

Abstract
Four ionic fullerene derivatives, which are relatively soluble in polar solvents, are shown to organize into morphologically different nanoscale structures. Spheres, nanorods, and nanotubules form in water depending on the side chain appendage of the fullerene spheroid. Images at different nanoscale structures were obtained via transmission electron microscopy. Also, computer simulations were used for investigating the relative spatial arrangements. The efficient method to fabricate almost perfect and uniformly shaped nanotubular crystals, which order spontaneously by self-assembly, opens the way to the possibility of exploiting the fullerene properties at the nanometer scale.
The ability to engineer distinct one-, two-, or three-dimensional patterns at the supramolecular level by modifying specific chemical components is a crucial step toward nanometer-sized technology (1, 2). Preliminary and successful methodologies have yielded a large variety of noncovalently bonded structures in solutions and crystals. However, the possibility of regulating size and shape of nanostructures in relation to function remains a current challenge of exceptional interest (3–9). It is therefore expected that the rules of nanopatterning of organic molecules, either spontaneous or induced, once understood, can play a major role in future and emerging technologies.
Fundamental requisites, accompanying the addition of well-designed chemical functionalities to drive self-assembly processes to (pre)determined mesoscopic shapes of controlled sizes and outer-shell structures, are the conservation, and possibly even the enhancement, of the molecular level properties. To this end, fullerenes are ideal candidates, both because of their excellent electronic properties—most of their derivatives have so far shown to be outstanding electron acceptors (10–15)—and because their amphiphilic derivatives were recently found to self-assemble at the nanometer scale to furnish nanorod and vesicle patterns (16–26). Fullerenes, in fact, have a strong tendency to form clusters of different sizes, especially in polar solvents (27, 28). Aggregation of C60 units may cause a significant change in their photochemical and photophysical properties, as compared with isolated molecules in solution (12). Of course, this change can have a profound influence on fullerene-based optical and electronic materials (29, 30). For instance, aggregation of fullerene spheroids was shown to play a crucial role in the preparation of photovoltaic cells (31).
Also, biological tests of fullerenes, which are usually carried out in aqueous solutions, are heavily affected by aggregation (32). Basically, dissolution of unmodified C60 or monofunctionalized organofullerenes is always accompanied by a high degree of clustering.
Here, we show that the hydrophobic fullerene core, combined with hydrophilic ammonium groups and also with other self-organizing groups, assembles into three fundamentally different low-dimensional shapes and, in particular, it is demonstrated that the linkage with a porphyrin macrocycle introduces a substantial morphological refinement at the mesoscopic level.
Materials and Methods
Compounds 1–3 were prepared according to our reported procedure (33).
Preparation of Compound 4.
A mixture of 20 mg of amino acid 5 (6.5 μmol) and 14 mg of porphyrin aldehyde 6 (21.8 μmol) was heated to reflux in toluene (40 ml) in the presence of 31 mg of C60 (43 μmol). After 3 h, the mixture was cooled to room temperature and the crude product was purified by flash chromatography (toluene/ethyl acetate 80/20) giving the fullerene derivative 7 (3.6 mg, 2.2 μmol), yield 10.3% based on starting aldehyde). Electrospray mass spectrum (THF/MeOH 1:1): m/z 1,607 (MH+); 1H NMR (CDCl3, 200 MHz): δ −2.9 (s, 2H), 1.41 (s, 9H), 2.95 (s, 1H), 3.34 (m, 2H), 3.61 (t, 2H), 3.80 (m, 4H), 4.12 (t, 4H), 5.00 (s, NH), 5.15 (s, 2H), 7.74 (s, 10H), 8.05 (m, 1H), 8.15 (s, 9H), 8.60 (m, 1H), 8.81 (s, 6H).
The fullerene salt 4 (3.24 mg, 2.1 μmol, yield 95%) was obtained by purging a solution of 7 in CH2Cl2 with HCl gas and was purified by washing the precipitate several times with toluene. Electrospray mass spectrum (MeOH): m/z 1,507 (M+).
Transmission Electron Microscopy (TEM)
A total of 0.5 mg of the fullerene salts 1–4 was dispersed in 1 ml of filtered water (Millex-HV filter, Millipore). The mixture was sonicated three times for 5 min in a ultrasonic water bath with 5-min intervals. The insoluble material was removed by centrifugation at 3,000 rpm (Hettich, model Eba 1Z), and one drop from the clear solution was transferred to a TEM grid (copper grid, 3.0 mm, 200 mesh, coated with formvar film) together with a drop of uranyl acetate (2% water solution). After air drying of the grid, TEM images were taken on a Philips TEM 208 at an accelerating voltage of 100 kV.
Computational.
Computer simulations (34–36) were performed with the MM3 model (37–39) using periodic boundary conditions and a combination of simulated annealing and geometry optimization techniques. The MM3 force field has been widely used in one of our laboratories and was developed by fitting both heats of formation and structural results in the gas phase and in crystals of simple organics (40–43). MM3 is specifically parameterized to describe H bonds in terms of dipole interactions and includes specific interatomic nonbonded potential energy functions that allow a quantitative treatment of the van der Waals and electrostatic interactions, which play an essential role in hydrogen bonding and in the π–π interactions between aromatic rings. In the treatment of the periodic boundary conditions, Ewald summation was used for the electrostatic interaction, thereby including all of the possible terms.
Results and Discussion
The 1,3-dipolar cycloaddition of azomethine ylides to C60 is a very powerful methodology for the functionalization of fullerenes, allowing the introduction of potentially any functional groups (44, 45). Compounds 1–3 (Scheme S1) were easily prepared by using the appropriately functionalized glycine and formaldehyde. The tert-butoxycarbonyl (Boc)-protected amino end groups were liberated with the use of gaseous HCl. In the case of 3, methylation of pyrrolidine nitrogen preceded amino group deprotection (33).
Scheme 1.

Porphyrin derivative 4 was synthesized by the same experimental procedure employing a porphyrin aldehyde (Scheme S2).
Scheme 2.

The four amphiphilic derivatives in Scheme S1 have a distinct hydrophobicity–hydrophilicity balance. They are all characterized by the presence of at least one positive charge, which makes them partially soluble in water, while, at the same time, the known fullerene–fullerene attraction forces are expected to cause for aggregation. Differences in the nature and chain length are also anticipated to be of significance.
A dispersion of 1 (0.5 mg) in distilled and filtered water (1 ml) was immersed in an ultrasonic bath three times with 5-min intervals. Most of the compound did not dissolve, therefore the heterogeneous mixture was centrifuged. The resulting homogeneous solution was then transferred onto a TEM grid. A representative image is shown in Fig. 1, which very clearly reveals perfectly round shapes. The spheres have very similar sizes, with diameters ranging from 500 nm to 1.2 μm, and tend to aggregate with each other. The size of these spheres falls within the wide range of the already observed fullerene vesicles, which can typically reach from 17 nm to 10 μm (16–25). It is therefore clear that the size of the spheres is determined by the different methods of preparation (i.e., time and type of sonication, presence of cosolvents, deposition, etc.) or the hydrophobic area left on the fullerene core (i.e., number and nature of the functional groups).
Figure 1.

TEM images of the spheres formed by fullerene derivative 1 (Upper), and closer view of the spheres (Lower).
However, when compound 2 was treated under identical conditions as 1, no spherical objects were observed by TEM. Instead, long uniform bundles consisting of tens of nanorods all perfectly aligned in parallel, with diameters of about 4 nm and lengths of several microns, appeared (Fig. 2). In other words, a templated growth affording completely different shapes evolves from the simple structural modification from 1 to 2. The reason must lie in the different size and nature of the chain. In the case of 1, a short chain favors the self-assembly in the form of spheres, probably as the result of the balance between attractive (fullerene–fullerene) and repulsive (between ammonium head groups) forces. In the case of 2, the longer oligoethylene glycol chain leads to rod-like structures. It is important to note that no nanorods were detected in the electron micrographs of 1, nor spheres were ever seen in the case of 2. Molecular tailoring of fullerene derivatives therefore induces completely different kinds of assembly!
Figure 2.

TEM images of a bundle of nanorods formed by compound 2 (Upper), and closer view of the bundle (Lower).
The two compounds 2 and 3 behave similarly, the only difference being an increase in the solubility after the introduction of a second charge in compound 3. Although 2 has a limited solubility in water (0.32 mg/ml), 0.5 mg of 3 dissolves completely (maximum solubility is 0.8 mg/ml). A representative example of a TEM picture obtained by using derivative 3 is reported in Fig. 3.
Figure 3.
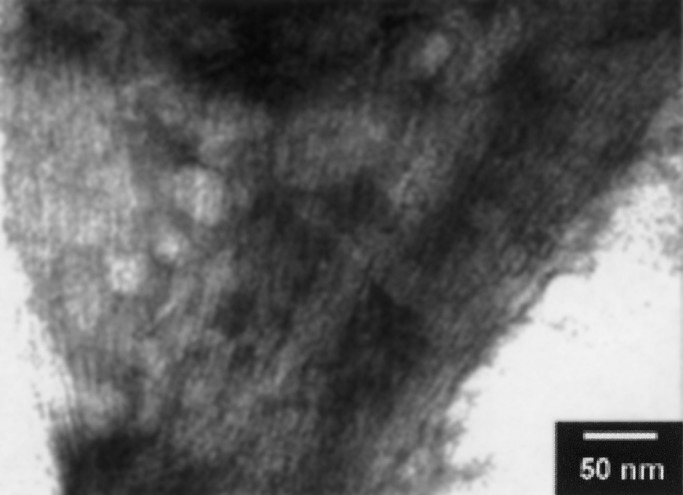
TEM image of a bundle of nanorods formed by compound 3.
A possible molecular structure of the nanorods was computer-generated, considering the fullerene–fullerene interactions similar to those occurring in the solid state (Fig. 4).
Figure 4.
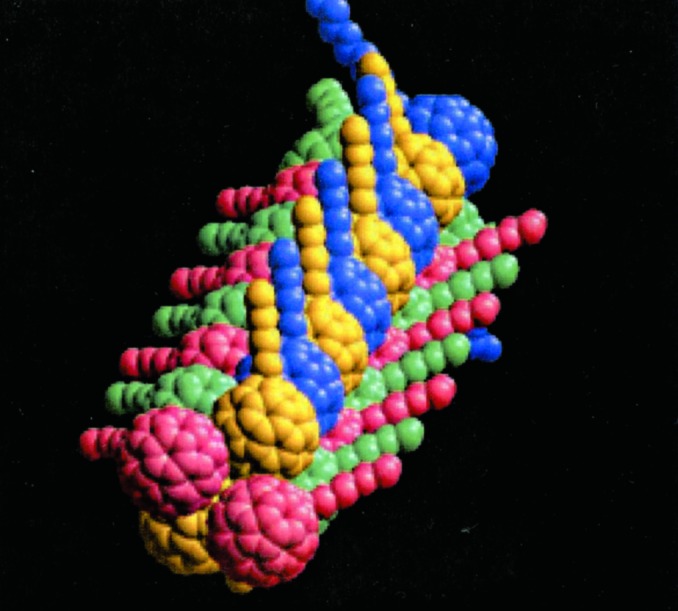
Pictorial representation of the nanorods formed by self-assembly of salt 2 in water,
To further tune the self-assembly process, and in the perspective of possible applications based on photoinduced electron-transfer, an additional element of ordering, namely a porphyrin macrocycle (a free base tetraphenylporphyrin), was linked to the skeleton of 2 to afford 4. It is known that porphyrins give rise to strong π-stacking interactions both among themselves and with fullerenes (46–48).
When 4 was sonicated in water under the same conditions as 1–3, individual, isolated tubules were formed with dimensions typically reaching 30 nm in diameter and 500 nm in length (Fig. 5). No other shapes—including bundles or spheres—were detected. Therefore, ligation of 2 with a porphyrin moiety promotes the formation of nanorods with a strongly improved morphology. Emphasis should be placed on the uniformity of diameters and the photo- and electroactive role that the new addend potentially plays, for instance, in photoconversion processes or in electronic nanocircuitry. On sonication, compound 4 can undergo a possible cooperative transformation into fibrous rods, whose structure is discernable in the supramolecularly resolved TEM images. The fact that the rods assemble in water suggests a hydrophilic contribution to the stabilization of the shape.
Figure 5.
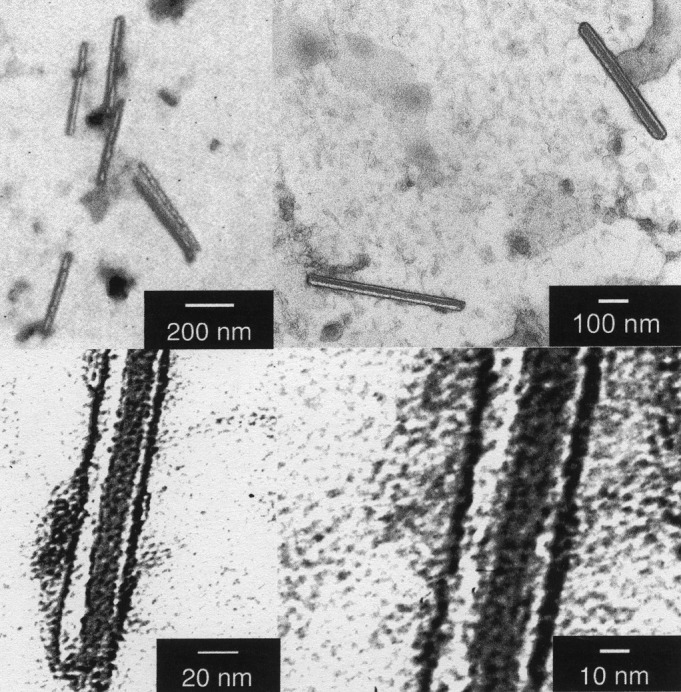
Nanotubules formed by compound 4 observed at different scales. The measures of the two focused tubules are 35 × 530 and 50 × 470 nm.
In a reference experiment, an aqueous suspension of 4, which was not subjected to sonication, failed to generate any nanorod-like structures.
The porphyrin–porphyrin, porphyrin–C60, and C60–C60 interactions are the driving forces of the self-assembly of these systems. Energetically, the porphyrin–porphyrin and the porphyrin–C60 interactions exceed the C60–C60 interactions by a factor of 4–6. Computers have been used here to investigate the relative spatial arrangement of the porphyrin and C60 units, first as a dimer complex and then as an oligomeric extension, and also using periodic boundary conditions. The structure of Fig. 6 Upper is more stable than dimer of Fig. 6 Lower by 6 kcal/mol. In 6 Lower, the stable porphyrin–porphyrin interaction is not fully exploited because the fullerenes act as spacers between them. One- and two-dimensional extensions of the dimeric structures (opposed to full three-dimensional crystal structures) can be obtained in a computer experiment by exploiting the different packing distance of porphyrins, ≈3.2 Å, and C60, ≈10 Å.
Figure 6.
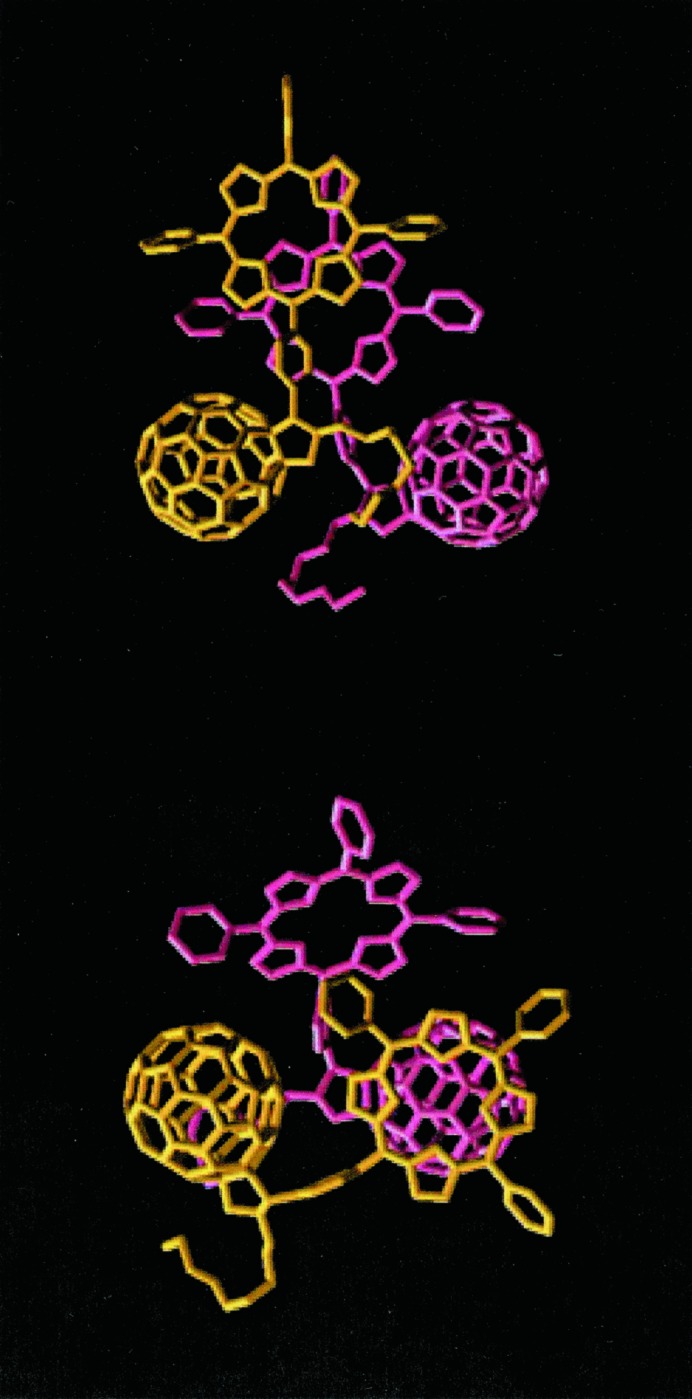
Most stable dimeric structures of 4. Note that Upper is 6 kcal/mol per molecule more stable than Lower.
Fig. 7 Upper shows the periodic resetting of the different packing distance by combining the two dimeric structures and replacing a porphyrin–porphyrin interacting subunit with a porphyrin–C60 interacting subunit. Fig. 7 Lower shows an even more stable structure (by 2 kcal/mol) where porphyrin–porphyrin interactions add up to reach the C60–C60 spacing. In silico construction of porphyrin–porphyrin complexes, in the presence of C60, easily organizes the molecules linearly and forfeits truly three-dimensional structures.
Figure 7.
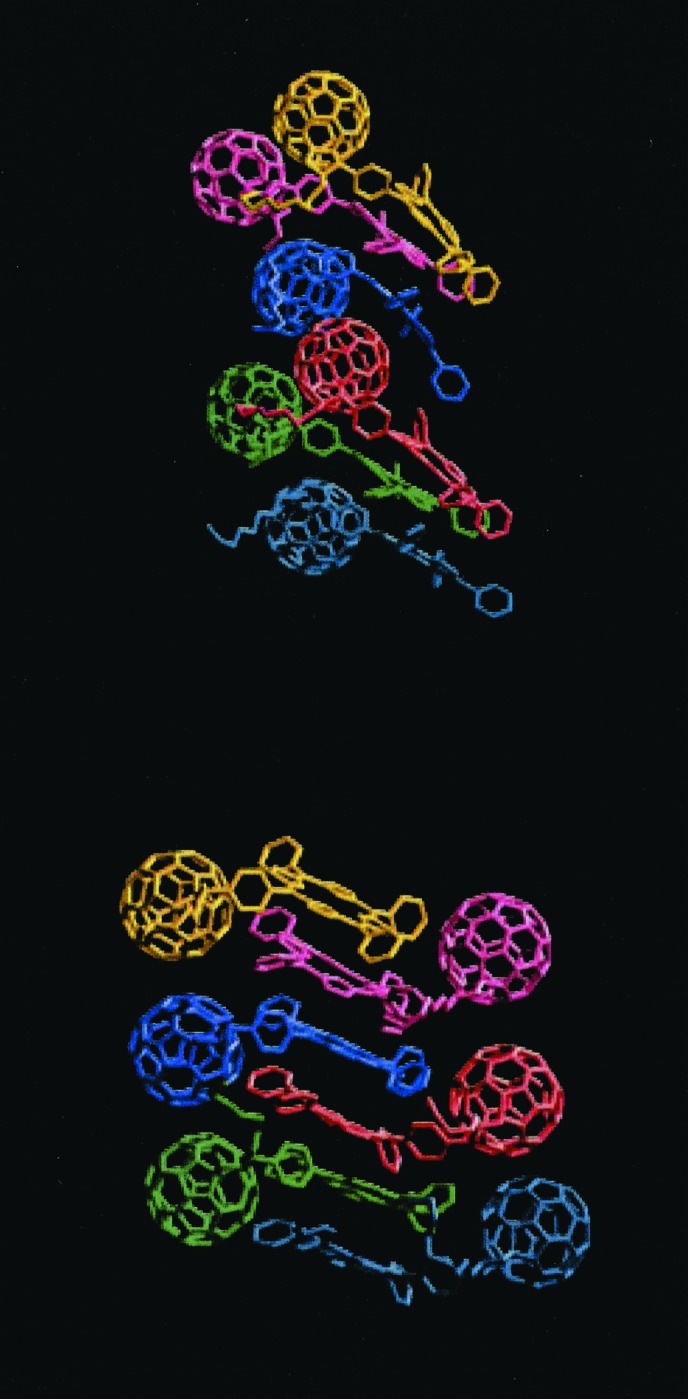
(Upper) Interlayered structure (see text). (Lower) Porphyrin stacked structure. Notice that the porphyrin stacked structure is 2 kcal/mol per molecule more stable than the interlayered structure.
Close-up inspection of the TEM images of the nanotubules (Fig. 5) shows that each tubule is formed by at least four slimmer rods with dimensions ranging between 3.3 and 3.7 nm, laying close together in the center of the tubule and two individual like walls. The width of these nanorods is slightly bigger than the size of a single derivative (about 2 nm), which suggests that the rods are supramolecularly assembled including at least two units. Importantly, the assembly of thinner rods of the type shown in Fig. 7 can revert the energy order of 7 Upper and 7 Lower. In particular, for 7 Upper the zigzag location of fullerenes, naturally offers a grip to establish porphyrin–fullerene interactions. Interestingly, the small bumps observed in Fig. 5 on the surface of the nanorods agree with this picture.
The driving force for the formation of each superstructure is the result of a subtle balance between hydrophobic interactions and specific, directional interactions between the molecules. The 1 to 4 compounds are all amphiphilic, but have very different solubilities in water. The least-soluble compound, 1, forms spheres or droplets, thus suggesting that the driving force for aggregation is hydrophobic and nondirectional (for a comprehensive discussion, see ref. 49). Formation of “infinitely” long rods, as in the case of 2 and 3, indicates a lesser role played by hydrophobicity. Indeed, the longer chains can improve solubilization of the terminal hydrophilic groups. The resulting superstructures can still occur without the presence of preferred, or directional, interactions (see 49). Presence of directionality is here positively introduced in 4 by the porphyrin ring (see Fig. 5) and causes the finite length of the nanotubules and their more regular structures (see 49).
In summary, the assembly methodology described here provides an excellent opportunity to develop tailoring architectural criteria for the formation of nanotubular structures. The addition of a porphyrin as a close-packing motif at the molecular level leads to a significant refinement of the tubular structure. The possibility of creating carbon-based tubular structures by spontaneous self-assembly is a very exciting prospect in the search of materials that exhibit nearly ideal electrical, optical, and mechanical properties.
Acknowledgments
We are deeply indebted to Mr. Claudio Gamboz and Prof. Maria Rosa Soranzo (CSPA, University of Trieste), for continuous help with TEM measurements. This work was carried out with partial support from the European Union Training and Mobility of Researchers Network “USEFULL” Contract FRMX-CT97–0126, Human Potential Network “FUNCARS” Contract HPRN-1999-00011, Ministero dell'Universitá e della Ricerca Scientifica e Tecnologica (PRIN 2000, MM03198284), Consiglio Nazionale delle Ricerche (Italy) Program “Materiali Innovativi (legge 95/95)”, and the Office of Basic Energy Sciences of the United States Department of Energy. This is document NDRL-4321 from the Notre Dame Radiation Laboratory.
Abbreviation
- TEM
transmission electron microscopy
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Lehn J-M. Supramolecular Chemistry. New York: VCH Press; 1995. [Google Scholar]
- 2.Whitesides G M, Mathias J P, Seto C T. Science. 1991;254:1312–1319. doi: 10.1126/science.1962191. [DOI] [PubMed] [Google Scholar]
- 3.Stupp S I, LeBonheur V, Walker K, Li L S, Huggins K E, Keser M, Amstutz A. Science. 1997;276:384–389. doi: 10.1126/science.276.5311.384. [DOI] [PubMed] [Google Scholar]
- 4.Zubarev E R, Pralle M U, Li L M, Stupp S I. Science. 1999;283:523–526. doi: 10.1126/science.283.5401.523. [DOI] [PubMed] [Google Scholar]
- 5.Zubarev E R, Pralle M U, Sone E D, Stupp S I. J Am Chem Soc. 2001;123:4105–4106. doi: 10.1021/ja015653+. [DOI] [PubMed] [Google Scholar]
- 6.Won Y-Y, Davis H T, Bates F S. Science. 1999;283:960–963. doi: 10.1126/science.283.5404.960. [DOI] [PubMed] [Google Scholar]
- 7.Johnson S A, Ollivier P J, Mallouk T E. Science. 1999;283:963–965. doi: 10.1126/science.283.5404.963. [DOI] [PubMed] [Google Scholar]
- 8.Piner R D, Zhu J, Xu F, Hong S, Mirkin C A. Science. 1999;283:661–663. doi: 10.1126/science.283.5402.661. [DOI] [PubMed] [Google Scholar]
- 9.Yokoyama T, Yokoyama S, Kamikado T, Okuno Y, Mashiko S. Nature (London) 2001;413:619–621. doi: 10.1038/35098059. [DOI] [PubMed] [Google Scholar]
- 10. Imahori, H. & Sakata, Y. (1999) Eur. J. Org. Chem., 2445–2457.
- 11. Guldi, D. M. (2000) Chem. Commun., 321–327.
- 12.Guldi D M, Prato M. Acc Chem Res. 2000;33:695–703. doi: 10.1021/ar990144m. [DOI] [PubMed] [Google Scholar]
- 13.Gust D, Moore T A, Moore A L. Acc Chem Res. 2001;34:40–48. doi: 10.1021/ar9801301. [DOI] [PubMed] [Google Scholar]
- 14.Echegoyen L, Echegoyen L E. Acc Chem Res. 1998;31:593–601. [Google Scholar]
- 15.Martín N, Sànchez L, Illescas B, Pérez I. Chem Rev. 1998;98:2527–2547. doi: 10.1021/cr9701039. [DOI] [PubMed] [Google Scholar]
- 16.Prassides K, Keshavarz M, Beer E, C, B, Gonzalez R, Murata Y, Wudl F, Cheetham A K, Zhang J P. Chem Mater. 1996;8:2405–2408. [Google Scholar]
- 17.Jenekhe S A, Chen X L. Science. 1998;279:1903–1907. doi: 10.1126/science.279.5358.1903. [DOI] [PubMed] [Google Scholar]
- 18.Hetzer M, Clausen-Schaumann H, Bayerl S, Bayerl T M, Camps X, Vostrowsky O, Hirsch A. Angew Chem Int Ed Engl. 1999;38:1962–1965. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1962::AID-ANIE1962>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 19.Cassell A M, Asplund C L, Tour J M. Angew Chem Int Ed Engl. 1999;38:2403–2405. doi: 10.1002/(sici)1521-3773(19990816)38:16<2403::aid-anie2403>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 20. Murakami, H., Shirakusa, M., Sagara, T. & Nakashima, N. (1999) Chem. Lett., 815–816.
- 21. Sawamura, M., Nagahama, N., Toganoh, M., Hackler, U. E., Isobe, H., Nakamura, E., Zhou, S.-Q. & Chu, B. (2000) Chem. Lett., 1098–1099. [DOI] [PubMed]
- 22.Sano M, Oishi K, Ishi-i T, Shinkai S. Langmuir. 2000;16:3773–3776. [Google Scholar]
- 23.Nakashima N, Ishii T, Shirakusa M, Nakanishi T, Murakami H, Sagara T. Chem Eur J. 2001;7:1766–1772. doi: 10.1002/1521-3765(20010417)7:8<1766::aid-chem17660>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 24. Shi, Z., Jin, J., Li, Y., Guo, Z., Wang, S., Jiang, L. & Zhu, D. (2001) New J. Chem., 670–672.
- 25.Zhou S Q, Burger C, Chu B, Sawamura M, Nagahama N, Toganoh M, Hackler U E, Isobe H, Nakamura E. Science. 2001;291:1944–1947. doi: 10.1126/science.291.5510.1944. [DOI] [PubMed] [Google Scholar]
- 26.Guldi D M, Maggini M, Mondini S, Guerin F, Fendler J H. Langmuir. 2000;16:1311–1316. [Google Scholar]
- 27.Sun Y P, Bunker C. Nature (London) 1993;365:398–401. [Google Scholar]
- 28.Alargova R, Deguchi S, Tsujii K. J Am Chem Soc. 2001;123:10460–10467. doi: 10.1021/ja010202a. [DOI] [PubMed] [Google Scholar]
- 29.Prato M. J Mater Chem. 1997;7:1097–1109. [Google Scholar]
- 30.Prato M. Top Curr Chem. 1999;199:173–188. [Google Scholar]
- 31.Brabec C, Sariciftci N, Hummelen J. Adv Funct Mater. 2001;11:15–26. [Google Scholar]
- 32. Da Ros, T. & Prato, M. (1999) Chem. Commun., 663–669.
- 33.Kordatos K, Da Ros T, Bosi S, Vàzquez E, Bergamin M, Cusan C, Pellarini F, Tomberli V, Baiti B, Pantarotto D, et al. J Org Chem. 2001;66:4915–4920. doi: 10.1021/jo015608k. [DOI] [PubMed] [Google Scholar]
- 34.Ponder J, Richards F. J Comput Chem. 1987;8:1016–1024. [Google Scholar]
- 35.Kundrot C, Ponder J, Richards F. J Comput Chem. 1991;12:402–409. [Google Scholar]
- 36.Dudek M, Ponder J. J Comput Chem. 1995;16:791–816. [Google Scholar]
- 37.Allinger N, Yuh Y, Lii J-H. J Am Chem Soc. 1989;111:8551–8566. [Google Scholar]
- 38.Lii J-H, Allinger N. J Am Chem Soc. 1989;111:8566–8575. [Google Scholar]
- 39.Lii J-H, Allinger N. J Am Chem Soc. 1989;111:8576–8582. [Google Scholar]
- 40.Deleuze M, Zerbetto F. J Am Chem Soc. 1999;121:5281–5286. [Google Scholar]
- 41.Bermudez V, Capron N, Gase T, FG, G, Kajzar F, Leigh D, Zerbetto F, Zhang S. Nature (London) 2000;406:608–611. doi: 10.1038/35020531. [DOI] [PubMed] [Google Scholar]
- 42.Leigh D, Parker S, Timpel D, Zerbetto F. J Chem Phys. 2001;114:5006–5011. [Google Scholar]
- 43.Cavallini M, Lazzaroni R, Zamboni R, Biscarini F, Timpel D, Zerbetto F, Clarkson G, Leigh D. J Phys Chem B. 2001;105:10826–10830. [Google Scholar]
- 44.Maggini M, Scorrano G, Prato M. J Am Chem Soc. 1993;115:9798–9799. [Google Scholar]
- 45.Prato M, Maggini M. Acc Chem Res. 1998;31:519–526. [Google Scholar]
- 46.Boyd P D W, Hodgson M C, Rickard C E F, Oliver A G, Chaker L, Brothers P J, Bolskar R D, Tham F S, Reed C A. J Am Chem Soc. 1999;121:10487–10495. [Google Scholar]
- 47.Zheng J-Y, Tashiro K, Hirabayashi Y, Kinbara K, Saigo K, Aida T, Sakamoto S, Yamaguchi K. Angew Chem Int Ed Engl. 2001;40:1858–1861. [PubMed] [Google Scholar]
- 48.Diederich F, Gomez Lopez M. Chem Soc Rev. 1999;28:263–277. [Google Scholar]
- 49.Israelachvili J N. Intermolecular Forces. 2nd Ed. London: Academic; 1992. pp. 349–360. [Google Scholar]