

Abstract
We present a DNA-containing polymeric nanocontainer using the self-assembled superstructure of amphiphilic block copolymers in aqueous solutions. To demonstrate that DNA translocation is possible across a completely synthetic block copolymer membrane, we have used a phage transfection strategy as a DNA-transfer model system. For this purpose the bacterial channel forming protein LamB was reconstituted in ABA-triblock copolymer vesicles. The outer membrane protein LamB is a specific transporter for maltodextrins but also serves as a receptor for λ phage to trigger the ejection of λ phage DNA. We demonstrate that the functionality of the LamB protein is fully preserved despite the artificial surrounding. This leads to a type of polymeric vehicle for DNA that could be useful for gene therapy.
It is well known that, similar to conventional lipids, appropriate amphiphilic block copolymers may self-assemble in aqueous media to membrane-like superstructures (e.g., vesicular aggregates; refs. 1–3). As a typical example, we introduced recently amphiphilic ABA-triblock copolymers consisting of a hydrophobic, highly flexible poly(dimethylsiloxane) (PDMS) middle block and two water-soluble poly(2-methyloxazoline) (PMOXA) side blocks that spontaneously form unilamellar vesicles in water (4). These block copolymer vesicles were found to be considerably more stable than, for example, conventional low molar mass lipid vesicles, reflected by the fact that they are stable over several years and do not show any changes in size or size distribution during that time within experimental error. Moreover, if the polymers carry additionally polymerizable end groups, these self-assembled superstructures can be frozen during a subsequent crosslinking polymerization. The resulting polymerized vesicles then have solid-state properties such as shape persistence because of their crosslinked nature and preserve their hollow sphere structure even after their isolation from aqueous phase (4).
Recently we demonstrated that despite their considerably higher thickness and mechanical stability the underlying polymer membranes can be regarded as mimetics of biological membranes that allow reconstitution of membrane proteins (5, 6). Interestingly the membrane proteins remained fully functional despite this artificial environment, even after a subsequent polymerization of their reactive block copolymer matrix. This combination leads to new types of polymer protein systems that are potentially interesting for applications in areas such as diagnostics or pharmacology.
The outside of such polymeric containers is covered completely by the hydrophilic PMOXA blocks of the triblock copolymers, which are known to prevent an unspecific protein adsorption (7). Therefore we expected, at least for long enough hydrophilic blocks, receptors in the walls of such vesicles to be hidden below a hydrophilic polymer layer such that larger ligands would not have access to them. This could be particularly interesting for use of such nanocontainers as intravasal drug delivery devices. We chose the bacterial receptor protein LamB as a model system for our investigations. This protein forms trimeric channels in the outer cell wall of Gram-negative bacteria that allow a specific transport of maltose and maltodextrins (8, 9). Simultaneously, it serves as a receptor for phage λ (10, 11). Because during infection the phages transfer their genome into the host cells, it has been proposed that the channels of LamB could be a major intrusion path for the viral DNA (12). Interestingly, it has been shown that although LamB from both Escherichia coli and Shigella sonnei induce binding of λ phages, only the Shigella protein efficiently triggers the ejection and successful translocation of DNA (13, 14).
Here we report the insertion of LamB into the artificial shells of nanometer-sized triblock copolymer vesicles and its binding to phage λ (Fig. 1). The transfer of DNA into the polymeric nanocontainers was quantified following a recently established procedure via labeling the viral DNA with the fluorescent dye YO-PRO-1.
Figure 1.

Schematic representation of a DNA-loaded PMOXA–PDMS–PMOXA vesicle. λ phage binds a LamB protein, and the DNA is transferred across the block copolymer membrane.
Materials and Methods
Preparation of Phage λ.
Phage λ was isolated from an infected culture of E. coli JM109, which was grown in Luria–Bertani medium supplemented with 0.5% maltose. After an overnight infection, uninfected E. coli cells and cell fragments were removed as described (15).
Phages were collected from the cleared supernatant by ultracentrifugation at 75,000 × g for 3 h at 4°C. The glassy pellet was resuspended in a buffer containing 10 mM MgCl2 and 10 mM Tris⋅HCl, pH 8. Typically this method yielded a concentrated phage stock of 1011–1012 plaque-forming units⋅ml−1.
Purification of LamB Protein.
The LamB from S. sonnei was produced using E. coli strain pop 154, a derivative of E. coli K12, carrying the malB gene of S. sonnei 3070 (14).
The protein was solubilized from the outer membrane by using octyl-polyoxyethylene (octyl-POE, Alexis, Läufelfingen, Switzerland; ref. 1) as the detergent and purified in a procedure analogous to that used for porins from E. coli (16).
Preparation of PMOXA–PDMS–PMOXA Triblock Copolymer Vesicles and Proteovesicles.
The PMOXA–PDMS–PMOXA triblock copolymer (Mn, PMOXA = 1,800 g⋅mol−1, Mn, PDMS = 5,400 g⋅mol−1; Mw/Mn = 1.7) used was described previously (4).The formation of small unilamellar vesicles from the triblock copolymer could be achieved according to the following procedure. The polymer was dissolved in ethanol to yield a clear, homogeneous solution containing 17 wt% polymer. This solution was added dropwise under vigorous stirring to the respective volume of a 100 mM KCl/5 mM MgCl2/1 mM CaCl2/10 mM Tris⋅HCl, pH 7.5 buffer solution. The procedure leads to a dispersion of triblock copolymer vesicles of a rather broad size distribution. Similar to lipid molecules, the vesicle size could be controlled by repeated extrusion through Nucleopore filters (Millipore) with defined pore size. For example, a pore diameter of 200 nm yields vesicles with a diameter of approximately 250 nm.
The vesicles containing the Shigella receptor were prepared by mixing a stock solution of the purified LamB (3 mg⋅ml−1 1% Octyl-POE/NaCl 100 mM/20 mM NaH2PO4, pH 7.5) with a 17 wt% solution of the triblock copolymer in ethanol. Afterward, this mixture was added to the buffer solution as described previously.
The non-incorporated proteins were removed by gel filtration chromatography on a 1 × 55-cm Sepharose 4 B (Sigma) column using the Tris⋅HCl buffer mentioned previously.
Fluorescence Experiments.
Ejection of phage DNA was measured with a spectrofluorimeter Jasco F-P-773 in a 1 × 0.4-cm cuvette thermostated at 37°C using a fluorescent quaternary ammonium dye derivative (YO-PRO-1, Molecular Probes), which intercalates into double-stranded DNA (17, 18). Excitation and emission wavelengths were set at 491 and 509 nm, respectively, and slits were 3 nm for excitation and 10 nm for emission. The final concentration of YO-PRO-1 was 8 μM, and the final volume of the sample was 1 ml.
Reconstituted LamB was mixed with 3.109 phage λ in 1 ml of Tris⋅HCl buffer containing 5 mM MgCl2, 1 mM CaCl2, and 8 μM YO-PRO-1. The increase of the fluorescence signal was recorded as a function of time. To solubilize the proteovesicles, octyl-POE was added to a final concentration of 1%. In one control experiment, vesicles devoid of LamB protein were used. In another control, 1 μg of detergent-solubilized LamB protein was used in 1% octyl-POE.
Transmission Electron Microscopy Experiments.
Five microliters of the phage–receptor complexes were adsorbed to carbon parlodion composite film on copper grids and negatively stained with 2% uranyl acetate solution as described by Engel and coworkers (19). The grids were allowed to dry in air and examined in a Phillips EM 400 with a magnification of ×55,000.
Langmuir Trough Experiments.
The protein–polymer and protein–lipid interactions were measured using the monolayer technique.
The surface pressure was measured by the Wilhemy plate method (20, 21).
Both triblock copolymer and the lipid monolayers were spread from a chloroform solution (1 mg⋅ml−1) at the air–water interface in a Teflon dish to give an initial surface pressure of 9 mN⋅m−1.
The LamB protein was injected with a microsyringe directly to the subphase composed of a 100 mM KCl/5 mM MgCl2/1 mM CaCl2/10 mM Tris⋅HCl, pH 7.5, buffer solution.
Measurements were started 30 min after protein injection. The resulting compression isotherm curves of lipids and triblock copolymers were recorded in the absence or presence of different amounts of purified LamB.
Results and Discussion
LamB Protein Insertion in a PMOXA–PDMS–PMOXA Triblock Copolymer Monolayer.
The interaction of the membrane protein LamB with the block copolymer was studied by injecting the protein beneath the block copolymer film and then measuring the consequent changes of monolayer surface pressure. In parallel we performed the same study with a lipid film to compare the LamB protein behavior in both systems. Because of the noncharged block copolymer characteristic, we chose ceramide to avoid interference of electrostatic effects.
In each experiment LamB was added to the subphase at an initial pressure of 9 mN⋅m−1. The addition of LamB to the subphase of the monolayer film induced a shift toward higher surface pressure and larger effective area per lipid per polymer molecule in the compression isotherms, indicative of LamB-monolayer interaction (data not shown). Furthermore, the surface-pressure increase was protein concentration-dependent (Fig. 2).
Figure 2.

Influence of LamB protein on a ceramide and triblock copolymer monolayer. The surface pressure increases as a function of protein concentration because of increasing protein insertion.
A maximum surface pressure of 7.7 mN/m was reached at 3.5 nM LamB for the ceramide films. In contrast, for the polymer film at the same concentration of LamB, only ≈2.5 mN/m was obtained. Thus in the latter case seven times more LamB protein had to be added to induce a similar surface-pressure increase. Compared with the ceramide film, a lower but still substantial fraction of protein inserts in the block copolymer film. Consequently, this lower insertion reflects directly the higher stability of block copolymer films. Indeed, the triblock copolymer films are considerably thicker than conventional low molecular weight lipid films. As described previously (22), the energy required for defect formation in the polymer membrane is approximately four times higher than for lipid membranes.
Reconstituted LamB in PMOXA–PDMS–PMOXA Triblock Copolymer Vesicles Still Functions as a Phage Receptor.
The LamB protein was reconstituted into the membrane of triblock copolymer vesicles. The nonincorporated proteins were removed by gel-filtration chromatography to avoid interaction of the phage with the free proteins. To check whether the phages are still able to bind to the LamB in the walls of the polymeric nanocontainers, we incubated the particles with phage λ (1010 particles). As shown directly by the transmission electron micrograph of Fig. 3, the phages clearly bind the receptors in the walls of the amphiphilic block copolymer nanocontainers via their tails.
Figure 3.
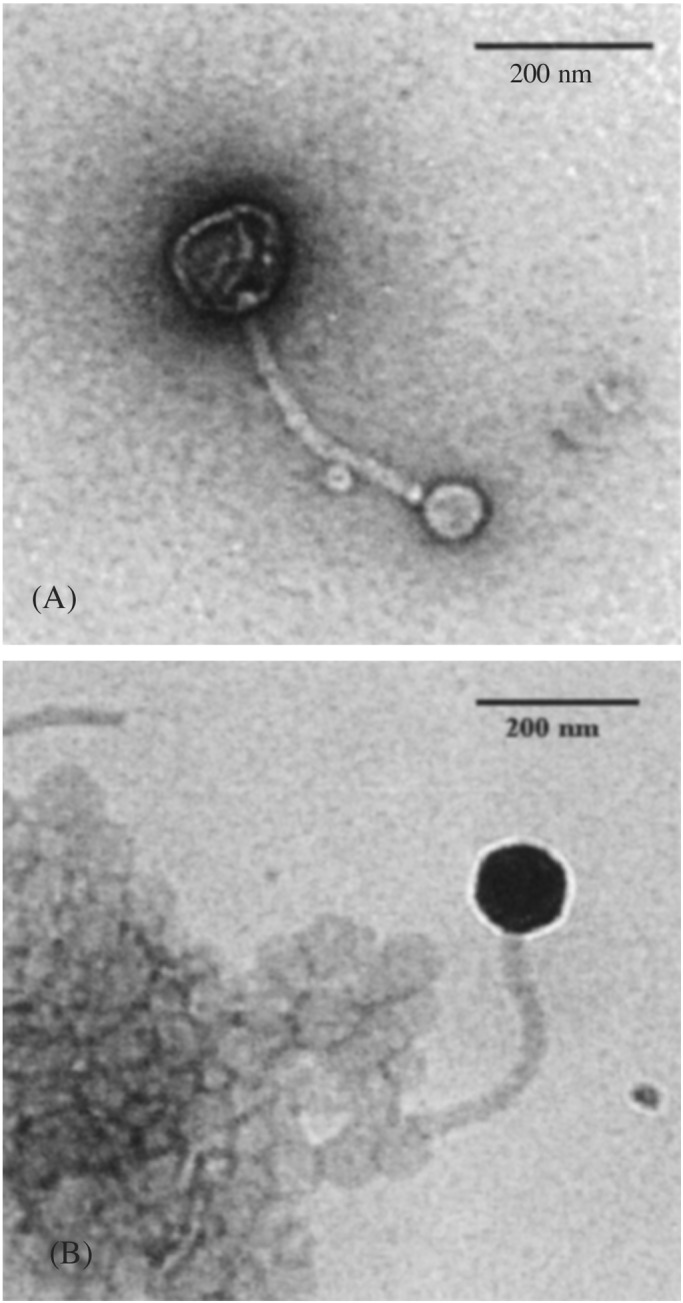
Electron micrograph of negatively stained complexes formed between λ phage and vesicles bearing LamB protein at 37°C. (A) λ phage is attached to one vesicle via its tail. (B) A λ phage bound to a vesicle aggregate bearing LamB protein.
Moreover, the enormous size difference of the phage as compared with the LamB protein–vesicle complex suggests that the interaction of both structures is sufficiently strong. No phage interaction was detected with non-LamB-containing vesicles (data not shown).
PMOXA–PDMS–PMOXA Triblock Copolymer Vesicles Can Be Loaded with DNA.
In nature, after receptor recognition, the phage λ genome is injected into the cell, which then is turned into a phage-producing machinery. This fact immediately raises the question of whether the functionality of the protein is preserved also in the artificial block copolymer membranes, i.e., whether it is still able to induce the injection of the viral DNA into the nanocontainers. The injection can be followed with the help of the dye YO-PRO-1, which intercalates into double-stranded DNA and forms a fluorescent complex (17, 18). After the addition of λ phage (3.109 particles), a steep increase in the fluorescence intensity was observed (Fig. 4). This increase can be attributed to the presence of naked DNA from lysed or broken phages (curves A and B). An additional increase is obtained with purified LamB (curve C), caused by ejected DNA after interaction of the phages with free receptor molecules. Although both control experiments (curves B and C) show a fast-establishing plateau, the signal of the proteovesicles (curve A) slowly increases. This obviously is because of ejection of phage DNA into the YO-PRO-1-containing medium caused by an inappropriate interaction between the phage and the receptor (23).
Figure 4.

Infection of λ phage DNA in PMOXA–PDMS–PMOXA triblock copolymer vesicles reconstituted with LamB receptor. The time course of the fluorescent dye YO-PRO-1 interacting with DNA is shown. Curve A corresponds to LamB-containing vesicles, curve B corresponds to a control experiment with vesicles without LamB, and curve C corresponds to a control with the detergent-solubilized LamB protein (no block copolymer was present in the solution). Arrow 1 corresponds to the addition of λ phage (1010 particles) into the respective buffer containing additional 8 μM fluorescent dye [buffer and vesicles bearing LamB or no LamB or the detergent-solubilized LamB (3 μg)]. Arrow 2 corresponds to solubilization of the vesicles by the addition of 1% octyl-POE, and arrow 3 indicates the addition of DNase I (15 μg/ml).
After the fluorescence intensity reached a plateau, we added the detergent Octyl-POE up to a concentration of 1% (see Fig. 4, arrow 2) to solubilize the vesicles. This treatment caused a rapid increase of fluorescence intensity by ≈40% (curve A). This increase is caused by the encapsulated DNA that is released into the medium after solubilization of the block copolymer vesicles and not by DNA released from detergent-solubilized phage heads. This cause is documented by the control experiment of curve B, which remained virtually unaffected. To prove that the detergent-induced increase was caused by released DNA, we subsequently added DNase I (Fig. 4, arrow 3). The fluorescence signal dropped dramatically (curves A) because of enzymatic hydrolysis of the free DNA in the medium. In both control experiments, the initially naked DNA present in the phage stock (curves B and C) and the ejected DNA (curve C) were equally well digested by DNase I treatment.
An additional treatment with the DNase did not affect the fluorescence intensity (data not shown). Because the fluorescence signal never reached the baseline, quantification of the DNA ejected in the medium proved to be difficult. This residual fluorescence, observed in all curves, has to be attributed to remaining oligonucleotides that cannot be hydrolyzed further by DNase I (24) but are still able to interact with YO-PRO-1.
Interestingly, the fluorescence intensity decrease for free, nonencapsulated DNA is considerably faster than for the DNA released from the interior of the nanocontainers. It seems that the small volume within the aqueous core of the block copolymer vesicles (diameter, ≈250 nm) accessible to the DNA forces it into a very compact, condensed conformation. After solubilization of the polymer shell, the released DNA has then first to “unfold” to be hydrolyzed by the DNase.
Alternatively, the DNA might be protected by interactions with the triblock copolymer. Pure phages (in a polymer- and protein-free solution) revealed similar kinetics to those of protein-free vesicles (data not shown).
The same preparation, vesicles containing LamB protein, was polymerized with UV light (data not shown). Because of their covalently crosslinked nature, the polymerized vesicles are resistant to detergent solubilization. Thus, after the addition of Octyl-POE no DNA was released. The small increase in fluorescence intensity probably is caused by DNA ejection induced by LamB proteins that have been released from the polymeric membrane during the crosslinking reaction. This result is a consequence of the internal stress occurring in the membrane during the polymer chain reaction, which may lead to a steric contraction of the PMOXA part of the polymer (4, 5). Similarly, part of the OmpF channel reconstituted into planar freestanding PMOXA–PDMS–PMOXA triblock copolymer membranes could be expulsed after UV crosslinking of the triblock copolymer (5).
DNA Inside PMOXA–PDMS–PMOXA Triblock Copolymer Vesicles Is Protected from Hydrolysis.
In another approach (Fig. 5 A and B), we added DNase I before solubilization of the vesicles (Fig. 5A, arrow 2) to remove the DNA present in the external solution. Thus DNase treatment caused a rapid decrease of the fluorescence signal. The DNA entrapped in the vesicles was not accessible to the large enzyme (which is not able to enter their interior). In a following step (Fig. 5A, arrow 3) we released the entrapped DNA from the vesicles by detergent solubilization. An instantaneous increase of the fluorescence was observed as the released DNA came in contact with the fluorescent dye. This fluorescence increase was followed immediately by a decrease that can be explained by the presence of active DNase I in the medium, which hydrolyses the released DNA.
Figure 5.

λ phage DNA release from the triblock copolymer vesicles. (A) Curve A corresponds to LamB-containing vesicles, and curve B is the control experiment with protein-free block copolymer vesicles. Arrow 1 corresponds to the addition of λ phage (1010 particles) to the vesicle dispersion in the presence of 8 μM fluorescent dye YO-PRO-1, arrow 2 corresponds to the addition of DNase I (15 μg/ml), and arrow 3 indicates the solubilization of the vesicles by the detergent octyl-POE (1%). (B) Curve A corresponds to LamB-containing vesicles, and curve B is the control. Arrow 1 corresponds to the addition of LamB in excess (1 μg) to the vesicles' dispersion in the presence of 8 μM fluorescent dye YO-PRO-1 and λ phage (1010 particles). Arrow 2 indicates the addition of DNase I (15 μg/ml), and arrow 3 corresponds to the solubilization of the vesicles by addition of 1% octyl-POE.
Part of the ejected DNA was DNase I-resistant. Nevertheless, the results show clearly that the phage DNA had been injected originally into the inner aqueous cavity of the vesicles where it is protected from externally added DNase. Subsequent disruption of the vesicles with detergent rendered the entrapped DNA sensitive to degradation by DNase.
The same experiment was done with an excess of LamB to prevent new phage binding and DNA ejection after vesicle solubilization. Thus, after vesicle solubilization (Fig. 5B, arrow 3) we observed an increase of the fluorescence intensity. This increase is only because of entrapped DNA in the vesicles.
Conclusion and Outlook
In conclusion, our investigations show that the conformation of the receptor LamB is not affected by the artificial block copolymer membrane used in this study, and its functionality is preserved fully. Similar observations have been made also with other membrane proteins. Here we demonstrated that DNA translocation mediated by the λ phage receptor LamB is possible across a completely synthetic block copolymer membrane. This phage-mediated loading of polymer nanocontainers with DNA represents a very stable model system to investigate the transport of foreign genes into eukaryotic cells. Possible applications of such DNA-loaded nanoparticles in the area of gene therapy are obvious. One major problem in this field is how to find efficient and biocompatible vectors that are able to transport the gene specifically into the desired target cells. Here particularly the high diversity of block copolymer chemistry (i.e., the possibility to modify their chemical constitution, block lengths, architecture, etc… ) could be interesting, because it allows for the optimization of these systems with respect to a desired application.
Acknowledgments
We thank Dr. T. Pugsley for providing the strain pop 154 and Pr. M. Winterhalter for the Monolayer setup. We also thank Y. Rohrer for his contribution to the experimental part. The Swiss National Science Foundation is gratefully acknowledged for financial support.
Abbreviations
- PDMS
poly(dimethylsiloxane)
- PMOXA
poly(2-methyloxazoline)
- octyl-POE
octyl-polyoxyethylene
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Wanka G, Hoffmann H, Ulbricht W. Macromolecules. 1994;27:4145–4149. [Google Scholar]
- 2.Svensson P, Alexandridis P, Linse P. Macromolecules. 1999;32:637–645. [Google Scholar]
- 3.Förster S, Zisenis M, Wenz E, Antonietti M. J Chem Phys. 1996;104:9956–9970. [Google Scholar]
- 4.Nardin C, Hirt T, Leukel J, Meier W. Langmuir. 2000;16:1035–1041. [Google Scholar]
- 5.Meier W, Nardin C, Winterhalter M. Angew Chem Int Ed Engl. 2000;39:4599–4602. [PubMed] [Google Scholar]
- 6.Nardin C, Widmer J, Winterhalter M, Meier W. Eur Phys J E. 2001;4:403–410. [Google Scholar]
- 7.Lasic D D. Liposomes: From Physics to Applications. Amsterdam: Elsevier Science; 1993. pp. 287–289. [Google Scholar]
- 8.Szmelcman S, Hoffnung M. J Bacteriol. 1975;124:112–118. doi: 10.1128/jb.124.1.112-118.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schirmer T. J Struct Biol. 1998;121:101–109. doi: 10.1006/jsbi.1997.3946. [DOI] [PubMed] [Google Scholar]
- 10.Randall-Hazelbauer L, Schwartz M. J Bacteriol. 1973;116:1436–1446. doi: 10.1128/jb.116.3.1436-1446.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartz M. J Mol Biol. 1976;103:521–536. doi: 10.1016/0022-2836(76)90215-1. [DOI] [PubMed] [Google Scholar]
- 12.Mackay D J, Bode V C. Virology. 1976;72:154–166. doi: 10.1016/0042-6822(76)90320-2. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz M, Le Minor L. J Virol. 1975;15:679–685. doi: 10.1128/jvi.15.4.679-685.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roa M, Scandella D. Virology. 1976;72:182–194. doi: 10.1016/0042-6822(76)90322-6. [DOI] [PubMed] [Google Scholar]
- 15.Sambrook J, Frisch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 16.Prilipov A, Phale P S, Van Gelder P, Rosenbusch J P, Koebnik R. FEMS Microbiol Lett. 1998;163:65–72. doi: 10.1111/j.1574-6968.1998.tb13027.x. [DOI] [PubMed] [Google Scholar]
- 17.Marie D, Vaulot D, Partensky F. Appl Environ Microbiol. 1996;62:1649–1655. doi: 10.1128/aem.62.5.1649-1655.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulanger P, le Maire M, Dubois S, Desmadril M, Letellier L. Biochemistry. 1996;35:14216–14224. doi: 10.1021/bi9608673. [DOI] [PubMed] [Google Scholar]
- 19.Ringler P, Heymann B J, Engel A. In: Membrane Transport. Baldwin S A, editor. Oxford: Oxford Univ. Press; 2000. pp. 229–268. [Google Scholar]
- 20.Martin P, Szablewski M. Tensiometers and Langmuir-Blodgett Throughs Operating Manual. Cranfield University Milton Keynes, England: Nima; 1995. [Google Scholar]
- 21.Demes R A. Methods Ezymol. 1974;32:539–545. [Google Scholar]
- 22.Nardin C, Winterhalter M, Meier W. Langmuir. 2000;16:7708–7712. [Google Scholar]
- 23.Plançon L, Chamis M, Letellier L. J Biol Chem. 1997;272:16868–16872. doi: 10.1074/jbc.272.27.16868. [DOI] [PubMed] [Google Scholar]
- 24.Holloman W K. J Biol Chem. 1973;248:8114–8119. [PubMed] [Google Scholar]