; 2000. [Google Scholar]
- 4.Swiegers G F, Malefetse T J. Chem Rev. 2000;100:3483–3537. doi: 10.1021/cr000023w. [DOI] [PubMed] [Google Scholar]
- 5.Leininger S, Olenyuk B, Stang P J. Chem Rev. 2000;100:853–908. doi: 10.1021/cr9601324. [DOI] [PubMed] [Google Scholar]
- 6.Caulder D L, Brückner C, Powers R E, König S, Parac T N, Leary J A, Raymond K N. J Am Chem Soc. 2001;123:8923–8938. doi: 10.1021/ja0104507. [DOI] [PubMed] [Google Scholar]
- 7.Caulder D L, Raymond K N. Acc Chem Res. 1999;32:975–982. [Google Scholar]
- 8. Caulder, D. L. & Raymond, K. N. (1999) J. Chem. Soc. Dalton Trans., 1185–1200.
- 9.Baxter P N W, Lehn J-M, Baum G, Fenske D. Chem Eur J. 1999;5:102–112. [Google Scholar]
- 10.Chambron J-C, Dietrich-Buchecker C, Sauvage J-P, editors. Comprehensive Supramolecular Chemistry. Oxford: Pergamon; 1996. [Google Scholar]
- 11.Campos-Fernández C S, Clérac R, Koomen J M, Russell D H, Dunbar K R. J Am Chem Soc. 2001;123:773–774. doi: 10.1021/ja002960r. [DOI] [PubMed] [Google Scholar]
- 12.Campos-Fernández C S, Clérac R, Dunbar K R. Angew Chem Int Ed Engl. 1999;38:3477–3479. doi: 10.1002/(sici)1521-3773(19991203)38:23<3477::aid-anie3477>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 13.Scherer M, Caulder D L, Johnson D W, Raymond K N. Angew Chem Int Ed Engl. 1999;38:1588–1592. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1587::AID-ANIE1587>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 14.Klausmeyer K K, Wilson S R, Rauchfuss T B. J Am Chem Soc. 1999;121:2705–2711. [Google Scholar]
- 15.McMorran D A, Steel P J. Angew Chem Int Ed Engl. 1998;37:3295–3297. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3295::AID-ANIE3295>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 16.Hasenknopf B, Lehn J-M, Boumediene N, Leize E, Van Dorsselaer A. Angew Chem Int Ed Engl. 1998;37:3265–3268. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3265::AID-ANIE3265>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 17.Fleming J S, Mann K L V, Carraz C-A, Psillakis E, Jeffrey J C, McCleverty J A, Ward M D. Angew Chem Int Ed Engl. 1998;37:1279–1281. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1279::AID-ANIE1279>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 18.Vilar R, Mingos D M P, White A J P, Williams D J. Angew Chem Int Ed Engl. 1998;37:1258–1261. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1258::AID-ANIE1258>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 19.Hasenknopf B, Lehn J-M, Boumediene N, Dupont-Gervais A, Van Dorsselaer A, Kniesel B, Fenske D. J Am Chem Soc. 1997;119:10956–10962. [Google Scholar]
- 20.Mann S, Huttner G, Zsolnai L, Heinze K. Angew Chem Int Ed Engl. 1996;35:2808–2809. [Google Scholar]
- 21.Hasenknopf B, Lehn J-M, Kniesel B O, Baum G, Fenske D. Angew Chem Int Ed Engl. 1996;35:1838–1840. [Google Scholar]
- 22.Beer P D, Gale P A. Angew Chem Int Ed Engl. 2001;40:486–516. [PubMed] [Google Scholar]
- 23.Schmidten F P, Berger M. Chem Rev. 1997;97:1609–1646. doi: 10.1021/cr9603845. [DOI] [PubMed] [Google Scholar]
- 24. Antonise, M. M. G. & Reinhoudt, D. N. (1998) Chem. Commun., 443–448.
- 25.Keefe M H, Benkstein K D, Hupp J T. Coord Chem Rev. 2000;205:221–243. [Google Scholar]
- 26.Chang S H, Chung K B, Slone R V, Hupp J T. Synth Metals. 2001;117:215–217. [Google Scholar]
- 27.Keefe M H, Slone R V, Hupp J T, Czaplewski K F, Snurr R Q, Stern C L. Langmuir. 2000;16:3964–3970. [Google Scholar]
- 28.Sun S-S, Lees A J. J Am Chem Soc. 2000;122:8956–8967. [Google Scholar]
- 29.Kang J, Rebek J., Jr Nature (London) 1997;385:50–52. doi: 10.1038/385050a0. [DOI] [PubMed] [Google Scholar]
- 30.Fujita M, Kwon Y J, Washizu S, Ogura K. J Am Chem Soc. 1994;116:1151–1152. [Google Scholar]
- 31.Fujita M, Nagao S, Ogura K. J Am Chem Soc. 1995;117:1649–1650. doi: 10.1021/ja00110a026. [DOI] [PubMed] [Google Scholar]
- 32.Hiraoka S, Fujita M. J Am Chem Soc. 1999;121:10239–10240. [Google Scholar]
- 33. Hiraoka, S., Kubota, Y. & Fujita, M. (2000) Chem. Commun., 1509–1510.
- 34.Ikeda A, Udzu H, Zhong Z, Shinkai S, Sakamoto S, Yamaguchi K. J Am Chem Soc. 2001;123:3872–3877. doi: 10.1021/ja003269r. [DOI] [PubMed] [Google Scholar]
- 35.Ikeda A, Yoshimura M, Udzu H, Fukuhara C, Shinkai S. J Am Chem Soc. 1999;121:4296–4297. [Google Scholar]
- 36.Sun W-Y, Fan J, Okamura T, Xie J, Yu K-B, Ueyama N. Chem Eur J. 2001;7:2557–2562. doi: 10.1002/1521-3765(20010618)7:12<2557::aid-chem25570>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 37. Liu, H.-K., Sun, W.-Y., Ma, D.-J., Yu, K.-B. & Tang, W.-X. (2000) Chem. Commun., 591–592.
- 38. Su, C.-Y., Cai, Y.-P., Chen, C.-L., Zhang, H.-X. & Kang, B.-S. (2001) J. Chem. Soc. Dalton Trans., 359–361.
- 39. Manimaran, B., Rajendran, T., Lu, Y.-L., Lee, G.-H., Peng, S.-M. & Lu, K.-L. (2001) Eur. J. Inorg. Chem., 633–636.
- 40. Lindner, E., Hermann, C., Baum, G. & Fenske, D. (1999) Eur. J. Inorg. Chem., 679–685.
- 41.Fujita M, Oguro D, Miyazawa M, Oka H, Yamaguchi K, Ogura K. Nature (London) 1995;378:469–471. [Google Scholar]
- 42.Fujita M, Yu S-Y, Kusukawa T, Funaki H, Ogura K, Yamaguchi K. Angew Chem Int Ed Engl. 1998;37:2082–2085. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2082::AID-ANIE2082>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 43.Sun X, Johnson D W, Raymond K N, Wong E H. Inorg Chem. 2001;40:4504–4506. doi: 10.1021/ic010154v. [DOI] [PubMed] [Google Scholar]
- 44.Sun X, Johnson D W, Caulder D L, Raymond K N, Wong E H. J Am Chem Soc. 2001;123:2752–2763. doi: 10.1021/ja0029376. [DOI] [PubMed] [Google Scholar]
- 45.Sun X, Johnson D W, Caulder D L, Powers R E, Raymond K N, Wong E H. Angew Chem Int Ed Engl. 1999;38:1303–1307. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1303::AID-ANIE1303>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 46.Manna J, Kuehl C J, Whiteford J A, Stang P J. Organometallics. 1997;16:1897–1905. [Google Scholar]
- 47.Olenyuk B O, Levin M D, Whiteford J A, Shield J E, Stang P J. J Am Chem Soc. 1999;121:10434–10435. [Google Scholar]
- 48.Kuehl C J, Huang S D, Stang P J. J Am Chem Soc. 2001;123:9634–9641. doi: 10.1021/ja0114355. [DOI] [PubMed] [Google Scholar]
- 49.Kuehl C J, Mayne C L, Arif A M, Stang P J. Org Lett. 2000;2:3727–3729. doi: 10.1021/ol006638x. [DOI] [PubMed] [Google Scholar]
- 50.Gallasch D P, Tiekink E R T, Rendina L M. Organometallics. 2001;20:3373–3382. [Google Scholar]
- 51.Fuss M, Siehl H-U, Olenyuk B, Stang P J. Organometallics. 1999;18:758–769. [Google Scholar]
- 52.Stang P J, Olenyuk B O, Arif A M. Organometallics. 1995;14:5281–5289. [Google Scholar]
- 53.Brown J M, Pérez-Torrente J J, Alcock N W. Organometallics. 1995;14:1195–1203. [Google Scholar]
- 54.Alcock N W, Brown J M, Pérez-Torrente J J. Tetrahedron Lett. 1992;33:389–392. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.