

Abstract
The self-organization of multicomponent tetrarosette assemblies into ordered nanostructures on graphite surfaces has been studied by atomic force microscopy (AFM). Real-space information on the level of individual molecules allowed us to analyze the underlying structure in unprecedented detail. In highly ordered nanorod domains, tetrarosettes 13⋅(DEB)12 arrange in the form of parallel rows with a spacing of 4.6 ± 0.1 nm. High resolution AFM revealed the internal packing of the tetrarosette assemblies in these rows, which can be described by an oblique lattice with a = 2.5 ± 0.3 nm, b = 5.0 ± 0.1 nm, and γ = 122 ± 3°. The results, together with recent improvements in synthetic approaches, contribute to the development of a general strategy to develop H-bonding-based nanostructures with molecular precision.
The self-assembly of small molecular building blocks into supramolecular aggregates by using non-covalent interactions is anticipated to open the path toward the realization of molecular devices and well-defined nanometer-scale structures and objects (1). Expanding on the profound knowledge in the fields of supramolecular chemistry (2–5) and intermolecular (surface) forces (6, 7), recent progress in nanostructuring has enabled several groups to arrange supramolecular aggregates in two dimensions on surfaces by clever design of the interactions and by using scanning probe microscopy approaches at variable temperatures (8–11). Other promising approaches include molecular beam epitaxy (12). As shown very recently, supramolecular assemblies in three dimensions serve as potentially valuable templates for the formation of, e.g., single crystal silver nanowires (13).
In this article, we report on our recent progress in the formation and structural analysis of nanometer-scale aggregates by using self-assembled rosette structures based on hydrogen-bonding (14). Our previous work on molecular boxes derived from the corresponding double rosettes showed that nanorod structures can be obtained under certain conditions on graphite surfaces (15). Because the synthetic pathways for selective functionalization of these molecules have been developed fully in recent years, it is possible to attach functional units, such as receptors and reporters, at virtually any preselected location in the molecules. If the lateral assembly of higher order structures, such as nanorods or crystals, can be controlled similarly well in two dimensions, it will be possible to position and pattern functional nanostructures by spontaneous self-assembly processes. These nanostructures possess, hence, considerable potential as templates, receptor arrays, versatile molecular print-boards, etc. Here, we focus on the necessary molecular level investigation of the structure, organization, and two-dimensional morphology of novel tetrarosette supramolecular nanostructures and their evolution in monolayers on graphite. The ultimate aim is the development of a generally applicable approach toward positioning of functional nanostructures with submolecular precision in well-defined arrays.
Methods
The synthesis of tetramelamine 1 was performed starting from a bis(chlorotriazine)calix [4]arene derivative via reaction with an excess of 2,2-dimethyl-1,3-propane diamine (16), followed by monoprotection of the corresponding amino group with di-tert-butyl dicarbonate and subsequent coupling with 1,4-diisocyanatobutane (17). The thin film samples were prepared by deposition of one drop of a dilute solution of 13⋅(DEB)12 in chloroform (3:12 ratio of tetramelamine 1:5,5-diethyl barbituric acid (DEB), c = 0.01 mg/ml) onto freshly cleaved highly oriented pyrolitic graphite (HOPG) in a near-saturated atmosphere of chloroform. After the solvent was slowly evaporated, the samples were treated for ≈15 min in oil pump vacuum to remove any traces of residual solvent. The tapping mode atomic force microscopy (AFM) data were acquired with a NanoScope III multimode AFM (Digital Instruments, Santa Barbara, CA) by using a 10-μm (E) scanner and microfabricated silicon tips/cantilevers (model TESP, resonance frequency ν0 ≈ 300 kHz, Nanosensors, Wetzlar, Germany). For the high resolution imaging, the AFM scan head was placed in an integrated acoustic/vibration isolation system (model VT-103-3K, Digital Instruments), and the system was thermally equilibrated over the period of typically 1 to 2 days by operating the AFM in contact mode with false engagement. The rms amplitude of the cantilever (≈0.8 V) and the amplitude damping (≈5%) were minimized to reduce the peak normal forces. Height, phase, and amplitude images were captured by using scan rates between 0.5 and ≈3.0 Hz. All data presented here have been subject to a first order plane fit to compensate for sample tilt. The time-resolved evolution of the layer structure at controlled temperature was followed in situ by using a miniaturized AFM hot stage (18).
Results and Discussion
The tetrarosette nanostructure 13⋅(DEB)12 shown in Scheme S1 forms spontaneously in chloroform solution as a result of 72 cooperative hydrogen bonds between tetramelamine 1 and 5,5-diethyl barbituric acid (DEB; ref. 19). The self-assembly of these tetrarosettes 13⋅(DEB)12 from a dilute solution on HOPG by slow solvent evaporation and subsequent vacuum treatment resulted in the formation of multiphase films (see below).
Scheme 1.

Tetrarosette 13⋅(DEB)12. (Left) Structure. (Center) Schematic structure. (Right) Gas phase minimized structure in top view (Upper) and side view (Lower) including sizes (Upper, diameter; Lower, height).
The most prominent features of these films are the ordered domains, which consist of parallel stripes (Fig. 1) as revealed by tapping mode AFM images. The periodic structures were clearly discernible in all three imaging modes; however, the phase images with their stiffness-based contrast were superior in image contrast. Thus, the tetrarosettes 13⋅(DEB)12 forms nanorod assemblies on HOPG, which are similar to the structures observed previously for double rosettes (15). The mutual orientation of rods in different domains is correlated with the symmetry directions of the graphite lattice, i.e., the directions of the rods in different domains possess relative angles of 0°, 60°, and 120°, respectively (Fig. 1). Hence, the alignment is determined by the substrate.
Figure 1.
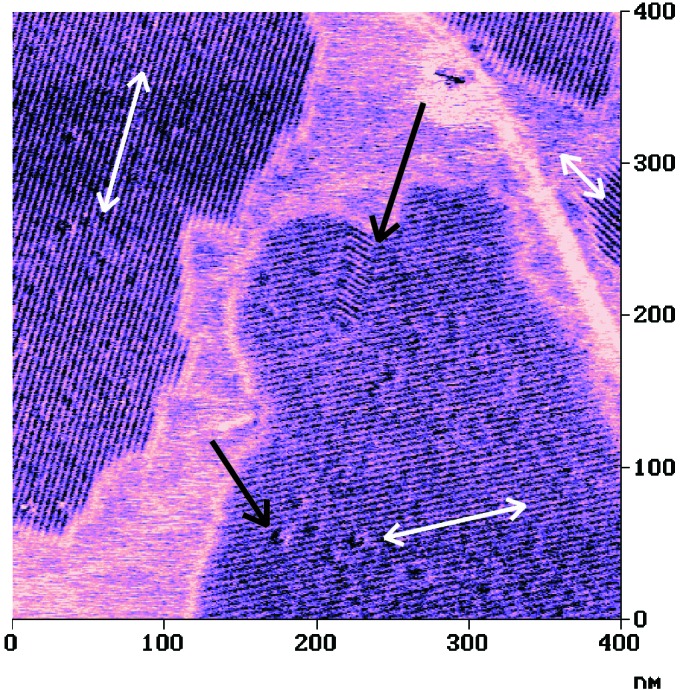
Tapping mode AFM phase image of nanorod domains of tetrarosettes 13⋅(DEB)12 on HOPG. The nanorod domains are correlated with the threefold symmetry of the HOPG substrate as shown by the three bright arrows. Distinct defects can be recognized as indicated by the dark arrows.
In addition to the mentioned domains of parallel nanorods, a bulk crystalline phase, a granular phase, and a featureless gas-like or liquid-like phase were observed, all of which are similar to those reported by Loi et al. of polyphenylene dendrimers on HOPG (20). In contrast to this dendrimer system, the tetrarosettes 13⋅(DEB)12 nanostructures were found to be (meta)stable in a limited temperature region (T < 40°C).
The nanorods observed by AFM are characterized by a highly reproducible interrow spacing of 4.6 ± 0.1 nm [heart to heart distance (15) measured normal to the row direction]. The distances were observed in more than 10 independent experiments for various concentrations and deposition conditions. Thus, the spacing appears to be an intrinsic property of the tetrarosette 13⋅(DEB)12 nanorod assembly on HOPG.
In higher resolution AFM images, a superstructure with smaller periodicity is also clearly present (Fig. 2). The raw data shown in Fig. 2A suggest the presence of inclined elongated features along the rows. The quantitative analysis of the two-dimensional fast Fourier transforms (Fig. 2A Inset) reveals an oblique lattice structure with a = 2.5 ± 0.3 nm, b = 5.0 ± 0.1 nm, and γ = 122 ± 3°. This unit cell, which has an area of 10.6 nm2 and contains probably one rosette nanostructure, is indicated in the Fourier filtered section shown in Fig. 2B.
Figure 2.

(A) Unfiltered high resolution tapping mode AFM phase image of tetrarosette 13⋅(DEB)12 nanorod domain structure; (Inset) two-dimensional fast Fourier transform. (B) Fourier filtered section of raw data shown in A and unit cell of the lattice structure.
Considering the known crystal structure of the corresponding double rosette assemblies (21) and the gas phase minimized structure of the tetrarosettes (see Scheme S1; ref. 22), the observed nanorods can be concluded to consist of rows of tetrarosette assemblies. Based on the gas phase minimized structure, the area requirement for such an arrangement is ≈8.6–9.9 nm2, depending on the orientation. This value compares reasonably well with 10.6 nm2 observed on HOPG (see above). If the possible spreading of the alkyl side chains of the rosettes because of the strong interaction of the methylene units with the graphite and the concomitant flattening of the nanostructures is taken into consideration, the area requirements would agree even better. Owing to the small differences in height and width of the 13⋅(DEB)12 nanostructure, no definite assignment of the orientation of this assembly in the nanorod domains on HOPG can be made based on the AFM data.
The perfect order of the rosettes self-assembled into a well-defined two-dimensional structure on HOPG, as shown above, would result in very versatile patterns for, e.g., templates, receptor arrays, or molecular print-boards. The current challenge is to significantly increase the typical domain sizes of ≈200–500 nm. In some cases, we have achieved virtually complete coverage of the HOPG substrate over distances exceeding several micrometers. The reproducibility of depositing these nanorod domains is still difficult because of various factors, such as concentration gradients and evaporation rates, which are difficult to control precisely, and the fact that these films are multiphase systems (see above).
The ordered arrays of supramolecular nanostructures are formed spontaneously by a self-assembly process. In addition to the intermolecular interactions between individual tetrarosette assemblies, the adsorption energy of these molecules to HOPG plays an important role in this process. This result can be concluded from the correlation of the nanorod structures with the symmetry directions of the underlying substrate. For methylene groups (in n-alkanes) and phenyl groups on HOPG, adsorption energies of Ead (CH2) ≈ 7 kJ/mol (23) and Ead (phenyl) ≈ 15 kJ/mol, respectively (24), have been reported. The tetrarosette adsorbates do not possess long alkyl-substituents; thus, the adsorption energies will be modest. This feature is responsible for the observed dynamic behavior of the multiphase films even at temperatures between room temperature and ≤40°C, as revealed by real-time AFM experiments at controlled temperature. For instance, the diffusion of granular domains and the formation of nanorod domains from a featureless gas-like or liquid-like phase of 13⋅(DEB)12 were observed by AFM. This result clearly indicates that the individual tetrarosette assemblies can diffuse on HOPG at these temperatures.
Considering the fact that longer alkyl substituents can easily be introduced or the size of the rosettes can be altered, these interactions can be optimized to stabilize the metastable nanorod structures. Hence, by expanding on the versatile synthetic chemistry developed for these and related compounds (19), it is possible to develop the series of double and tetrarosettes to higher order complex structures, such as hexa- and octarosettes (19). It is anticipated that the stability of the adsorbed nanorod structures can be tailored by means of changing the size of the primary unit.
Furthermore, it is possible to attach, for instance, metal (e.g., gold) atoms and functional units on these building blocks and, by means of the adsorption to HOPG as described here, to position them with molecular precision in space. The application of these arrays of functionalized nanorods as templates for the deposition of metals and other materials warrants further investigation.
Conclusions
The structure of self-assembled multicomponent tetrarosette-based nanostructures on graphite surfaces has been unveiled by AFM. Highly ordered nanorod domains with a spacing of 4.6 ± 0.1 nm were observed. Molecularly resolved tapping mode AFM images yielded direct evidence for the internal structure composed of stacked tetrarosette building blocks. The results, together with recent improvements in synthetic approaches, form the basis for a general strategy to develop nanostructures with molecular precision.
Acknowledgments
H.S. gratefully acknowledges financial support by the Council for Chemical Sciences of the Netherlands Organization for Scientific Research (CW-NWO) in the framework of the priority program materials (PPM project number 96PPM039), the Deutsche Akademischer Austauschdienst (DAAD) in the framework of the “Hochschulsonderprogramm III,” and the National Science Foundation Materials Research Science and Engineering Center (NSF MRSEC) Center on Polymer Interfaces and Macromolecular Assemblies (CPIMA) under DMR 9808677. The research of M.C.-C. has been made possible by a fellowship of the Royal Netherlands Academy of Arts and Sciences.
Abbreviations
- DEB
diethyl barbituric acid
- HOPG
highly oriented pyrolitic graphite
- AFM
atomic force microscopy
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Lehn J-M. Supramolecular Chemistry: Concepts and Perspectives. New York: VCH; 1995. [Google Scholar]
- 2.Lehn J-M. Angew Chem Int Ed Engl. 1988;27:89–112. [Google Scholar]
- 3.Cram D J. Angew Chem Int Ed Engl. 1988;27:1009–1020. [Google Scholar]
- 4.Pedersen C J. Angew Chem Int Ed Engl. 1988;27:1021–1027. [Google Scholar]
- 5.Reinhoudt D N, editor. Supramolecular Materials and Technologies. New York: Wiley; 1999. [Google Scholar]
- 6.Israelachvili J. Intermolecular and Surface Forces. London: Academic; 1991. [Google Scholar]
- 7.Schönherr H, Beulen M W J, van Veggel F C J M, Bügler J, Huskens J, Reinhoudt D N, Vancso G J. J Am Chem Soc. 2000;122:4963–4967. [Google Scholar]
- 8.Yokoyama T, Yokoyama S, Kamikado T, Okuno Y, Mashiko S. Nature (London) 2001;413:619–621. doi: 10.1038/35098059. [DOI] [PubMed] [Google Scholar]
- 9.Neu B, Meyer G, Rieder K H. Mod Phys Lett. 1995;9:963–969. [Google Scholar]
- 10.Jung T A, Schlittler R R, Gimzewski J K, Tang H, Joachim C. Science. 1996;271:181–184. [Google Scholar]
- 11.Gimzewski J K, Joachim C. Science. 1999;283:1683–1688. doi: 10.1126/science.283.5408.1683. [DOI] [PubMed] [Google Scholar]
- 12.Barth J V, Weckesser J, Cai C, Günter P, Bürgi L, Jeandupeux O, Kern K. Angew Chem Int Ed. 2000;39:1230–1234. doi: 10.1002/(sici)1521-3773(20000403)39:7<1230::aid-anie1230>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 13.Hong B H, Bae S C, Lee C-W, Jeong S, Kim K S. Science. 2001;294:348–351. doi: 10.1126/science.1062126. [DOI] [PubMed] [Google Scholar]
- 14.Prins L J, Reinhoudt D N, Timmerman P. Angew Chem Int Ed. 2001;40:2382–2426. doi: 10.1002/1521-3773(20010702)40:13<2382::aid-anie2382>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 15.Klok H-A, Jolliffe K-A, Schauer C L, Prins L J, Spatz J P, Möller M, Timmerman P, Reinhoudt D N. J Am Chem Soc. 1999;121:7154–7155. [Google Scholar]
- 16.Kerckhoffs J M C A, Crego-Calama M, Luyten I, Timmerman P, Reinhoudt D N. Org Lett. 2000;2:4121–4124. doi: 10.1021/ol000244o. [DOI] [PubMed] [Google Scholar]
- 17.Prins L J. Ph.D. thesis. Enschede, The Netherlands: Univ. of Twente; 2001. [Google Scholar]
- 18.Schönherr H, Bailey L E, Frank C W. Langmuir. 2002;18:490–498. [Google Scholar]
- 19.Paraschiv V, Crego-Calama M, Fokkens R H, Padberg C J, Timmerman P, Reinhoudt D N. J Org Chem. 2001;66:8297–8301. doi: 10.1021/jo0105742. [DOI] [PubMed] [Google Scholar]
- 20. Loi, S., Wiesler, U.-M., Butt, H.-J. & Müllen, K. (2000) J. Chem. Soc. Chem. Comm., 1169–1170.
- 21.Timmerman P, Vreekamp R H, Hulst R, Verboom W, Reinhoudt D N, Rissanen K, Udachin K A, Ripmeester J. Chem Eur J. 1997;3:1823–1832. [Google Scholar]
- 22. Timmerman, P., Weidmann, J.-L., Jolliffe, K. A., Prins, L. J., Reinhoudt, D. N., Shinkai, S., Frish, L. & Cohen, Y. (2000) J. Chem. Soc. Perkin Trans. 2, 2077–2089.
- 23.Martin D S, Weightman P, Gauntlett J T. Surf Sci. 1998;417:390–405. [Google Scholar]
- 24.Vernov A, Steele W A. Langmuir. 1991;7:2817–2820. [Google Scholar]