

Abstract
A (R)-binaphthol tethered bis-hexameric oligo(m-phenylene ethynylene) foldamer was examined in 30 solvents to correlate the unfolded–folded conformational equilibrium to bulk solvent properties and specific solvent–chain interactions. The oligomer is soluble in a variety of solvents of intermediate polarity, with the majority of these solvents being helicogenic. The amphiphilic nature of the chain allows the solvophobic backbone to be solubilized in a wide range of solvents through the polar triethylene glycol side chains. As demonstrated through UV and CD spectroscopic experiments, the helical conformation is increasingly stabilized with increasing solvent polarity in the absence of specific solvent–chain interactions. Surprisingly, very few solvents are capable of fully denaturing the helix, indicating the strength of the solvophobic driving forces in this cooperative system. The folding reaction for this amphiphilic oligomer can be described as a compromise in solubility properties, where chains collapse intramolecularly into helical conformations to minimize solvent–backbone contacts while maintaining favorable solvent–side chain interactions for solvation. In terms of mimicking the properties of biomacromolecules, foldamers using solvophobic driving forces must be tempered with functionalities that promote solubility of the folded state while at the same time allowing access to the unfolded state through the use of denaturants.
The conformation of biological and synthetic macromolecules is determined by both the accessible torsional states—dictated by the covalent framework—and the balance between chain–chain versus solvent–chain contacts. Solvent–solute interactions play a key role in modulating the strength of chain–chain interactions that determine the structure of the collapsed state (1–3). When solvent–chain interactions are favorable (i.e., solvents of good quality), the unfolded chain adopts expanded random conformations. Synthetic macromolecules in poor solvents, especially homopolymers, adopt an ensemble of collapsed globular states in dynamic equilibrium (4–6). In contrast, biological macromolecules such as proteins adopt collapsed structures, often globular folded states in water with a dense hydrophobic core and a hydrophilic periphery. Consequently, the spatial arrangement of the functionalities in the collapsed state imparts function in biomacromolecules, such as molecular recognition, binding, and catalysis.
Foldamer research aims at developing chain molecules that adopt well-defined collapsed conformations analogous to biomacromolecules through the use of flexible unnatural backbones, where the folded state is stabilized by various intramolecular noncovalent interactions (7, 8). Central to understanding the forces involved in the conformational organization of a macromolecular backbone is the folding reaction, wherein the unfolded and folded states are observable in solution through various spectroscopic techniques. As with biological macromolecules (9), solvent-induced equilibrium shifting from the folded to the unfolded state involves disruption of these noncovalent interactions through either competitive solvation or changes in the bulk properties of the medium. In the foldamer field, the impact of solvent on foldable chains has been addressed only recently, and of these studies, only a limited scope of solvents has been explored (7, 10–17). This fact is surprising considering the ease with which this experimental variable can be modulated and the information that can be obtained about the nature of the driving forces involved in the folding reaction. Therefore, understanding how the conformational states of the chain respond to the surrounding media, a major focus already existing in the fields of biological and polymer science, is the key to ascertaining the sensitivity of a foldamer backbone to solvent as well as improving the design of foldable chains.
We have recently described oligomeric m-phenylene ethynylenes bearing polar triethylene glycol (Tg) side chains (18, 19). Like many biomacromolecules, the solubility characteristics of the backbone segments are rather different from the side chains. Whereas for biomacromolecules the backbone is polar and the side chains are hydrophobic (nucleic acids) or amphiphilic (proteins), these m-phenylene ethynylenes have apolar backbones and polar side chains. Specifically, the parent hydrocarbon backbone by itself is poorly soluble in most common solvents when the chain consists of more than a few repeat units (20). In contrast, the Tg side chains are strong solubility promoters, especially in polar solvents, as suggested by the miscibility of poly(ethylene glycol)s in water (21). Covalent attachment of Tg side chains to the m-phenylene ethynylene backbone has led to an intriguing type of amphiphilic macromolecule that expresses its supramolecular properties intramolecularly. These oligomers are of interest because they exhibit solvent-dependent conformational extremes ranging from organized compact structures to disordered states. In the dilute solution limit (micromolar concentrations), a unimolecular transition between these two limiting states is observable, depending on solvent composition (22) or temperature (23). These chains exist as an ensemble of random conformations in chloroform, whereas in acetonitrile, the backbone adopts a helical conformation stabilized by aromatic–aromatic interactions between nonadjacent segments of the backbone. Evidence in support of the helical conformation has been obtained from a variety of solution (18, 19, 24, 25) and solid-state studies (26).
A more exhaustive survey of solvents allowed us to better understand the solvophobic driving force behind the folding reaction and to generalize how these weak nonspecific supramolecular interactions can be used to create ordered conformations. To study solvent–chain interactions, a (R)-binaphthol tethered bis-hexameric phenylene ethynylene oligomer 1 was chosen (Fig. 1) (27). In the solvophobic collapse of 1, the binaphthol moiety induces a twist sense bias in the backbone, as evidenced by the large Cotton effect and exciton couplet (28) in the CD spectrum. For those solvents capable of solubilizing the oligomer, we hoped to determine general characteristics or specific parameters of the solvents that promote compact helical conformations. Specifically, are most solvents helix promoting (helicogenic) or helix disrupting? Here we answer this question by examining the conformational behavior of 1 in a variety of solvents. The oligomer is soluble in a wide range of solvents, yet most solvents are helicogenic and incapable of fully unfolding the chain through destabilization of the helical conformation. We were surprised to discover that denaturing solvents are much more uncommon than helicogenic solvents, revealing the inherent foldability of this backbone. Furthermore, these studies demonstrate on a fundamental level how solvent can impact self organization through preferential solvation of chain molecules whose backbone and side chain segments have dichotomous solubility characteristics. On the basis of these results, we discuss the generality of amphiphilicity as an important component in foldamer design.
Figure 1.

The chemical structure of 1: a bis-hexameric phenylene ethynylene tethered through (R)-binaphthol (Inset); the side chains promote solubility in a wide range of solvents. The solvent-dependent folding reaction of 1: the unfolded state contains both transoid and cisoid backbone conformations, which become all-cisoid in the folded state. The result is a helical conformation stabilized by intramolecular aromatic–aromatic contacts whose strength is modulated by solvent. The (R)-binaphthol moiety induces a twist sense bias in the resulting helix. Side chains have been omitted for clarity. The helical conformation shown is an energy minimized structure.§
Methods
The synthesis of oligomer 1 has been previously reported (27). Spectrophotometric or anhydrous‡ grade solvents were used without further purification for the dissolution of 1. The absorption and CD spectra of 1 were recorded on an OLIS (Jefferson, GA) Cary-17 UV/CD spectrophotometer by using 1-cm quartz cells at 3–4 μM concentrations. This instrument was routinely calibrated by using holmium oxide for UV and (+)-camphor sulfonic acid for CD, according to the two-point methodology described by Chen and Yang (29). The presence of aggregates was monitored by varying the concentration over the range 0.5–5 μM. For solvents of low to intermediate polarity (from CCl4 to dimethylformamide), no aggregates of 1 were detected over this concentration range. The CD spectra, recorded in millidegrees (θ), were converted to Δɛ using the equation Δɛ = θ/(32982cl) (30), where Δɛ is the difference in molar absorptivity for oppositely polarized light in M−1⋅cm−1, c is the concentration of the sample in mol⋅liter−1, and l is the path length through the cell in centimeters.
Results
Absorption and CD spectroscopy have provided reliable signatures of backbone conformational order in oligomer 1 (27). Absorption ratios and CD intensities of 1 in pure solvents are provided in Table 1, where reported values are averages of two independently prepared solutions with the intensities deviating by less than 5%. A qualitative solubility indicator of the oligomer at the experimental micromolar concentration is also indicated in Table 1 for each solvent. In the absorption spectra, extinction coefficients of 1 at 295 nm were determined over the range of solvents to be between 2 and 3⋅105 M−1⋅cm−1 with the absorption maxima deviating by only ±1.0 nm. These results are consistent with previous spectroscopic studies on oligo(m-phenylene ethynylene)s too short to adopt helical conformations where extinction coefficients are generally independent of solvent (23). Additionally, isosbestic and isodichroic points were observed with 1 over the range of nonaggregating solvents. Typical binaphthol π→π* and ester n→π* extinction coefficients are significantly lower in this spectral region, and therefore the observed spectra can be predominantly attributed to the transoid and cisoid conformational states of the phenylene ethynylene chromophore (Fig. 1). In the CD spectra, the window of strong Cotton effect coincides with the phenylene ethynylene absorption, revealing a chiral environment of the backbone chromophores as expected for the folded helical conformation.
Table 1.
UV absorption ratios and CD intensities of 1 in various solvents correlated to solvent polarity parameter
| Solvent | E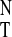
|
A313/A295 | Δɛ322 | Solubility* |
|---|---|---|---|---|
| Cyclohexane | 0.006 | − | − | I |
| NEt3 | 0.043 | − | − | I |
| CCl4 | 0.052 | 0.779 | −191.8 | S |
| Di-n-Bu ether | 0.071 | − | − | I |
| Diethyl ether | 0.117 | − | − | I |
| t-Bu Me ether | 0.148 | − | − | I |
| 1,4-Dioxane | 0.164 | 0.811 | −189.0 | S |
| 1,1,1-TCE | 0.170 | 0.793 | −251.7 | S |
| THF | 0.207 | 0.860 | −121.9 | S |
| EtOAc | 0.228 | 0.719 | −203.8 | S |
| 1,2-DME | 0.231 | 0.782 | −257.9 | S |
| TEGDME | 0.253 | 0.752 | −247.6 | S |
| CHCl3 | 0.259 | 0.938 | −20.5 | S |
| MeOAc | 0.287 | 0.739 | −353.3 | S |
| CH2Cl2 | 0.309 | 0.958 | −57.7 | S |
| 1,2-DCE | 0.327 | 0.903 | −102.7 | S |
| Triacetin | 0.330 | 0.744 | −287.8 | S |
| NMP | 0.355 | 0.800 | −243.6 | S |
| t-BuOH | 0.389 | − | − | I |
| DMAC | 0.401 | 0.758 | −210.4 | S |
| DMF | 0.404 | 0.752 | −215.8 | S |
| DMSO | 0.444 | 0.679 | −321.0 | S |
| CH3CN | 0.460 | 0.672 | −398.8 | S |
| IPA | 0.546 | − | − | I |
| EtOH | 0.654 | − | − | I |
| MeOH | 0.762 | 0.611 | −932.0 | P |
| Ethylene glycol | 0.799 | − | − | I |
| 1,1,1-TFE | 0.898 | 0.679 | −325.1 | S |
| H2O | 1.000 | − | − | I |
| HFIP | 1.068 | 0.784 | −187.9 | S |
Solubility indicator: S, soluble; I, insoluble; P, partial solubility.
TCE, 1,1,1-trichloroethane; THF, tetrahydrofuran; DME, dimethyl ether 1,2-dimethoxyethane; TEGDME, triethylene glycol DME; 1,2 DCE, 1,2 dichloroethane; DMF, dimethylformamide; IPA, isopropanol; TFE, trifluoroethanol; HFIP, hexfluoroisopropanol.
The UV and CD spectra of 1 in chloroform and acetonitrile are shown in Fig. 2. Two absorption maxima of similar intensity are observed at 295 and 313 nm in the UV of 1 in chloroform. In acetonitrile, a decrease in the overall band intensity is observed (hypochromicity), especially for the band centered at 313 nm, suggesting that chromophores interact with one another in a stacked conformation. A decrease in the A313/A295 absorption ratio signals a conformational change in acetonitrile consistent with previously interpreted solvent-dependent transoid–cisoid equilibrium shifts of the backbone that drive the folding reaction of the chain. In chloroform, a random distribution of the chromophore conformations is suggested, whereas in acetonitrile the backbone adopts predominantly cisoid conformations producing the helical state (Fig. 1) (27). The CD spectrum of 1 in chloroform shows a weak signal attributed to the binaphthol moiety within the oligomer chain. In acetonitrile, a strong Cotton effect is observed with the greatest intensity at 322 nm, corresponding to electronic transitions in the backbone chromophores interacting in a chiral stacked arrangement. For all data described below, similar bandshapes were obtained in each of the solvents, except in higher polarity solvents (e.g., DMSO), where blue-shifted bands appeared in both the UV and CD and are attributed to the formation of aggregates. Oligo(m-phenylene ethynylene)s have been shown to associate intermolecularly in solvent compositions of high polarity, as evidenced by nonlinear effects in CD intensities (31).
Figure 2.

UV absorption and CD spectra of 1 (3.0⋅10−6 M at ambient temperature) in CHCl3 (solid) and CH3CN (dashed).
Fig. 3 shows a plot of UV
A313/A295 ratios of
1 in various solvents versus normalized empirical solvent
polarity parameter E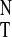 from Table 1. The
E
from Table 1. The
E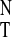 index has found acceptance in the
literature as a reliable and convenient measurement of solvochromatic
effects (32). Solvents chosen for this study are those that span the
polarity range without absorbing strongly in the wavelength region
where the phenylene ethynylene chromophores absorb. Hence, no
aromatic solvents were investigated in this study because of their
overlapping absorptions with the phenylene ethynylene backbone.
However, previous studies by 1H NMR on
macrocyclic phenylene ethynylenes have shown aromatic solvents to have
intermediate solvophobic effects on the intermolecular association
through similar aromatic–aromatic stacking interactions that stabilize
the helical conformation of the oligomers (12). For the UV data, a
general decrease in the
A313/A295 absorption
ratio is observed with increasing solvent polarity, consistent with an
increase in the population of cisoid conformations,
stabilized through polar solvophobic interactions. Surprisingly, the
solvents inducing the highest degree of unfolding as measured by
A313/A295 were not the
least polar, as would be expected from strict hydrophobic
considerations, but were of intermediate polarities. The three solvents
with the highest
A313/A295 ratios (most
unfolded) were chlorohydrocarbon solvents (chloroform, methylene
chloride, and 1,2-dichloroethane). Consistent with these results, the
CD spectra of 1 follow a similar trend as the absorption
spectra where an increase in signal intensity at
Δɛ322 is observed with increasing solvent
polarity (Fig. 4). Again, the
chlorohydrocarbon solvents are outliers to this trend, providing the
lowest degree of chiral induction within the backbone chromophores. For
all solvents, including the chlorohydrocarbons, a good linear
correlation exists between
A313/A295 and
Δɛ322 (excluding the protic
solvents).¶ Thus, these two
independent spectroscopic observables measure foldability in a
consistent way.
index has found acceptance in the
literature as a reliable and convenient measurement of solvochromatic
effects (32). Solvents chosen for this study are those that span the
polarity range without absorbing strongly in the wavelength region
where the phenylene ethynylene chromophores absorb. Hence, no
aromatic solvents were investigated in this study because of their
overlapping absorptions with the phenylene ethynylene backbone.
However, previous studies by 1H NMR on
macrocyclic phenylene ethynylenes have shown aromatic solvents to have
intermediate solvophobic effects on the intermolecular association
through similar aromatic–aromatic stacking interactions that stabilize
the helical conformation of the oligomers (12). For the UV data, a
general decrease in the
A313/A295 absorption
ratio is observed with increasing solvent polarity, consistent with an
increase in the population of cisoid conformations,
stabilized through polar solvophobic interactions. Surprisingly, the
solvents inducing the highest degree of unfolding as measured by
A313/A295 were not the
least polar, as would be expected from strict hydrophobic
considerations, but were of intermediate polarities. The three solvents
with the highest
A313/A295 ratios (most
unfolded) were chlorohydrocarbon solvents (chloroform, methylene
chloride, and 1,2-dichloroethane). Consistent with these results, the
CD spectra of 1 follow a similar trend as the absorption
spectra where an increase in signal intensity at
Δɛ322 is observed with increasing solvent
polarity (Fig. 4). Again, the
chlorohydrocarbon solvents are outliers to this trend, providing the
lowest degree of chiral induction within the backbone chromophores. For
all solvents, including the chlorohydrocarbons, a good linear
correlation exists between
A313/A295 and
Δɛ322 (excluding the protic
solvents).¶ Thus, these two
independent spectroscopic observables measure foldability in a
consistent way.
Figure 3.

UV absorption ratios (A313/A295) of
1 from Table 1 (ranging from CCl4 to
CH3CN). The linear fit [excluding data from the
three chlorohydrocarbon (Δ) solvents] provided the following
relationship: A313/A295 =
0.834–0.259⋅E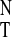 .
.
Figure 4.

CD intensities at Δɛ322 of 1 from Table 1
(ranging from CCl4 to
CH3CN). The linear fit [excluding data from the
three chlorohydrocarbon (Δ) solvents] provided the following
relationship: Δɛ322 =
−167.5–3,121⋅E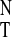 .
.
Discussion
To successfully elucidate the impact of solvent on the folding reaction of an amphiphilic chain, two criteria must be met: (i) the solvent must promote the solubility of the chain, and (ii) the folded and unfolded states must be clearly defined. Although the first criterion seems trivial, it highlights the importance of solvent–chain contacts with foldamers in solution where weaker interactions produce the solubilized folded state, and stronger interactions stabilize the unfolded state, as is the case with denaturing solvents (9). A range of solubilities for the oligomer, therefore, is desirable to observe both conformational extremes and to satisfy the second criteria. Previous studies on 1 showed folded conformations in acetonitrile and unfolded states in chloroform through both UV and CD spectroscopy (Fig. 2) (27). To gain better insight into the forces responsible for the folding reaction of 1, the interpretation of solvent effects on chain conformation will be considered, first in terms of bulk solvent effects and then by specific solvent–chain interactions.
To elucidate bulk solvent effects on solute conformations,
multiparameter analyses are used to correlate solvent parameters and
thermodynamic data, such as the equilibrium position in the folding
reaction. The position of 1 was approximated by assigning
the extreme values of the
A313/A295 absorption
ratios to reflect 100% unfolded and 100% folded conformations. This
estimate is reasonable since binary mixtures of these two
solvents show sigmoidal curves with plateaus approaching pure solvent
limits (23). The spectrum in methyl acetate was chosen as the
spectroscopic reference of the helical conformation, because in this
solvent the greatest absorption ratios and the strongest CD intensities
were obtained, whereas there is no indication of intermolecular
association. From these two conformational endpoints, equilibrium
constants for 1 in each solvent were estimated by principal
component analysis (33,
34).‖ This analysis assumes
a two-state model (35), in which chains exist in either folded or
unfolded states, and where the observed spectra are linear combinations
of these two extremes (36). Although the analysis revealed only two
components in solution, we were unable to reliably fit all
of these equilibrium constants (data not shown) to any of the more
commonly used multiparameter equations for bulk solvent correlations
(32, 37). However, with the exclusion of the chlorohydrocarbon
solvents, trend lines for both plots (Figs. 3 and 4) could be
reasonably fit to a single polarity parameter,
E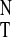 . From purely geometric considerations,
the helical conformation shown in Fig. 1 generates a pseudocylindrical
cavity with an interior surface area of approximately 75
Å2 and a free volume of approximately 100
Å3. Assuming that these dimensions are
independent of solvent, there is no clear correlation between solvent
size relative to cavity size and the spectroscopic trends in Figs. 3
and 4. Although stoichiometric binding of small organic molecules has
been demonstrated for related oligo(phenylene ethynylene)s (25), more
involved studies would be necessary to elucidate whether this
association was operative with specific binding of solvent molecules.
Together these two plots reveal a general correlation for both the
absorbance and CD to solvent polarity and hence the conformational
equilibrium of the unfolded and folded states.
. From purely geometric considerations,
the helical conformation shown in Fig. 1 generates a pseudocylindrical
cavity with an interior surface area of approximately 75
Å2 and a free volume of approximately 100
Å3. Assuming that these dimensions are
independent of solvent, there is no clear correlation between solvent
size relative to cavity size and the spectroscopic trends in Figs. 3
and 4. Although stoichiometric binding of small organic molecules has
been demonstrated for related oligo(phenylene ethynylene)s (25), more
involved studies would be necessary to elucidate whether this
association was operative with specific binding of solvent molecules.
Together these two plots reveal a general correlation for both the
absorbance and CD to solvent polarity and hence the conformational
equilibrium of the unfolded and folded states.
A more thorough examination of solvent effects reveals unique features of the chain solvation that impact the conformational equilibrium of 1. Surprisingly, complete denaturation of the helical conformation was possible only in chlorohydrocarbon solvents (CHCl3, CH2Cl2, and 1,2-dichloroethane). The exact nature of the interaction cannot be determined for certain, but it is reasonable that more specific solvent–backbone contacts, such as CH–π interactions (38), are responsible for the unfolded state in these denaturants. Additionally, 1 was not only soluble in the polarizable nonpolar solvents, carbon tetrachloride (CCl4) and 1,1,1-trichloroethane (1,1,1-TCE), but the oligomer adopted moderate to high degrees of folded conformations, respectively. These results suggest that favorable dipole interactions in the solvation of the backbone are not operative because CCl4 has a zero dipole moment, whereas TCE has a dipole moment comparable to water. The insolubility of 1 in other nonpolar solvents (cyclohexane, diethyl ether, di-n-butyl ether, t-butyl methyl ether, and NEt3) is presumably due to the polar Tg side chains, because m-phenylene ethynylene backbones bearing alkyl side chains have been shown to be soluble in heptane, adopting folded conformations (39). With compositions similar to the Tg side chains, 1 adopts nearly identical degrees of folded conformations in 1,2-dimethoxyethane and triethylene glycol dimethyl ether, suggesting that the local chemical environment of the side chains may encourage backbone–backbone contacts. In the cyclic ethers (tetrahydrofuran and 1,4-dioxane), the oligomer readily dissolves and populates significant degrees of unfolded conformations, potentially solvating the backbone through favorable van der Waals contacts between their hydrophobic faces and the aromatic backbone. Solvents of higher polarity, including the ester-, amide-, and nitrile-containing solvents, favor the folded conformation of 1 to varying degrees, whereas in DMSO the oligomer is considerably folded and aggregrated.
Considering the structure of 1, it seems possible that protic solvents may be able to solvate the oligomer through hydrogen-bonding interactions with the Tg side chains, whereas disfavorable solvent–backbone interactions would induce folded conformations of the backbone. Of the protic solvents investigated, 1 was partially soluble in methanol alone (9.0⋅10−7 M), adopting folded and highly aggregated conformations. This result was surprising given that ethanol, isopropanol, and tert-butanol are less polar because of their higher hydrocarbon compositions. These results suggest that these solvents do not participate in specific solvent–solute interactions, although favorable interactions likely occur between the side chains and methanol. In water, the oligomer does not dissolve to any detectable amount. Interestingly, the fluorine-containing alcohols trifluoroethanol and hexfluoroisopropanol both solubilize 1 showing absorption ratios greater than acetonitrile, indicating an increased destabilization of the helical conformation. Recent studies of α-peptides in mixtures of water and fluorinated solvents demonstrated stabilization of helical conformations, wherein the fluorine segments of the solvents created solvation shells around the hydrophobic solute (40, 41). Similar solvent–backbone interactions may be operative with 1, in this case destabilizing the folded conformation, because fluorine-containing alcohols can solvate the oligomer, whereas their hydrogen-containing solvent counterparts cannot.
Implications for Foldamer Research.
To characterize the folding reaction of amphiphilic chain molecules, helicogenic and denaturing solvents must be identified to access both the folded and unfolded states, respectively. Denaturants are solvents capable of solvating all segments along a foldamer, unfolding the chain through specific interactions that overcome chain–chain contacts (9). A survey of chain conformations in a wide range of solvents should become standard protocol in foldamer research. As discovered in the study described herein, finding denaturants may be more difficult than expected, depending on the strength of the noncovalent forces involved in the folding reaction. Hence, in the design of foldamers, chain molecules must be endowed with amphiphilicity, incorporating side chains that promote solubility and backbones whose strength of intramolecular contacts can be modulated by environmental conditions. As the methodologies for the conformational analysis of foldamers progress, techniques for assessing the folding reaction of chain molecules will be essential to mimicking biomacromolecular machinery.
Acknowledgments
We thank Mary S. Gin (University of Illinois) for generously providing oligomer 1 and Marissa B. Schelstraete (University of Illinois) for aiding in data collection. This material is based on work supported by the U.S. Department of Energy, Division of Materials Sciences, under Award No. DEFG02-91ER45439, through the Frederick Seitz Materials Research Laboratory at the University of Illinois at Urbana–Champaign. This research was supported by a grant from the National Science Foundation (NSF CHE 00-91931).
Abbreviation
- Tg
triethylene glycol
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Recent solvent studies have described the effect of deleterious water present in solvents on foldamer conformations.(see ref. 13). Results from our laboratory suggest at least 10% water composition in acetonitrile is necessary to cause significant spectroscopic shifts (see ref. 23).
Minimizations were performed by using macromodel 5.5 and the OPLS* GB/SA force field. Monte Carlo searches from several different initial conformations yielded the folded structure shown in Fig. 1 as the putative global minimum.
Linear fit of the absorption ratios vs. Δɛ322 (excluding protic solvents) provided the following relationship: Δɛ322 = 1,086(A313/A295) − 1,074 (where R = 0.901 and SD = 45.5).
Principal component analysis was calculated using the software package mathematica 4.0 (Wolfram Research, Inc., Champaign, IL 61820).
References
- 1.Kauzman W. Adv Protein Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 2.Williams C, Brochard F, Frisch H L. Annu Rev Phys Chem. 1981;32:433–451. [Google Scholar]
- 3.Dill K A. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 4.Chan H S, Dill K A. Macromolecules. 1989;22:4559–4573. [Google Scholar]
- 5.Camacho C J, Thirumalai D. Phys Rev Lett. 1993;71:2505–2508. doi: 10.1103/PhysRevLett.71.2505. [DOI] [PubMed] [Google Scholar]
- 6.Dill K A, Bromberg S, Yue K, Fiebig K M, Yee D P, Thomas P D, Chan H S. Protein Sci. 1995;4:561–602. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill D J, Mio M J, Prince R B, Hughes T H, Moore J S. Chem Rev. 2001;101:3893–4012. doi: 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- 8.Gellman S H. Acc Chem Res. 1998;31:173–180. [Google Scholar]
- 9.Schellman J A. Biophys Chem. 1990;37:121–140. doi: 10.1016/0301-4622(90)88013-i. [DOI] [PubMed] [Google Scholar]
- 10.Tanatani A, Yamaguchi K, Azumaya I, Fukutomi R, Shudo K, Kagechika H. J Am Chem Soc. 1998;120:6433–6442. [Google Scholar]
- 11.Sindkhedkar M D, Mulla H R, Cammers-Goodwin A. J Am Chem Soc. 2000;122:9271–9277. [Google Scholar]
- 12.Lahiri S, Thompson J L, Moore J S. J Am Chem Soc. 2000;122:11315–11319. [Google Scholar]
- 13.Berl V, Huc I, Khoury R G, Lehn J-M. Chem Eur J. 2001;7:2810–2820. doi: 10.1002/1521-3765(20010702)7:13<2810::aid-chem2810>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 14.Berl V, Huc I, Khoury R G, Lehn J-M. Chem Eur J. 2001;7:2798–2809. doi: 10.1002/1521-3765(20010702)7:13<2798::aid-chem2798>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 15.Yagi S, Morinaga T, Nomura T, Takagishi T, Mizutani T, Kitagawa S, Ogoshi H. J Org Chem. 2001;66:3848–3853. doi: 10.1021/jo001770w. [DOI] [PubMed] [Google Scholar]
- 16.Sakurai S-i, Goto H, Yashima E. Org Lett. 2001;3:2379–2382. doi: 10.1021/ol016189g. [DOI] [PubMed] [Google Scholar]
- 17.Cubberley M S, Iverson B L. J Am Chem Soc. 2001;123:7560–7563. doi: 10.1021/ja015817m. [DOI] [PubMed] [Google Scholar]
- 18.Nelson J C, Saven J G, Moore J S, Wolynes P G. Science. 1997;277:1793–1796. doi: 10.1126/science.277.5333.1793. [DOI] [PubMed] [Google Scholar]
- 19.Prince R B, Saven J G, Wolynes P G, Moore J S. J Am Chem Soc. 1999;121:3114–3121. [Google Scholar]
- 20.Zhang J. Ph. D. thesis. Ann Arbor, MI: Univ. of Michigan; 1993. [Google Scholar]
- 21.Harris J M, Zalipsky S, editors. Poly(ethylene glycol): Chemistry and Biological Applications. Washington, DC: Am. Chem. Soc.; 1997. [Google Scholar]
- 22.Nelson J C. Ph.D. thesis. Urbana, IL: Univ. of Illinois; 1997. [Google Scholar]
- 23.Prince R B. Ph. D. thesis. Urbana, IL: Univ. of Illinois; 2000. [Google Scholar]
- 24.Prince R B, Okada T, Moore J S. Angew Chem Int Ed. 1999;38:233–236. [Google Scholar]
- 25.Prince R B, Barnes S A, Moore J S. J Am Chem Soc. 2000;122:2758–2762. [Google Scholar]
- 26.Mio M J, Prince R B, Moore J S, Kuebel C, Martin D C. J Am Chem Soc. 2000;122:6134–6135. [Google Scholar]
- 27.Gin M S, Yokozawa T, Prince R B, Moore J S. J Am Chem Soc. 1999;121:2643–2644. [Google Scholar]
- 28.Johnson W C., Jr Methods Biochem Anal. 1985;31:61–163. doi: 10.1002/9780470110522.ch2. [DOI] [PubMed] [Google Scholar]
- 29.Chen G C, Yang J T. Anal Lett. 1977;10:1195–1207. [Google Scholar]
- 30.Rodger A, Nordén B. Circular Dichroism and Linear Dichroism. Oxford: Oxford Univ. Press; 1997. pp. 6–7. [Google Scholar]
- 31.Brunsveld L, Meijer E W, Prince R B, Moore J S. J Am Chem Soc. 2001;123:7978–7984. doi: 10.1021/ja010751g. [DOI] [PubMed] [Google Scholar]
- 32.Reichardt C. Solvents and Solvent Effects in Organic Chemistry. New York: VCH; 1990. [Google Scholar]
- 33.Ohta N. Anal Chem. 1973;45:553–557. [Google Scholar]
- 34.Takatsuki M, Yamaoka K. J Sci Hiroshima Univ Ser A. 1976;40:387–415. [Google Scholar]
- 35.Lumry R, Biltonen R, Brandts J F. Biopolymers. 1966;4:917–944. doi: 10.1002/bip.1966.360040808. [DOI] [PubMed] [Google Scholar]
- 36.Pace C N. In: Methods in Enzymology. Hirs C H W, Timasheff S N, editors. Vol. 131. New York: Academic; 1986. pp. 266–280. [DOI] [PubMed] [Google Scholar]
- 37.Gajewski J J. J Org Chem. 1992;57:5500–5506. [Google Scholar]
- 38.Nishio M, Hirota M, Umezawa Y. The CH/π Interaction: Evidence, Nature and Consequences. New York: Wiley–VCH; 1998. [Google Scholar]
- 39.Brunsveld L, Prince R B, Meijer E W, Moore J S. Org Lett. 2000;2:1525–1528. doi: 10.1021/ol0056877. [DOI] [PubMed] [Google Scholar]
- 40.Cammers-Goodwin A, Allen T J, Oslick S L, McClure K F, Lee J H, Kemp D S. J Am Chem Soc. 1996;118:3082–3090. [Google Scholar]
- 41.Walgers R, Lee T C, Cammers-Goodwin A. J Am Chem Soc. 1998;120:5073–5079. [Google Scholar]