

Abstract
There is considerable interest in synthetic ionophores with high affinity and selectivity for Li+. But so far, compounds that selectively bind Li+ in the presence of other alkali and alkaline earth metal ions are rare and current approaches toward this goal are often accompanied with substantial synthetic efforts. Here we describe a trinuclear ruthenium metallamacrocyclic complex (1) that was obtained by self-assembly of ruthenium halfsandwich complexes and 3-hydroxy-2-pyridone ligands. This complex was shown to be an extremely potent receptor for LiCl with an affinity high enough to extract LiCl from water. The selectivity of this receptor is exceptional: even in the presence of a large excess of Na+, K+, Cs+, Ca2+, and Mg2+, Li+ was extracted exclusively. The Li+/Na+ selectivity ratio was determined to be higher than 1,000:1. Compared with other synthetic ionophores, the receptor 1 offers two additional advantages: (i) the synthesis can be accomplished in one step by using simple starting materials; and (ii) the presence of lithium ions can be detected electrochemically. Complex 1 is therefore a very attractive candidate for the construction of a Li+-specific chemosensor.
Lithium salts have found various applications in technology and medicine. Methods to selectively sequester and detect Li+ are therefore of importance. Li2CO3, for example, is one of the most important drugs for the treatment of manic depression (1, 2). Because of its narrow therapeutic range, the Li+ concentration in the blood of the patients needs to be controlled during the treatment. Thus, a small amount Li+ has to be detected in the presence of other metal ions such as Na+, K+, Ca2+, and Mg2+ (3).
To selectively sequester Li+, ionophores have been considered early on. But the design and the synthesis of molecules that display a high affinity and selectivity for Li+ is a challenging task (4–9). Although considerable progress has been made in this field, the synthesis of such ionophores often requires substantial synthetic efforts. This difficulty is nicely illustrated by probably the best Li+ receptor known so far, a spherand developed by Cram and his colleagues (10). Because of the perfect preorganization of six oxygen donor atoms, a very high affinity combined with an excellent selectivity for Li+ is observed. The synthesis, however, requires several steps and gives less than 7% overall yield.
Here we describe a redox-responsive metallamacrocycle (1; see Scheme S1) that was obtained by self-assembly (for self-assembled metallacrowns see ref. 11). With this receptor we were able to selectively extract LiCl from an aqueous solution containing an excess of NaCl, KCl, CsCl, CaCl2, and MgCl2. The presence of LiCl can subsequently be detected by electrochemical means.
Scheme 1.

Synthesis of metallamacrocycle 1.
Materials and Methods
General.
The synthesis of all complexes was performed under an atmosphere of dry dinitrogen, by using standard Schlenk techniques. The 1H, 13C, and 7Li spectra were recorded on a JEOL EX 400 or a GSX 270 spectrometer using the residual protonated solvents as internal standards (1H, 13C) or LiCl in D2O as the external standard. The M+ and M2+ concentrations were determined by inductively coupled plasma atomic emission spectroscopy with a Varian Vista inductively coupled plasma atomic emission spectrometer. Potentials were measured against a Ag/AgCl reference electrode by using a glassy carbon working electrode at a scan rate of 4,015 mV/s. The receptor 1 or its LiCl adduct was dissolved in dry CH3CN/CH2Cl2 (1:1) (5 mM) containing tetra-n-butylammonium perchlorate as the supporting electrolyte (0.1 M) under a dinitrogen atmosphere. The molecular weights were determined with a JEOL MStation JMS 700 mass spectrometer in the fast atom bombardment (FAB) mode by using m-nitrobenzyl alcohol (NBA) as the matrix.
Complex 1.
A suspension of [(C6H5CO2Et)RuCl2]2 (322 mg, 0.50 mmol), 3-hydroxy-2-pyridone (111 mg, 1.00 mmol), and Cs2CO3 (815 mg, 2.50 mmol) in degassed CH2Cl2 (30 ml) was stirred for 20 h at room temperature. After filtration, hexane (20 ml) was added, and the solvent was evaporated to give an orange powder (yield: 320 mg, 89%). Crystals were obtained by vapor diffusion of pentane into a solution of 1 in benzene. IR (KBr): ν (cm−1) = 1,720 (s), 1,630 (br, m), 1,598 (m, sh), 1,541 (s), 1,453 (br, s); 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.28 (d, 3J = 7 Hz, 9 H, CH3), 4.26–4.34 (m, 6 H, CH2), 5.53 (pt, 3J = 6 Hz, 3 H, CH, C6H5CO2Et), 5.64 (pt 3J = 6 Hz, 3 H, CH, C6H5CO2Et), 5.68 (dd, 3J = 6 Hz 3J = 7 Hz, 3 H, CH, C6H3NO2), 5.78 (pt, 3J = 6 Hz, 3 H, CH, C6H5CO2Et), 6.17 (dd, 3J = 7 Hz, 4J = 2 Hz, 3 H, CH, C6H3NO2), 6.45 (d, 3J = 6 Hz, 3 H, CH, C6H5CO2Et), 6.51 (dd, 3J = 6 Hz, 4J = 2 Hz, 3 H, CH, C6H3NO2), 6.69 (d, 3J = 6 Hz, 3 H, CH, C6H5CO2Et); 13C NMR (101 MHz, CDCl3): δ (ppm) = 14.29 (CH3), 62.13 (CH2), 76.00, 77.49, 79.48, 87.04, 88.00, 89.20 (C6H5CO2Et), 110.78, 115.33, 131.99, 156.21, 166.61, 170.63 (C6H3NO2 + CO2); Mcalc, = 1,082, MS (FAB+): (m/z): 1,083 [M + H]+; elemental analysis (%) calculated for C42H39N3O12Ru3⋅H2O: C 45.90, H 3.76, N 3.82; found: C 45.60, H 3.72, N 3.74.
Complex 1⋅LiCl.
A solution of the receptor 1 (60 mg, 55 μmol) and an excess of LiCl in ethanol was stirred for 1 h. After evaporation of the solvent under reduced pressure, the LiCl adduct was extracted with CH2Cl2. Evaporation of the solvent gave the products as an orange powder (yield: 51 mg, 80%). Crystals were obtained by vapor diffusion of pentane into a solution of 1⋅LiCl in benzene. IR (KBr): ν (cm−1) = 1,723 (s), 1,641 (w), 1,595 (m), 1,548 (s), 1,450 (s), 1,438 (s); 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.24 (d, 3J = 7 Hz, 9 H, CH3), 4.16–4.30 (m, 6 H, CH2), 5.89 (dd, 3J = 6 Hz, 3J = 7 Hz, 3 H, CH, C6H3NO2), 6.19 (pt, 3J = 6 Hz, 3 H, CH, C6H5CO2Et), 6.29 (pt, 3J = 6 Hz, 3 H, CH, C6H5CO2Et), 6.39 (dd, 3J = 7 Hz, 4J = 2 Hz, 3 H, CH, C6H3NO2), 6.44 (pt, 3J = 6 Hz, 3 H, CH, C6H5CO2Et), 6.50 (d, 3J = 6 Hz, 3 H, CH, C6H5CO2Et), 6.58 (dd, 3J = 6 Hz, 4J = 2 Hz, 3 H, CH, C6H3NO2), 7.13 (d, 3J = 6 Hz, 3 H, CH, C6H5CO2Et); 13C NMR (101 MHz, CDCl3): δ (ppm) = 14.27 (CH3), 62.29 (CH2), 75.23, 78.77, 79.53, 88.70, 89.10, 90.26 (C6H5CO2Et), 113.36, 118.38, 132.68, 155.54, 165.61, 167.52 (C6H3NO2 + CO2); Mcalc = 1,123, MS (FAB+): (m/z): 1,089 [M − C−]; elemental analysis (%) calculated for C42H39ClLiN3O12Ru3⋅2H2O: C 43.51, H 3.74, N 3.62; found: C 43.31, H 3.34, N 3.56.
Complex 1⋅NaCl.
The synthesis was performed analogous to that of 1⋅LiCl. Crystals were obtained by vapor diffusion of pentane into a solution of 1 × NaCl in benzene (yield: 56 mg, 75%). IR (KBr): ν (cm−1) = 1,725 (s), 1,636 (w), 1,596 (m), 1,544 (s), 1,453 (s), 1,437 (s, sh); 1H NMR (400 MHz, CDCl3): δ (ppm) = 1.24 (d, 3J = 7 Hz, 9 H, CH3), 4.17–4.24 (m, 6 H, CH2), 5.83 (pt, 3J = 7 Hz, 3 H, CH, C6H3NO2), 6.04 (pt, 3J = 6 Hz, 3 H, CH, C6H5CO2Et), 6.18–6.25 (m, 6 H, CH, C6H5CO2Et), 6.32 (dd, 3J = 7 Hz, 4J = 1 Hz, 3 H, CH, C6H3NO2), 6.53–6.56 (m, 6 H, CH, C6H3NO2 + C6H5CO2Et), 6.88 (d, 3J = 5 Hz, 3 H, CH, C6H5CO2Et); 13C NMR (101 MHz, CDCl3): δ (ppm) = 14.22 (CH3), 62.48 (CH2), 77.30, 78.79, 79.76, 86.74, 87.77, 88.60 (C6H5CO2Et), 112.88, 117.74, 132.59, 155.42, 165.24, 168.48 (C6H3NO2 + CO2); Mcalc = 1139, MS (FAB+): (m/z): 1104 [M − Cl−]; elemental analysis (%) calculated for C42H39ClN3NaO12Ru3⋅2C6H6⋅3H2O: C 48.05, H 4.26, N 3.11; found: C 47.71, H 4.36, N 3.15.
Crystal Structures.
Analysis of 1⋅2H2O: Nonius Kappa
charge-coupled device, Mo Kα radiation, λ = 0.71073 Å,
crystal size 0.30 × 0.10 × 0.03 mm. The crystal was
embedded in oil and mounted with Lithelen, T = 200(2)
K, orange platelet, triclinic, space group P − 1,
a = 10.23390(10), b = 13.9785(2),
c = 14.9933(2) Å, α = 74.8537(6)°, β =
86.9735(7)°, γ = 87.5378(8)°, V = 2066.52(5)
Å3, Z = 2,
ρcalc = 1.795
Mg⋅m−3, μ = 1.156
mm−1. Data collection: 2θ from 2.82 to 48.00,
−11 ≤ h ≤ 11, −15 ≤ k ≤
15, −17 ≤ l ≤ 17, 25,509 reflections collected,
6,472 independent reflections, 576 parameters, 6 restraints, numerical
absorption correction, maximum/minimum transmission 0.9518/0.7840,
R1 = 0.0433,
wR2 = 0.1116, goodness of fit
(F2 = 1.164, largest difference peak
0.756 e Å−3, largest difference hole −1.294 e
Å−3, the weighting scheme is
w−1 =
σ2F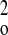 +
(0.0654P)2 + 1.0411P
with P = (F
+
(0.0654P)2 + 1.0411P
with P = (F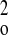 +
2F
+
2F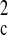 )/3. The structure was solved
with direct methods (SIR97) (12) and was refined by
full-matrix least-square methods on F2
(SHELXL97) (13). A riding model was used for the
hydrogen atoms. Restraints were used for the H atoms of the
water molecules. Analysis of
1⋅LiCl⋅H2O: Nonius Kappa
charge-coupled device, Mo Kα radiation, λ = 0.71073 Å,
crystal size 0.18 × 0.08 × 0.03 mm. The crystal was
embedded in oil and mounted with Lithelen, T = 200(2)
K, orange prismatic crystal, monoclinic, space group
P21/c, a
= 10.49560(10), b = 21.3138(2), c =
18.5551(2) Å, β = 92.0417(4)°, V = 4148.16(7)
Å3, Z = 4,
ρcalc = 1.828
Mg⋅m−3, μ = 1.214
mm−1. Data collection: 2θ from 2.92 to 48.00,
−12 ≤ h ≤ 12, −2 ≤ k ≤
24, −21 ≤ l ≤ 21, 59,142 reflections collected,
6,502 independent reflections, 578 parameters, 3 restraints, numerical
absorption correction, maximum/minimum transmission 0.9640/0.8440,
R1 = 0.0603,
wR2 = 0.1345, goodness of fit
(F2 = 1.122, largest difference peak
0.936 e Å−3, largest difference hole −1.271 e
Å−3, the weighting scheme is
w−1 =
σ2F
)/3. The structure was solved
with direct methods (SIR97) (12) and was refined by
full-matrix least-square methods on F2
(SHELXL97) (13). A riding model was used for the
hydrogen atoms. Restraints were used for the H atoms of the
water molecules. Analysis of
1⋅LiCl⋅H2O: Nonius Kappa
charge-coupled device, Mo Kα radiation, λ = 0.71073 Å,
crystal size 0.18 × 0.08 × 0.03 mm. The crystal was
embedded in oil and mounted with Lithelen, T = 200(2)
K, orange prismatic crystal, monoclinic, space group
P21/c, a
= 10.49560(10), b = 21.3138(2), c =
18.5551(2) Å, β = 92.0417(4)°, V = 4148.16(7)
Å3, Z = 4,
ρcalc = 1.828
Mg⋅m−3, μ = 1.214
mm−1. Data collection: 2θ from 2.92 to 48.00,
−12 ≤ h ≤ 12, −2 ≤ k ≤
24, −21 ≤ l ≤ 21, 59,142 reflections collected,
6,502 independent reflections, 578 parameters, 3 restraints, numerical
absorption correction, maximum/minimum transmission 0.9640/0.8440,
R1 = 0.0603,
wR2 = 0.1345, goodness of fit
(F2 = 1.122, largest difference peak
0.936 e Å−3, largest difference hole −1.271 e
Å−3, the weighting scheme is
w−1 =
σ2F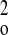 +
(0.0852P)2 with
P = (F
+
(0.0852P)2 with
P = (F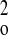 +
2F
+
2F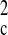 )/3. The structure was solved
with direct methods (SIR97) (12) and was refined by
full-matrix least-square methods on F2
(SHELXL97) (13). A riding model was used for the
hydrogen atoms. Restraints were used for the H atoms of the water
molecule. Analysis of
1⋅NaCl⋅H2O⋅2C6H6:
Nonius Kappa charge-coupled device, Mo Kα radiation, λ =
0.71073 Å, crystal size 0.28 × 0.14 × 0.04 mm. The crystal
was embedded in oil and mounted with Lithelen, T =
200(2) K, orange platelet, triclinic, space group P −
1, a = 10.7683(1), b = 15.6801(2),
c = 16.8725(2) Å, α = 70.3112(7)°, β =
85.9480(7)°, γ = 76.1555(5)°, V = 2604.25(5)
Å3, Z = 2,
ρcalc = 1.675
Mg⋅m−3, μ = 0.987
mm−1. Data collection: 2θ from 2.0 to 48.0,
−12 ≤ h ≤ 12, −17 ≤ k ≤
17, −19 ≤ l ≤ 19, 34,130 reflections collected,
8,183 independent reflections, 639 parameters, 15 restraints, numerical
absorption correction, maximum/minimum transmission 0.9624/0.7903,
R1 = 0.0694,
wR2 = 0.1723, goodness of fit
(F2 = 1.148, largest difference peak
1.430 e Å−3, largest difference hole −1.546 e
Å−3, the weighting scheme is
w−1 =
σ2F
)/3. The structure was solved
with direct methods (SIR97) (12) and was refined by
full-matrix least-square methods on F2
(SHELXL97) (13). A riding model was used for the
hydrogen atoms. Restraints were used for the H atoms of the water
molecule. Analysis of
1⋅NaCl⋅H2O⋅2C6H6:
Nonius Kappa charge-coupled device, Mo Kα radiation, λ =
0.71073 Å, crystal size 0.28 × 0.14 × 0.04 mm. The crystal
was embedded in oil and mounted with Lithelen, T =
200(2) K, orange platelet, triclinic, space group P −
1, a = 10.7683(1), b = 15.6801(2),
c = 16.8725(2) Å, α = 70.3112(7)°, β =
85.9480(7)°, γ = 76.1555(5)°, V = 2604.25(5)
Å3, Z = 2,
ρcalc = 1.675
Mg⋅m−3, μ = 0.987
mm−1. Data collection: 2θ from 2.0 to 48.0,
−12 ≤ h ≤ 12, −17 ≤ k ≤
17, −19 ≤ l ≤ 19, 34,130 reflections collected,
8,183 independent reflections, 639 parameters, 15 restraints, numerical
absorption correction, maximum/minimum transmission 0.9624/0.7903,
R1 = 0.0694,
wR2 = 0.1723, goodness of fit
(F2 = 1.148, largest difference peak
1.430 e Å−3, largest difference hole −1.546 e
Å−3, the weighting scheme is
w−1 =
σ2F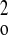 +
(0.1175P)2 with P =
(F
+
(0.1175P)2 with P =
(F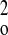 +
2F
+
2F )/3. The structure was solved
with direct methods (SIR97) (12) and was refined by
full-matrix least-square methods on F2
(SHELXL97) (13). A riding model was used for the
hydrogen atoms. Restraints were used for the H atoms of the water
molecule and for the benzene molecules. Two ester groups and the
benzene molecules are twofold disordered.
)/3. The structure was solved
with direct methods (SIR97) (12) and was refined by
full-matrix least-square methods on F2
(SHELXL97) (13). A riding model was used for the
hydrogen atoms. Restraints were used for the H atoms of the water
molecule and for the benzene molecules. Two ester groups and the
benzene molecules are twofold disordered.
Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre [CDS reference nos. CCDC-167800 (1), CCDC-167801 (1⋅LiCl), and CCDC-167802 (1⋅NaCl). Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, United Kingdom (E-mail: deposit@ccdc.cam.ac.uk).
Results and Discussion
In elaboration of a synthetic scheme which we have reported recently (14, 15), the trinuclear metallamacrocycle 1 was obtained by reaction of commercially available 3-hydroxy-2-pyridone with [(C6H5CO2Et)RuCl2]2 in the presence of base (Scheme S1). The chloro-bridged ruthenium complex used in this reaction is a common starting material in organometallic chemistry and can easily be obtained from RuCl3⋅(H2O)n and ethyl-1,4-cyclohexadiene-3-carboxylate (16).
The metallamacrocycle 1 was characterized by IR and NMR spectroscopy (1H, 13C), elemental analysis, mass spectrometry, and single-crystal x-ray analysis (Fig. 1). The three (arene)RuII-fragments are connected by dianionic 3-oxo-2-pyridonate ligands with the ruthenium atoms being 5.26 Å apart form each other [for other trimeric Cp*Rh- and arene-Ru complexes see refs. 17–21 (Cp* represents pentamethylcyclopentadienyl)]. The bond length and angles are within the expected range (15). The complex cocrystallizes with two molecules of water, one of which is found inside the cavity. Complex 1 displays a good solubility in a variety of organic solvents such as chloroform, methanol, and benzene.
Figure 1.

Molecular structure of 1 in the crystal together with the water molecule found inside the cavity.
The ethyl ester side chains on the arene ligands were introduced for two reasons. First, the electron-withdrawing character of these groups should increase the stability of the complex toward oxidation. And indeed, a solution of 1 can be exposed to air for several hours without decomposition, whereas an orange solution of the analogous benzene complex [(C6H6)Ru(C5H3NO2)]3 (15) rapidly turns brown. This qualitative result was confirmed by cyclic voltammetry (see below). Second, the oxygen atoms are potentially suited to participate in cation binding and can therefore facilitate complexation reactions.
To test whether complex 1 is able to bind Li+, we have investigated the reaction of 1 with an excess of LiCl in ethanol. After removal of the solvent and extraction with chloroform, the adduct 1⋅LiCl was obtained. The presence of the guest molecule results in pronounced differences in the 1H NMR spectrum of the macrocycle: the signals of the pyridonate protons as well as of the arene protons are shifted toward lower field (up to 0.66 ppm). Because the exchange of LiCl is slow compared with the NMR time scale (CDCl3 or CD3OD), NMR spectroscopy is an ideal method to quantify binding of LiCl under different conditions.
The expected coordination of Li+ to the three adjacent O atoms of the macrocyclic receptor was confirmed by a single-crystal x-ray analysis of 1⋅LiCl (Fig. 2). The lithium ion has a tetrahedral coordination geometry with a terminal chloride anion occupying the fourth site [Li–O range = 1.917(10) to 1.948(11) Å, Li—Cl = 2.372(9) Å]. The chloride anion is closely encapsulated by the arene ligands resulting in three short CH⋅⋅⋅Cl distances (2.70, 2.73, and 2.83 Å). The ester groups point away from the center and do not participate directly in cation binding.
Figure 2.
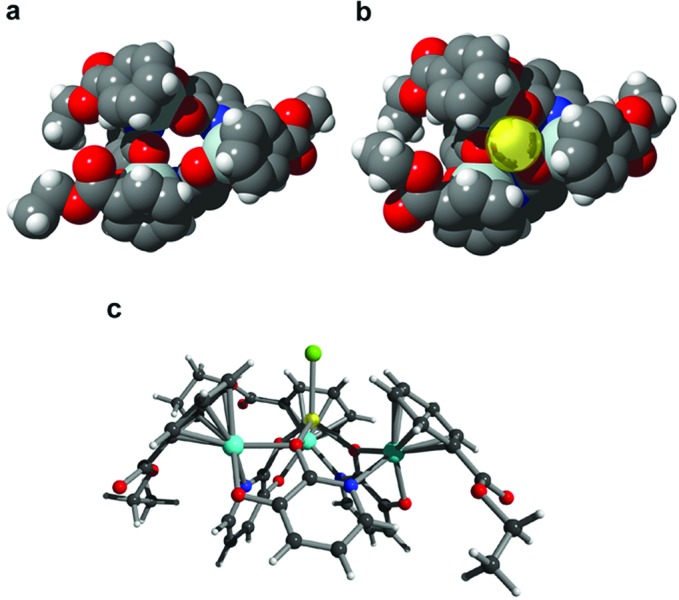
Corey–Pauling–Koltun representation of the receptor 1 the corresponding Li+ complex (b) (view along the pseudo C3 axis). The Li+ ion is located inside the cavity of the macrocycle and is bound to the three adjacent O atoms. The chloride anion is not shown. (c) Ball-and-stick representation of 1⋅LiCl (side view).
Na+ is likewise able to coordinate to the three adjacent O atoms of 1. Thus, we were able to synthesize 1⋅NaCl, which displays a structure in the crystal very similar to that of 1 × LiCl [Na–O range = 2.210(4) to 2.286(4) Å, Na–Cl = 2.581(3) Å, CH⋅⋅⋅Cl = 2.87, 2.90, and 2.95 Å]. The 1H NMR spectrum of 1⋅NaCl shows pyridonate signals that are shifted toward lower field, but the spectrum is clearly distinguishable from that of 1⋅LiCl. For K+ no complexation could be observed.
The affinity and selectivity of receptor 1 for LiCl was determined in extraction experiments. First, a CDCl3 solution of 1 (10 mM) was stirred vigorously with an aqueous solution of LiCl (2 M). After 24 h, the quantitative formation of 1⋅LiCl was observed by 1H NMR. It should be noted that LiCl is very difficult to extract from water because of the high enthalpy of hydration of Li+ (−521 kJ/mol) and Cl− (−363 kJ/mol) (22). There are only a few synthetic ionophores that have a high enough affinity for Li+ to extract LiCl from water to a significant extent (10, 23). Consequently, lithium salts with more lipophilic anions are generally used in experiments of this kind (4–9). The high affinity of 1 for LiCl can be attributed to several facts. (i) The three oxygen donor atoms are perfectly preorganized to bind Li+ (on binding of LiCl, the average O–O′ distance is reduced by only 0.07 Å). (ii) The energetic costs for the desolvation of the donor atoms are very low because only one molecule of water can fit inside the binding cavity (see Fig. 1). (iii) The lithium salt is bound as an ion pair and unfavorable charge separation is thus avoided.
The selectivity of 1 (CDCl3, 10 mM) was determined in competition experiments using an aqueous solution of LiCl (50 mM) containing a large excess of NaCl, KCl, CsCl, CaCl2, and MgCl2 (1 M each). Only LiCl was extracted by receptor 1: the 1H NMR spectrum of the CDCl3 phase showed exclusively the characteristic signals of 1⋅LiCl (>95%). The preferential extraction of LiCl was also confirmed by inductively coupled plasma atomic emission spectroscopy. The high selectivity for Li+ over Na+, K+, and Cs+ is remarkable, especially because the enthalpy of hydration is significantly smaller for the other alkali metal ions.
It is interesting to compare our receptor with the metallamacrocyclic ionophores that we have reported recently (15). Competitive extraction experiments described above were performed by using the structurally related trinuclear complex [(cymene)Ru(C5H3NO2)]3 (cymene = p-iPr-C6H4-CH3) (2). With the cymene complex 2, Na+ instead of Li+ was extracted dominantly (1H NMR analysis: Na+/Li+ = 6:1). Apparently, the replacement of the para-alkyl groups with one ethyl ester group changes the selectivity dramatically. In view of the fact that the macrocyclic cores of the two receptors are essentially the same, this effect is remarkable and unexpected. With the triethylbenzene complex [(1,3,5-C6H3Et3)Ru(C5H3NO2)]3 (3), on the other hand, neither metal ion is extracted to a significant extent. Although this receptor was shown to be selective for Li+ (15), its affinity is too small to extract LiCl.
Because NaCl is entrapped by receptor 1 under favorable conditions, we were interested whether selective LiCl extraction is also possible in the presence of very large amounts of NaCl. Therefore, we have performed a competition experiment similar to those described above by using a nearly saturated NaCl solution (5 M) that contained only 1% LiCl (50 mM). Again, the dominant formation of 1 × LiCl (>90%) was observed by 1H NMR revealing a Li+/Na+ selectivity of at least 1,000:1. The reduced stability of 1⋅NaCl as compared with 1⋅LiCl may partially be attributed to the fact that Na+ prefers higher coordination numbers (for a metallacrown complex with a tetrahedral sodium ion see ref. 24). But steric and/or electronic effects of the ester groups are clearly of central importance considering the pronounced difference in selectivity of 1 and 2 of more than three orders of magnitude. The very low affinity of the receptor 1 for K+ and Cs+ is most likely caused by the three arene ligands, which effectively block these larger ions from entering the binding pocket (Fig. 2).
We have also studied the redox behavior of the macrocyclic receptor 1 and of the complex 1⋅LiCl by using cyclic voltammetry. For 1, three irreversible oxidations are observed at 683, 963, and 1,150 mV (against Ag/AgCl). These values are higher than those found for the [(arene)Ru(C5H3NO2)]3 complexes previously described (15), indicating an increased stability of 1 toward oxidation. In the presence of LiCl, the first peak potential is shifted by more than 350 mV toward anodic potential. Thus, 1 can be used to detect Li+ electrochemically (25).
Conclusions
The metallomacrocyclic complex 1 represents a very potent receptor for Li+, allowing the quantitative extraction of LiCl from water. Furthermore, the receptor shows an outstanding selectivity that is comparable to that of the best Li+ ionophores described so far. Control experiments with structurally related complexes have shown that the selectivity is modulated to a surprising extent by subtle steric and electronic modifications. Applications in Li+ sensing can be envisioned, especially because the synthesis of 1 can be accomplished in a simple self-assembly process and because the binding of the alkali metal ion can be detected electrochemically. The results highlight the potential of supramolecular coordination chemistry to build functional devices such as highly selective chemosensors.
Acknowledgments
We thank Prof. Dr. W. Beck for his generous support, E. Karaghiosoff and I. C. Sielaff for technical assistance, and Dr. G. Hilt for the cyclic voltammetry studies. Financial funding from the Deutsche Forschungsgemeinschaft is gratefully acknowledged.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, Cambridge CB2 1EZ, United Kingdom [CSD reference nos. CCDC-167800 (1), CCDC-167801 (1⋅LiCl), and CCDC-167802 (1⋅NaCl)].
References
- 1.Johnson F N. Depression and Mania: Modern Lithium Therapy. Oxford: IRL Press; 1989. [Google Scholar]
- 2.Friedrich M J. J Am Med Assoc. 1999;281:2271–2273. doi: 10.1001/jama.281.24.2271. [DOI] [PubMed] [Google Scholar]
- 3.Christian G D. J Anal Biomed Anal. 1996;14:899–908. doi: 10.1016/s0731-7085(96)01743-8. [DOI] [PubMed] [Google Scholar]
- 4.Bartsch R A, Ramesh V, Bach R O, Shono T, Kimura K. In: Lithium Chemistry. Sapse A-M, von Ragué Schleyer P, editors. New York: Wiley; 1995. pp. 393–476. [Google Scholar]
- 5.Kobiro K. Coord Chem Rev. 1996;148:135–149. [Google Scholar]
- 6.Paquette L A, Tae J. J Am Chem Soc. 2001;123:4974–4984. doi: 10.1021/ja010103x. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Yang F, Gong S. Tetrahedron Lett. 2000;41:4815–4818. [Google Scholar]
- 8.Paquette L A, Tae J, Hickey E R, Rogers R D. Angew Chem Int Ed Engl. 1999;38:1409–1411. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1409::AID-ANIE1409>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 9.Inokuma S, Takezawa M, Satoh H, Nakamura Y, Sasaki T, Nishimura J. J Org Chem. 1998;63:5791–5796. doi: 10.1021/jo9801521. [DOI] [PubMed] [Google Scholar]
- 10.Cram D J. Angew Chem Int Ed Engl. 1986;25:1039–1057. [Google Scholar]
- 11.Pecoraro V L, Stemmler A J, Gibney B R, Bodwin J J, Wang H, Kampf J W, Barwinski A. Prog Inorg Chem. 1997;45:83–177. [Google Scholar]
- 12.Altomare A, Burla M C, Camalli M, Cascarano G L, Giacovazzo C, Guagliardi A, Moliterni A G G, Polidori G, Spagna R. J Appl Crystallogr. 1999;32:115–119. [Google Scholar]
- 13.Sheldrick G M. SHELXL97, Program for Crystal Structure Refinement. Göttingen, Germany: Univ. of Göttingen; 1997. [Google Scholar]
- 14.Piotrowski H, Polborn K, Hilt G, Severin K. J Am Chem Soc. 2001;123:2699–2700. doi: 10.1021/ja005804t. [DOI] [PubMed] [Google Scholar]
- 15.Piotrowski H, Hilt G, Schulz A, Mayer P, Polborn K, Severin K. Chem Eur J. 2001;7:3196–3208. doi: 10.1002/1521-3765(20010803)7:15<3196::aid-chem3196>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 16.Therrien B, Ward T R, Pilkington M, Hoffmann C, Gilardoni F, Weber J. Organometallics. 1998;17:330–337. [Google Scholar]
- 17.Smith D P, Baralt E, Morales B, Olmstead M M, Maestre M F, Fish R H. J Am Chem Soc. 1992;114:10647–10649. [Google Scholar]
- 18.Chen H, Olmstead M M, Smith D P, Maestre M F, Fish R H. Angew Chem Int Ed Engl. 1995;34:1514–1517. [Google Scholar]
- 19.Annen P, Schildberg S, Sheldrick W S. Inorg Chim Acta. 2000;307:115–124. [Google Scholar]
- 20. Korn, S. & Sheldrick, W. S. (1997) J. Chem. Soc. Dalton Trans., 2191–2199.
- 21.Korn S, Sheldrick W S. Inorg Chim Acta. 1997;254:85–91. [Google Scholar]
- 22.Holleman A F, Wiberg E, Wiberg N. Lehrbuch der Anorganischen Chemie. Berlin: Walter de Gruyter; 1995. 34 Ed., p. 161. [Google Scholar]
- 23.Cram D J, Kaneda T, Helgeson R C, Brown S B, Knobler C B, Maverick E, Trueblood K N. J Am Chem Soc. 1985;107:3645–3657. [Google Scholar]
- 24.Saalfrank R W, Bernt I, Hampel F. Angew Chem Int Ed Engl. 2001;40:1700–1703. doi: 10.1002/1521-3773(20010504)40:9<1700::aid-anie17000>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 25.Beer P D. Chem Soc Rev. 1989;18:409–450. [Google Scholar]