

Abstract
The design of polymers and oligomers that mimic the complex structures and remarkable biological properties of proteins is an important endeavor with both fundamental and practical implications. Recently, a number of nonnatural peptides with designed sequences have been elaborated to provide biologically active structures; in particular, facially amphiphilic peptides built from β-amino acids have been shown to mimic both the structures as well as the biological function of natural antimicrobial peptides such as magainins and cecropins. However, these natural peptides as well as their β-peptide analogues are expensive to prepare and difficult to produce on a large scale, limiting their potential use to certain pharmaceutical applications. We therefore have designed a series of facially amphiphilic arylamide polymers that capture the physical and biological properties of this class of antimicrobial peptides, but are easy to prepare from inexpensive monomers. The design process was aided by molecular calculations with density functional theory-computed torsional potentials. This new class of amphiphilic polymers may be applied in situations where inexpensive antimicrobial agents are required.
A large group of defense peptides, such as the magainins, are produced in eukaryotic systems and provide a first line of defense against bacterial infections (1–4). These peptides adopt facially amphiphilic conformations, in which positively charged and hydrophobic groups segregate onto opposite faces of a helix, sheet, or tertiary structure (Fig. 1) (5, 6). This structural feature is believed to be responsible for their ability to kill cells by disrupting phospholipid membranes.
Figure 1.
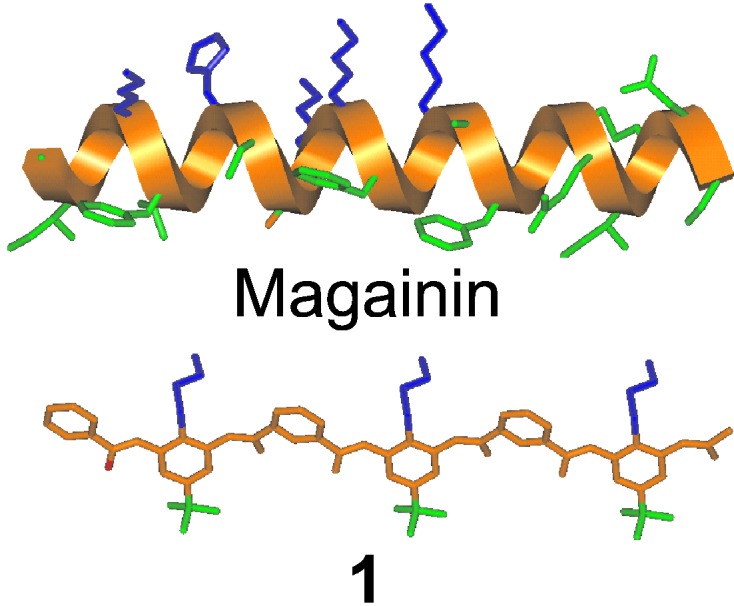
Amphiphilic structure of 1 and magainin. Hydrophobic side chains are shown in green, and hydrophilic side chains are in blue.
The activity of this class of peptides depends on its overall physicochemical properties rather than the precise details of its structures (6). Thus, it has been possible to design β-peptides (7–9) and self-assembling cyclic peptides (10) that mimic the biological and physical properties of antimicrobial peptides. An important consideration in the design of antimicrobial β-peptides was to match the hydrophobic period of the sequence with that of the secondary structure adopted by the peptide. For example, C3-substituted β-peptides adopt a “14-helix” with an approximate 3-residue repeat (11, 12). Thus, oligomers based on a repeating tripeptide sequence, Hb-Hb-Hp (Hb = hydrophobic, Hp = hydrophilic), adopt facially amphiphilic conformations in which apolar and polar side chains segregate onto opposite faces of the helix (7–9, 13). An important extension of this work would be to design inexpensive polymers and oligomers that adopt amphiphilic secondary structures. Such surface-active polymers could be used for a variety of purposes, such as antimicrobial materials and surfaces (14). The fundamental knowledge obtained in these studies would also have important implications for the design of sequence-specific oligomers with well defined three-dimensional structures and biological properties.
Materials and Methods
2,6-Dinitro-4-t-butyl-phenyl (4-methyl)-benzenesulfonate (8).
2,6-Dinitro-4-t-butyl-phenol (80 mmol) and tosyl chloride (80 mmol) were dissolved in 300 ml of CH2Cl2. Diisopropylethylamine (DIEA; 80 mmol) was added to the solution. The mixture was stirred at room temperature for 2 h. The solution was washed with 10% citric acid, saturated aqueous NaCl (sat. NaCl), and dried with MgSO4. The solvent was removed under reduced pressure, and the product was obtained as a bright yellow solid in quantitative yield. MP 145–146°C 1H NMR (500 MHz, CDCl3): δ = 8.12 (s, 2H), 7.80 (d, 2H), 7.40 (d, 2H), 2,51(s, 3H), 1.41 (s, 9H). Electrospray ionization-MS: m/z: 417.07 (calcd); 417.2 (M + Na+).
2,6-Dinitro-4-t-butyl-1-(2-t-butoxycarbonylaminoethyl)-sulfanylbenzene (9).
Compound 8 (13 mmol), 2-boc-aminoethanethiol (16 mmol), and DIEA (13 mmol) were dissolved in 50 ml of chloroform. The solution was stirred under nitrogen for 12 h. The solution was washed with 0.5 M NaOH, 10% citric acid, saturated (sat.) Na2CO3 and sat. NaCl, and dried with MgSO4. The solution volume was reduced to 15 ml by rotary evaporation. The product crystallized as a bright yellow solid after addition of 80 ml of hexane. Yield: 94%. MP 127–129°C 1H NMR (500 MHz, CDCl3): δ = 7.81 (s, 2H), 4.87 (s, 1H), 3.31 (t, 2H), 3.10 (t, 2H), 1.44 (s, 9H), 1.39 (s, 9H). Electrospray ionization-MS: m/z: 422.45 (calcd); 422.4 (M + Na+).
2,6-Diamino-4-t-butyl-1-(2-t-butoxycarbonylaminoethyl)sulfanylbenzene (10).
Dinitro compound 9 (20 mmol) and sodium acetate (200 mmol) were added to 50 ml of EtOH. The mixture was heated to 78°C, and the solid dissolved completely. SnCl2⋅2H2O (200 mmol) was added to the solution, and the reaction mixture was stirred at 78°C for 35 min. After removal of solvent under reduced pressure, the residue was dissolved in 800 ml of EtOAc and washed with 40% K2CO3. The solvent was reduced by rotary evaporation, and the product was purified by column chromatography (silica gel, CH2Cl2/MeOH 100:1 to 95:5). Yield: 93%. MP 111–113°C 1H NMR (500 MHz, CDCl3): δ = 6.21 (s, 2H), 5.41 (s, 1H), 4.35 (br, 4H), 3.21 (t, 2H), 2.75 (t, 2H), 1.35 (s, 9H), 1.24 (s, 9H). Electrospray ionization-MS: m/z: 340.51 (calcd); 340.5 (MH+).
General Method of Polymerization.
Diamine 10 (0.1 mmol) was dissolved in 3 ml of dimethylformamide (DMF). Isophthaloyl dichloride (0.1 mmol), triethylamine (0.2 mmol) and N,N-dimethylethylenediamine (0.02 mmol for 4 and 0 mmol for 5) were added while stirring. The mixture was stirred under nitrogen for 18 h. After the volume of solvent was reduced to 1 ml, water was added to precipitate the polymer. The polymer was collected and dried under vacuum. The Boc group was removed by treatment with trifluoroacetic acid (TFA; 3 ml) for 1 h. The deprotected polymer was dried under vacuum overnight.
Solid-Phase Synthesis of Oligomers 2 and 3.
Fmoc-PAL-PEG-resin (0.1 mmol) was swelled in DMF. Then the Fmoc was removed with 20% piperidine in DMF for 20 min. The oligomer was then built up by alternately coupling 10 equivalents of isophthalic acid or diamine 10. In each case, the couplings were carried out in DMF with 10 equivalents each of 2-(1H-benzotriazole-1-yl)-1,1,3,3,-tetramethyluronium hexafluorophosphate (HBTU) and N-hydroxybenzotriazole hydrate (HOBt), and 20 equivalents of DIEA for 24 h at room temperature. The oligomers were cleaved from the resin by treatment with TFA/anisole (95:5) for 1 h. Pure oligomers were obtained by HPLC on a reverse-phase C4 column, with a linear gradient from 30% to 80% solvent B in 50 min (solvent A, 0.1% TFA in water; solvent B, acetonitrile/water/TFA 900:99:1). Matrix-assisted laser desorption ionization–time-of-flight MS: 2: 755.99 (calcd); 756.5 (M + H+); 3: 1125.47 (calcd); 1125.6 (M + H+).
Antimicrobial Testing.
The compounds were dissolved in DMSO/water to make series of stocks of 2-fold dilution and were diluted 10-fold to cell culture on 96-well plates. Minimal inhibitory concentrations (MICs) were obtained by incubating the compounds with the bacteria for 18 h at 37°C and measuring cell growth at OD590.
Vesicle Leakage Assay.
The leakage of calcein from large unilamellar vesicles was measured as described (9). The polymer was dissolved in DMSO at concentrations from 300 μg/ml to 37.5 μg/ml. The vesicles were prepared by reverse-phase evaporation in 10 mM sodium phosphate buffer (pH 7) followed by a single extrusion through a 0.2-μm-pore-size polycarbonate filter. The nontrapped calcein was removed by eluting through a size-exclusion Sephadex G-25 column, with 90 mM sodium chloride/10 mM sodium phosphate (pH 7). The leakage process was monitored by following the increase of calcein fluorescence intensity at 515 nm (excitation at 490 nm, slit width 3.8 nm) after 100 μl of polymer stock was added to 1.9 ml of vesicle solution. Complete leakage was achieved by addition of 10 μl of 0.2% Triton X-100 to the 2-ml solution, and the corresponding fluorescence intensity was used as 100% leakage for the calculation of leakage fraction.
Results
Design.
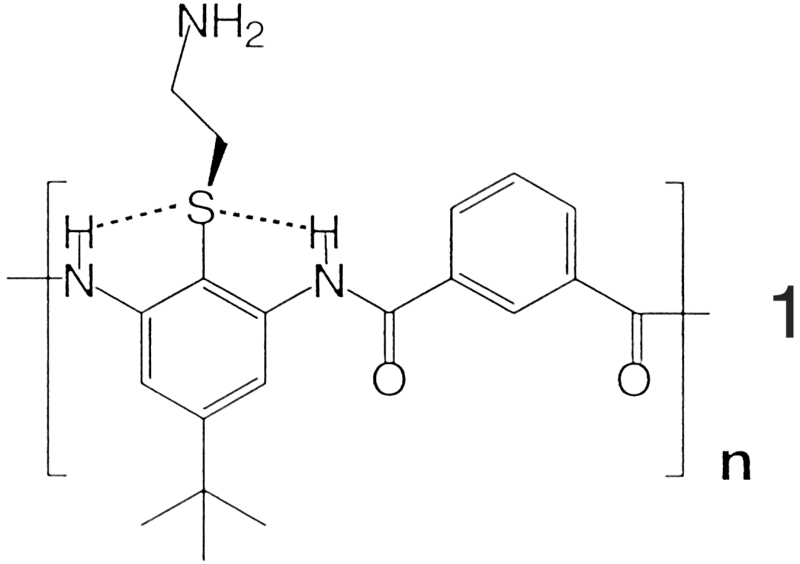
Our initial attempts to design facially amphiphilic structures have
focused on AB arylamide polymers typified by 1 (shown
below)
 .
.
The diamine was chosen for its ease of synthesis and its expected conformational properties. The choice of a thioether was based on the expectation that the methylene bonded to the thioether group would prefer to lie out of the plane of the ring, allowing weak hydrogen bonding to both amide protons. This interaction should stiffen the conformation and help to eliminate the formation of intermolecular hydrogen-bonded aggregates with low solubility.
Computational Studies.
Critical to the design of folded polymers is the development of accurate computational methods, analogous to those developed for peptide and protein structures that can predict low-energy conformations of the backbone. Density functional calculations of suitable model compounds were conducted to rigorously determine the torsional barriers for rotation about bonds within the arylamide system. The torsional potential profile (Fig. 2a) for a model compound, 2-methylthioacetanilide [C6H4(SCH3)(NHCOCH3)], exemplifies the importance of the designed hydrogen bond on the molecular flexibility of the backbone. [The barrier C C N (H) C(O) height is 8 kJ/mol higher for the CC N(H) C(O), CN(H) C(O) C, N(H)C(O)C C, and C C S C model compound with the methyl-thioether group than for the one without this substituent (Fig. 2a), and the overall torsional barrier for the former reaches 63 kJ/mol when the carbonyl O is directed in plane toward S.] Density functional calculations also confirmed that the methylene attached to the thioether sulfur would lie out of the plane of the phenyl ring (data not shown). These torsions were then used together with existing parameters (Fig. 2) to provide an improved molecular mechanics force field for the polymer.
Figure 2.

Computational calculations for aryl amide oligomers. Density functional theory based methods were used to generate torsional potentials, which were then combined with CHARMM (18) bond-stretching and -bending potentials, and TRAPPE (19) and OPLS (20) van der Waals and electrostatic potentials for MD simulations of the designed oligomers. (a) Relative energy of different conformations of C6H4(N(H)C(O)CH3)(X), X = H or SCH3, in which one C-C-N(H)-C(O) torsional angle is held approximately fixed by a Lagrangian multiplier, but all other parameters are relaxed [using the CPMD (19) program with the HCTH (22) density functional]. Zero degrees is defined as the angle that projects the amide proton toward the thioether S. The thioether methyl was initially placed perpendicular to the benzene ring plane, near the minimum conformation for the compound (∥). (b) Crystal structure of a model amide. (Left) X-ray structure. (Right) Simulated structure [from an MD calculation at T = 298 K and P = 1 atm (1 atm = 101.3 kPa)]. The top two images are for a 2 × 4 × 2 crystal lattice. Color notation: carbon (green), oxygen (red), nitrogen (blue), polar hydrogen (white), and sulfur (yellow); nonpolar hydrogens are not shown. The x-ray crystal structure vs. calculated results are cell lengths [Å] a: 15.34, 14.62; b: 8.28, 8.90; c: 23.16, 23.23; cell angles [°] α: 90, 90.00; β: 90.78, 94.58; γ: 90, 89.99. (c) Snapshots from two sets of MD calculations of oligomer 3 at the n-octane/water interface. One started with the entire oligomer immersed in the water phase (Right) and the other (Left) inside the n-octane phase. The initial configurations were generated from a Monte Carlo simulation with configurational-bias Monte Carlo techniques (22–24), and then the dynamic evolution of the polymer was monitored with MD in the isothermal-isobaric ensemble at T = 298 K and P = 1 atm. Color notation: n-octane (green), water (oxygen: red; hydrogen: white), and chlorine counterion (magenta); oligomer 3 is colored as in b.
Importantly, model compounds can be synthesized and their structures determined by x-ray diffraction as a test of the new force field. The crystal structure of an appropriate model thioether amide displays the conformational properties expected from the design (Fig. 2b). Also, molecular dynamics (MD) simulations of the crystal structure are in excellent agreement with experiment, providing support for the computed torsional potentials (Fig. 2b). Encouraged by this agreement, we conducted MD simulations on an oligomer 3 containing six aromatic units in an n-octane/water interfacial system. Irrespective of the initial starting configurations, the amphiphilic polyamide rapidly moved to the interface and exposed its polar amine functions into the water phase and its alkyl side chains into the n-octane phase (Fig. 2c).
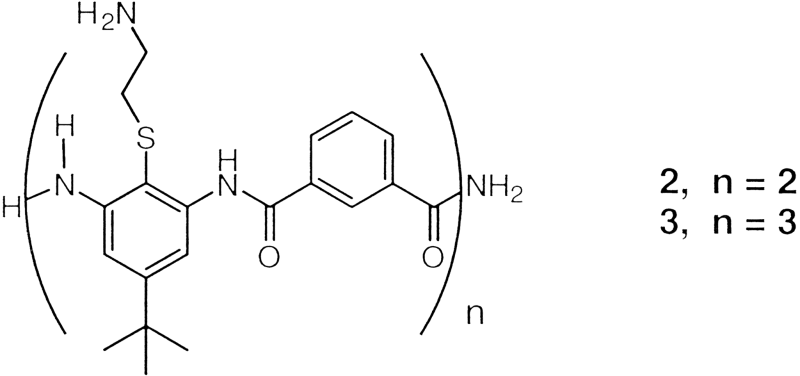
Synthesis.
Polymers based on the repeat, 1, are prepared by
polycondensation of the diamine with isophthaloyl chloride. The diamine
was derived from the commodity chemical 4-tert-butylphenol
and was synthesized from commercially available
2,6-dinitro-4-tert-butyl-phenol and other inexpensive
starting materials in several high-yielding steps (>90%) (Scheme
S1). Oligomers 2 and
3 (shown below) containing two or three AB units were also
prepared by solid-phase methods and purified to
homogeneity
 .
.
Scheme 1.

(i) TsCl, DIEA, CH2Cl2; (ii) Boc-NH(CH2)2SH, DIEA, CH2Cl2; (iii) SnCl2, EtOH 78°C; (iv) isophthaloyl chloride, triethylamine, dimethylethylenediamine, DMF; (v) TFA.
We also prepared analogous oligoureas by using phenyl 1,3-diisocyanate in place of isophthaloyl chloride. However, these molecules had low aqueous solubility, possibly because of intermolecular hydrogen bond formation involving the additional urea NH groups that are unable to form intramolecular hydrogen bonds.
Antimicrobial Activity.
The antimicrobial activity of the arylamides was measured with Klebsiella pneumoniae and Escherichia coli as Gram-negative strains and Bacillus subtilis as a Gram-positive strain (Table 1). Considering the simplicity of their structures, the short oligomers and polymers have good activity when compared with other molecules, including a synthetically modified magainin II derivative [MIC = 3.2 μg/ml vs. E. coli JM109 and 1.6 μg/ml vs. B. subtilis BR151 (8)] or the antimicrobial cyclic peptides very recently reported by Ghadiri and coworkers (10). There is a trend toward higher activity for the short polymers and oligomers, with an optimal length of ≈8 repeat units in this nonexhaustive survey of activity vs. chain length. The inactivity of the longer polymer may arise from reduced solubility, lower molar concentration (for a given concentration in μg/ml), or inability to penetrate through the proteoglycan layer. Also, acetylation of the highly active polymer with n = 8 (Table 1) eliminates the antimicrobial activity, indicating that the positively charged aminoethyl groups are required for activity.
Table 1.
Antibacterial activities of polyamides
| Oligomer | n | R | MIC,
μg/ml§
|
||
|---|---|---|---|---|---|
| E. coli | K. pneumoniae‡ | B. subtilis¶ | |||
 |
2 | NH3+ | 19 | 66 | 12 |
| 3 | NH3+ | <19 | NA | 19 | |
| 8*† | NH3+ | 7.5–15 | 31–50 | 16 | |
| 60* | NH3+ | >200 | — | — | |
| 8*† | NH-Ac | >500 | 250 | >500 | |
The average chain length is determined by the Flory equation, and the polymeric nature of the products is confirmed by gel permeation chromatography with methods similar to those described earlier (11). Polydispersity index: 1.10 (4), 1.64 (5).
The end groups were N,N-dimethylethylenediamine amides obtained by using N,N-dimethylethylenediamine as a terminator.
Mueller–Hinton medium.
MICs were obtained by incubating 2-fold dilution series of polymers with the bacteria for 18 h at 37 °C and measuring cell growth at OD590.
LB medium.
To determine whether the oligomers were bactericidal or bacteriostatic, the E. coli were incubated with the polymer (n = 8) overnight and then plated in the absence of the polymer. The number of colonies was decreased by at least 100-fold relative to that expected from the initial value, indicating that the polymer was acting as a bactericidal agent. One of the hallmarks of the host defense peptides is their activity against a broad spectrum of bacteria (6, 15, 16). The most active polymer was tested against a panel of a number of pathogenic bacteria (Table 2). The MICs were found to be below 50 μg/ml for each of these strains.
Table 2.
Activity of 4 against six different bacterial strains
| Strain | MIC, μg/ml§ |
|---|---|
| E. coliK91 | 12–50¶ |
| E. coli D31* | 7.5–15‖ |
| K. pneumoniae 10 | 31–50‖ |
| Salmonella typhimuniumS5† | <3.75** |
| Pseudomonas aeruginosa 10 | 31–62‖ |
| Enterococcus faecium‡ | 15–25‡‡ |
Ampicillin- and streptomycin-resistant.
Tetracycline-resistant.
Gram-positive.
Minimal inhibitory concentration at 18 h.
Minimal medium.
Quarter strength Mueller–Hinton (MH) medium.
Half strength MH medium.
Quarter strength Bushnell–Hass medium.
To confirm that the polymers and oligomers are indeed able to interact with and disrupt phospholipid bilayers, we measured the ability of 2 to induce leakage of a dye, calcein, entrapped within the interiors of large unilamellar vesicles of mixed phosphatidylserine and phosphatidylcholine lipids (SOPS/SOPC 1:9). The extent of leakage of encapsulated calcein was detected by its fluorescence at 515 nm (Fig. 3). Oligomer 2 gave rise to calcein leakage in a dose-dependent manner. As was observed for antimicrobial peptides (9), the value of IC50 for this assay was approximately 10-fold lower than the MIC values for the E. coli.
Figure 3.

Calcein efflux induced by oligomer 2. Fraction of leakage was calculated from the fluorescence intensity at 515 nm, with 100% leakage calibrated by addition of 0.2% Triton X-100.
Conclusions
Here we describe a class of facially amphiphilic polymers, in which hydrophobic and hydrophilic groups segregate onto opposite sides of a low-energy, repeating conformation (secondary structure). Amphiphilic polymers have many applications in colloid and surface science but they are generally formed by random copolymerization of hydrophobic and hydrophilic monomers, or by combining a hydrophilic block with a hydrophobic block to provide a detergent-like structure. Indeed, many amphiphilic block polymers form micellar and bilayer phases. By contrast, the facially amphiphilic polymers described in this work should be able to mimic a variety of surface-active peptides and proteins. By varying the charge and hydrophobicity of the side chains one might be able to mimic the properties of toxins such as melittin, selective antimicrobials, or apolipoproteins, all of which have been successfully mimicked by using β-peptides of defined length and sequence (7–9, 13). Thus, although compounds 2–5 show hemolytic activity, we expect this feature can be eliminated in subsequent rounds of design.
By preparing both oligomers of defined length as well as the corresponding polymers, one can combine the advantages of studying both classes of compounds. Defined oligomers are homogeneous organic compounds that are useful for structural studies, thermodynamic analyses, and provide well defined structure-activity relationships. However, polymers are easy to prepare in large quantities and are the products of choice for applications in which the cost of production is an issue.
In summary, this study provides a one-pot synthetic strategy to produce polymers with a specified facially amphiphilic secondary structure and broad antibacterial activity. These polymers differ from earlier dendrimeric and cationic antimicrobial polymers (14, 17) in that they mimic the essential features of a large class of antimicrobial peptides and proteins. By systematically varying the side chains and possibly also the polymeric backbone it should be possible to fine-tune the antimicrobial selectivity and toxicity of the polymers in a manner analogous to antimicrobial peptides and β-oligomers (7, 9).
Acknowledgments
We gratefully acknowledge stimulating discussions with Simone Raugei and Sanjoy Bandyopadhyay. The computations were supported in part by the National Science Foundation NPACI program, and the experimental work was funded by National Science Foundation Award no. 9905566 (to W.F.D.).
Abbreviations
- DIEA
diisopropylethylamine
- DMF
dimethylformamide
- TFA
trifluoroacetic acid
- MD
molecular dynamics
- MIC
minimal inhibitory concentration
Footnotes
cpmd/hcth-isolated molecule calculations were performed in a 10 Å × 12 Å × 12 Å box with valence electron wave functions expanded in plane waves with a 70-Rydberg cutoff and Trouiller–Martins normconserving pseudopotentials.
References
- 1.Zasloff M. Nature (London) 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 2.Zasloff M. Curr Opin Immunol. 1992;4:3–7. doi: 10.1016/0952-7915(92)90115-u. [DOI] [PubMed] [Google Scholar]
- 3.Boman H G. Immunol Rev. 2000;173:5–16. doi: 10.1034/j.1600-065x.2000.917301.x. [DOI] [PubMed] [Google Scholar]
- 4.Hancock R E, Lehrer R. Trends Biotechnol. 1998;16:82–88. doi: 10.1016/s0167-7799(97)01156-6. [DOI] [PubMed] [Google Scholar]
- 5.DeGrado W F. Adv Protein Chem. 1988;39:51–124. doi: 10.1016/s0065-3233(08)60375-7. [DOI] [PubMed] [Google Scholar]
- 6.Tossi A, Sandri L, Giangaspero A. Biopolymers. 2000;55:4–30. doi: 10.1002/1097-0282(2000)55:1<4::AID-BIP30>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 7.Hamuro Y, Schneider J P, DeGrado W F. J Am Chem Soc. 1999;121:12200–12201. [Google Scholar]
- 8.Porter E A, Wang X, Lee H S, Weisblum B, Gellman S H. Nature (London) 2000;404:565. doi: 10.1038/35007145. [DOI] [PubMed] [Google Scholar]
- 9.Liu D, DeGrado W. J Am Chem Soc. 2001;123:7553–7559. doi: 10.1021/ja0107475. [DOI] [PubMed] [Google Scholar]
- 10.Fernandez-Lopez S, Kim H S, Choi E C, Delgado M, Granja J R, Khasanov A, Kraehenbuehl K, Long G, Weinberger D A, Wilcoxen K M, Ghadiri M R. Nature (London) 2001;412:452–455. doi: 10.1038/35086601. [DOI] [PubMed] [Google Scholar]
- 11.Gellman S H. Acc Chem Res. 1998;31:173–180. [Google Scholar]
- 12.Seebach D, Ciceri P E, Overhand M, Jaun B, Rigo D. Helv Chim Acta. 1996;79:2043–2066. [Google Scholar]
- 13.Werder M, Hauser H, Abele S, Seebach D. Helv Chim Acta. 1999;82:1774–1783. [Google Scholar]
- 14.Tiller J C, Liao C J, Lewis K, Klibanov A M. Proc Natl Acad Sci USA. 2001;98:5981–5985. doi: 10.1073/pnas.111143098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simmaco M, Mignogna G, Barra D. Biopolymers. 1999;47:435–451. doi: 10.1002/(SICI)1097-0282(1998)47:6<435::AID-BIP3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 16.Andreu D, Rivas L. Biopolymers. 1998;47:415–433. doi: 10.1002/(SICI)1097-0282(1998)47:6<415::AID-BIP2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 17.Chen C, Beck-Tan N, Dhurjati P, van Dyk T, LaRossa R, Cooper S. Biomacromolecules. 2000;1:473–480. doi: 10.1021/bm0055495. [DOI] [PubMed] [Google Scholar]
- 18.MacKerell Jr A D. In: Computational Biochemistry. Beker O M, MacKerell A D Jr, Roux B, Watanabe M, editors. New York: Dekker; 2001. pp. 7–38. [Google Scholar]
- 19.Chen B, Potoff J J, Siepmann J I. J Phys Chem. 2001;B:105. , 3093–3104. [Google Scholar]
- 20.Jorgensen W L. In: Encyclopedia of Computational Chemistry. Schleyer P V R, editor. New York: Wiley; 1998. pp. 1986–1989. [Google Scholar]
- 21.Hutter J, Alavi A, Deutsch T, Bernsconi M, Goedecker S, Marx D, Tuckerman M, Parrinello M. cpmd [MPI (Stuttgart) and IBM Research Laboratory (Zurich)] 1999. [Google Scholar]
- 22.Hamprecht F A, Cohen A J, Tozer D J, Handy N C. J Chem Phys. 1998;109:6264–6271. [Google Scholar]
- 23.Siepmann J I, Frenkel D. Mol Phys. 1992;75:59–70. [Google Scholar]
- 24.Martin M G, Siepmann J I. J Phys Chem. 1999;B:103. , 4508–4517. [Google Scholar]