

Abstract
Design, synthesis, and study of a synthetic barrel-stave supramolecule with p-octiphenyl “staves,” β-sheet “hoops,” and hydrophobic exterior as well as internal carboxylate clusters are reported. Ion transport experiments indicate the formation of transmembrane pores at 5 < pH < 7 with nanomolar activity. Blockage of dye efflux from spherical bilayers by external Mg(OAc)2 and internal 8-aminonaphthalene-1,3,6-trisulfonate is suggestive for weakly cooperative (n = 1.16) formation of aspartate-Mg2+-8-aminonaphthalene-1,3,6-trisulfonate complexes within the barrel-stave supramolecule (KD = 2.9 mM). Corroborative evidence from structural studies by circular dichroism spectroscopy is provided and discussed with emphasis of the importance of internal charge repulsion for pore formation and future applications toward binding and catalysis within supramolecular synthetic pores.
The perspective of vectorial control over chemical processes that take place within transmembrane pores is captivating. The development of design strategies for the construction of easily variable active sites at the inner surface of transmembrane pores is the first step toward such organic chemistry within confined anisotropic space. In sharp contrast to breathtaking progress being made with biotechnologically modified natural pores (1), reliable and general design strategies for synthetic ion channels and pores with easily variable internal active sites do not exist (2–15). Reasonably straightforward covalent synthesis of barrel-like macromolecules with the required ≈3.5-nm height and variably functionalizable interior of ≥1.0 nm diameter is not (yet) possible. The more straightforward noncovalent syntheses (16–20) applied to barrel-like supramolecules with ion channel activity is, with one possible exception (15), limited by apparent difficulties to position functional groups at internal concave surfaces (2–15). However, we have recently found that this trend toward “peripheral crowding” in supramolecular synthesis (16–20) can be bypassed in artificial β-barrels with p-octiphenyl “staves” and have used their rationally designed hydrophobic, ionophoric, and cationic interior to encapsulate guests with complementary characteristics in water and bilayer membranes (21, 22). Here we report design, synthesis, and selected characteristics of p-octiphenyl β-barrels with anionic interior formed by multiple aspartate residues (i.e., 14, Fig. 1). We further show that anionic p-octiphenyl β-barrels 14 form doubly pH-gated pores in bilayer membranes and bind inorganic cations and organic anions.
Figure 1.

Design and synthesis of barrel-stave supramolecule 14. β-Strands are depicted as arrows pointing to the C terminus in the schematic side-view cutaway structure of 14 and solid (backbone) and dotted lines (hydrogen bonds) in the axial views of 14. α-Hydroxy and α-amino acid residues (one-letter abbreviation, G = -OCH2CO-) pointing toward the barrel exterior are depicted black on white, internal residues white on black. Axial views roughly reproduce dimension of molecular models; pertinent distances are indicated. Structures on gray: Commercial starting materials 2 a–b, 3 a–c. (a) p-Octiphenyl synthesis in nine steps (4.9% overall yield), see refs. 23–25. (b) Pentapeptide synthesis in eight steps (50.4% overall yield), see supporting information. (c) O-Benzotriazolyl-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU), triethylamine (TEA), 40%. (d) Trifluoroacetic acid (TFA), quantitative. (e) Self-assembly (see text and Fig. 10). The indicated 50% protonation of internal aspartates in 14 is an approximate assumption (see text).
Materials and Methods
General experimental procedures have been described in refs. 23–25.
Synthesis.
p-Octiphenyl 2 was synthesized from 2a and 2b following the original protocol (24) with minor changes published (23, 25) or to be published elsewhere. Details on the synthesis of 3 and 1 are published as supporting information on the PNAS web site, www.pnas.org.
Dye Leakage Experiments.
Egg yolk phosphatidylcholine-small unilamellar vesicles (EYPC-SUVs, 68 ± 4 nm) were prepared with a dialytic detergent removal technique by using a Mini Lipoprep (Sialomed, Columbia, MD) (26). A solution of EYPC (50 mg in 50 μl of EtOH; Avanti Polar Lipids) and sodium cholate (22.4 mg; Sigma) in 950 μl of buffer A was dialyzed against 50 ml of buffer A [>6 h, 5 mM N-[Tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid (TES) (Sigma)/12.5 mM 8-aminonaphtalene-1,3,6-trisulfonate (ANTS)/45.0 mM p-xylenebis(pyridinium)bromide (DPX) (Molecular Probes)/20 mM KCl, pH 7.0 (27)] and 1,000 ml of buffer B [>12 h, 5 mM TES/100 mM KCl, pH 7.0 (27)], purified by gel filtration (Sephadex G-50, buffer B), and diluted to 6 ml. ANTS/DPX-efflux was recorded as a function of time by measuring changes in emission intensity It [λem = 510 nm, λex = 353 nm (27, 28), FluoroMax-2; Jobin-Yvon-Spex, Longjumeau, France] after addition of these EYPC-SUVs (10 mM EYPC, 50 μl) to stirred thermostated buffer C (1.95 ml/10 mM Mes/100 mM KCl, pH 4.5–7.0) to give emission intensity I0. Concentrated stock solutions of 1 (in 20–40 μl of MeOH, final monomer concentrations 0 nM–10 μM), Mg(OAc)2 (in 20–40 μl of buffer C, final concentrations 0–50 mM), and second populations of EYPC-SUVs (10 mM EYPC, 50 μl) were added after defined periods of times and in defined sequences to give the specified concentrations indicated in the legends of all figures. Lysis (100%) was determined with Triton X-100 (10% aqueous, 50 μl) to give emission intensity I∞. Multichannel controls [λem = 510 nm, λex (1) = 360 nm, λex (2) = 340 nm, λex (3) = 320 nm] were run during each experiment to eliminate potential (but not observed) contributions from effects other than dye efflux. Flux curves were normalized to percent leakage ILt by using Eq. 1:
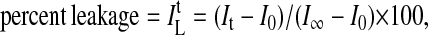 |
1 |
using It, I0, and I∞ as defined above (26). In case of a second addition of ANTS/DPX-loaded EYPC-SUVs equivalent to the initial concentration, the fractional percent leakage ILt/2 (%) is recorded and reported. Dependence on pH, concentration, and external blockers was determined by plotting the percentage leakage ILt (%) after a meaningful period of time (usually 250 sec), i.e., IL (%), as a function of the parameter of interest. Controls showed that plot of initial velocities or initial rate constants as a function of the parameter of interest gave roughly identical results. Percent blockage was calculated from Eq. 2
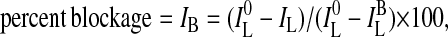 |
2 |
where IL0 is the percent leakage IL from Eq. 1 without blocker, and ILB is the percent leakage IL observed without pore. The fractional saturation Y was derived from Eq. 3
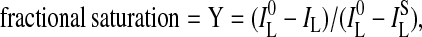 |
3 |
where IL0 is the percent leakage IL from Eq. 1 without blocker and ILS is the percent leakage IL at saturation with blocker. Hill plots for dose-response isotherms were calculated by using Eq. 4
 |
4 |
where n is the Hill coefficient (indicative for positive cooperativity if n > 1), and KD is the apparent dissociation constant in M (29).
Circular Dichroism (CD) and UV-Vis Spectroscopy.
EYPC-SUVs without entrapped ANTS/DPX were prepared as described above by using buffer C for all procedures. UV-Vis spectra were recorded on a Varian Cary 1 Bio spectrophotometer after addition of these EYPC-SUVs (10 mM EYPC, 25 μl), 1 [in 20–40 μl MeOH, ɛ319 = 28.6 mM−1cm−1 (26), final monomer concentrations 5.0 μM if not otherwise indicated], Mg(OAc)2 (in 20–40 μl of buffer C, final concentrations 0–20 mM), and ANTS [in 20–40 μl of buffer C, ɛ352 = 7.2 mM−1cm−1 (28), final concentrations 0–20 mM] to stirred thermostated buffer C (1.00 ml/10 mM Mes/100 mM KCl, pH 4.5–7.0). Samples for CD measurements were adjusted to A < 1.0 for the maxima of interest by using either cells with a pathlength of 1.0 (usually for 1) or 0.1 cm (usually for ANTS) as appropriate. CD spectra were recorded on a Jasco (Easton, MD) 710 spectropolarimeter. Δɛ values were calculated for monomeric p-oligophenyl and ANTS chromophores.
Results
The “negative design” of rigid-rod β-barrels is based on covalent p-oligophenyl “staves” (21, 22, 30). Preorganization of cooperative barrel self-assembly by both their arene–arene torsion angles ω ≠ 0° and their minimized axial molar entropy seem equally important to “bend” otherwise planar β-sheets and to maximize energy gains from β-sheet formation, respectively (21, 31).
The “positive design” of rigid-rod β-barrels is based on supramolecular β-sheet “hoops.” According to molecular models of rigid-rod β-barrels (25), p-octiphenyl 1 with eight lateral pentapeptides with a Gla-N terminus of the sequence -Gla-Leu-Asp-Leu-Asp-Leu-NH2 (i.e., -GLDLDL-NH2) is likely to self-assemble into a tetrameric barrel 14 with all leucine residues at the outer surface and all aspartate residues pointing toward the central channel (Gla, G, -OCH2CO-; H-G-OH, glycolic acid). The planar rectangles of ≈5 Å × ≈7 Å formed by four neighboring aspartates are considered as minimal internal active sites and named “D-quartets.”
Barrel-stave supramolecule 14 (molecular weight = 23.0 kDa) was prepared in overall 20 synthetic steps from five commercially available organics of ≤0.4 kDa, namely Fast Blue B Salt 2a (available from Fluka for 0.684 SFr/g) and α-hydroxy and α-amino acid derivatives 2b and 3 a–c (Fig. 1). The procedures relevant for the synthesis of p-octiphenyl-(G-OH)8 2 from biphenyl 2a have been published (24). More recent, minor improvements up to overall 4.7% yield have been (23, 25) and will be reported elsewhere. Pentapeptide 3 was prepared from 3 a–c by routine solution-phase peptide synthesis. Coupling of p-octiphenyl-(G-OH)8 2 and pentapeptide 3 followed by aspartate deprotection gave the desired p-octiphenyl-(GLDLDL-NH2)8 rod 1.
The capacity of p-octiphenyl-(GLDLDL-NH2)8 rod 1 to self-assemble into membrane-spanning pores 14 was assessed with the ANTS/DPX-leakage assay (27, 28). For this purpose, SUVs composed of EYPC were loaded with the anionic fluorophore ANTS and the cationic quencher DPX. Addition of nanomolar concentrations of p-octiphenyl-(GLDLDL-NH2)8 1 to ANTS/DPX-entrapped EYPC-SUVs caused the increase in ANTS-fluorescence expected for rapid efflux of either fluorophore ANTS or (and) quencher DPX in presence of membrane-spanning pores 14 (Fig. 2). The activity of pore 14 depended strongly on pH (Figs. 2 and 3A). The remarkably narrow pH profile was consistent with minimal activity for full protonation (pH < 5) and full deprotonation (pH > 7) as well as maximal activity at partial protonation of the internal aspartates of pore 14.
Figure 2.

Relative emission intensity
I of ANTS at
λem = 510 nm (λex = 353 nm) as a
function of time during addition of p-octiphenyl
1 (t ≈ 60 sec, final c = 50 nM) and
detergent (t ≈ 300 sec) to 50 μl of
ANTS/DPX-entrapped EYPC-SUVs (10 mM EYPC) in 1950 μl of buffer (10
mM Mes/100 mM NaCl) at pH 5.0, 6.0, and 7.0 with schematic indication
of possible processes [i.e., (a) ANTS/DPX-entrapped
EYPC-SUVs; (b) leakage through pore
14; (c) complete ANTS/DPX
“release” by lysis of EYPC-SUVs].
of ANTS at
λem = 510 nm (λex = 353 nm) as a
function of time during addition of p-octiphenyl
1 (t ≈ 60 sec, final c = 50 nM) and
detergent (t ≈ 300 sec) to 50 μl of
ANTS/DPX-entrapped EYPC-SUVs (10 mM EYPC) in 1950 μl of buffer (10
mM Mes/100 mM NaCl) at pH 5.0, 6.0, and 7.0 with schematic indication
of possible processes [i.e., (a) ANTS/DPX-entrapped
EYPC-SUVs; (b) leakage through pore
14; (c) complete ANTS/DPX
“release” by lysis of EYPC-SUVs].
Figure 3.

pH dependence of activity (A) and (supra)structure (B) of pore 14. (A) Percent ANTS/DPX leakage IL from EYPC-SUVs mediated by 50 nM p-octiphenyl 1 as a function of pH (exemplified in Fig. 2). (B) Changes of the first Cotton effect in the CD spectrum of p-octiphenyl 1 in presence (●) and absence (○) of EYPC-SUVs as a function of pH (exemplified in Fig. 4).
The CD spectra of p-octiphenyl-(GLDLDL-NH2)8 1 in water at pH = 5.0 (dashed line), 6.0 (solid line), and 7.0 (dotted line) reveal decreasing magnitude for the first three Cotton effects with increasing pH (Fig. 4A). In the presence of bilayer membranes, the CD spectrum at pH = 6.0 (Fig. 4B, solid) reveals a strong negative first Cotton (Δɛ307 = −25.6 M−1cm−1) similar to that in membrane-free acidic water (Fig. 4A, dashed line, Δɛ303 ≥ −21.0 M−1cm−1). A dominant positive second Cotton (Δɛ274 = +4.7 M−1cm−1) was observed in presence of EYPC-SUVs at pH = 5 (Fig. 4B, dashed line). The differences of the CD spectra at pH ≤ 5 suggest that favorable incorporation of “preformed” barrel 14 into the bilayer at “full” protonation of the internal aspartates is followed by substantial (supra)structural change, whereas binding of partially protonated barrels to bilayer membranes at 5 ≤ pH ≤ 6 causes little (supra)structural change. The CD spectra at pH ≥ 7, presumably originating from highly charged, aqueous, or interfacial monomers 1, were clearly less pronounced in presence and absence of spherical lipid bilayers (see Discussion).
Figure 4.

CD spectra of p-octiphenyl 1 in absence (A) and presence (B) of EYPC-SUVs in buffer (10 mM Mes/100 mM NaCl) at pH 5.0 (dashed line), 6.0 (solid line), and 7.0 (dotted line), and with 20 mM Mg(OAc)2 at pH 6.0 (○). Cell, l = 1.0 cm.
The first Cotton at ≈305 nm, originating from L transitions of the chiral p-octiphenyl chromophore, was taken as relevant measure to extract the pH profiles for (supra)structural changes of p-octiphenyl-(GLDLDL-NH2)8 1. In the absence of bilayer membranes, the pH profile revealed reversible transitions between two distinct chiral conformers/self-assemblies centered at pH = 6.0 (Fig. 3B, ○). The pH profile in the presence of EYPC-SUVs revealed maximal magnitude of the first Cotton at pH = 6.0 (Fig. 3B, ●). The bell-shaped pH profiles for structure (Fig. 3B, ●) and activity (Fig. 3A) of pore 14 were nearly identical and clearly different from the sigmoidal pH profile for structure in water (Fig. 3B, ○).
The activity of pore 14 depended linearly on the concentration of monomeric p-octiphenyl-(GLDLDL-NH2)8 (Fig. 5). Detectable activity down to ≈25 nM monomer concentration (i.e., ≈6 nM 14) identified pore 14 as the most potent pore constructed so far with rigid-rod molecules (21, 22). Inactivity at monomer concentrations of ≈15 nM rather than 0 nM obtained from linear curve fit indicated biphasic behavior (Fig. 5, dotted lines). The first four data points at pH = 6 followed a fourth-order dependence reasonably well (Fig. 5, solid lines). This biphasic behavior was indicative for a critical assembly concentration (CAC) ≈ 30 nM of pore 14 in water and different pore formation mechanisms above and below the CAC, and in weak support of a tetrameric suprastructure (refs. 2 and 31; see Discussion).
Figure 5.

Dependence of ANTS/DPX leakage IL on the concentration of monomer 1 at pH 5.3 (□), 6.0 (●), and 7.0 (■) with curve fits for linear (dotted lines) and fourth-order dependence (solid lines).
Intervesicular pore transfer was assessed by addition of ANTS/DPX-entrapped EYPC-SUVs to ANTS/DPX-released EYPC-SUVs containing pore 14 (Fig. 6). About a two-fold reduction of activity was found for second compared with first SUV population (Fig. 6, solid lines). A 50% reduced activity indicated 50% (i.e., quantitative) intravesicular transfer of pore 14 from first to second SUV population, supporting complete incorporation of pore 14 into the first SUV population as suggested by CD spectroscopy (Fig. 4).
Figure 6.

Relative emission intensity
I /2 of ANTS
at λem = 510 nm (λex = 353 nm) as
a function of time during addition of p-octiphenyl
1 (t ≈ 60 sec, final c = 100 nM),
Mg(OAc)2 [t ≈ 375 sec, final
c = 0 mM (solid line), and, with decreasing activity between 450
and 900 sec, 5 mM (dotted line), 10 mM (dashed line), and 20 mM (dotted
line)], 50 μl of ANTS/DPX-entrapped EYPC-SUVs (10 mM EYPC,
t ≈ 400 sec), and detergent (t ≈
900 sec) to 50 μl of ANTS/DPX-entrapped EYPC-SUVs (10 mM EYPC) in
1950 μl of buffer (10 mM Mes/100 mM NaCl, pH 6.0) with schematic
indication of possible processes [(i.e., a)
ANTS/DPX-entrapped EYPC-SUVs; (b) leakage through pore
14; (c) binding of
Mg2+ within 14;
(d) addition of new ANTS/DPX-entrapped EYPC-SUVs;
(e) intervesicular transfer of
14 with/without
Mg2+; (f) leakage
through pore 14 in new EYPC-SUVs; and
(g) complete ANTS/DPX “release” by lysis of
EYPC-SUVs].
/2 of ANTS
at λem = 510 nm (λex = 353 nm) as
a function of time during addition of p-octiphenyl
1 (t ≈ 60 sec, final c = 100 nM),
Mg(OAc)2 [t ≈ 375 sec, final
c = 0 mM (solid line), and, with decreasing activity between 450
and 900 sec, 5 mM (dotted line), 10 mM (dashed line), and 20 mM (dotted
line)], 50 μl of ANTS/DPX-entrapped EYPC-SUVs (10 mM EYPC,
t ≈ 400 sec), and detergent (t ≈
900 sec) to 50 μl of ANTS/DPX-entrapped EYPC-SUVs (10 mM EYPC) in
1950 μl of buffer (10 mM Mes/100 mM NaCl, pH 6.0) with schematic
indication of possible processes [(i.e., a)
ANTS/DPX-entrapped EYPC-SUVs; (b) leakage through pore
14; (c) binding of
Mg2+ within 14;
(d) addition of new ANTS/DPX-entrapped EYPC-SUVs;
(e) intervesicular transfer of
14 with/without
Mg2+; (f) leakage
through pore 14 in new EYPC-SUVs; and
(g) complete ANTS/DPX “release” by lysis of
EYPC-SUVs].
The presence of Mg2+ reduced the apparent activity of pore 14. Extravesicular addition of 20 mM Mg(OAc)2 before (Fig. 7, dotted line), during (Fig. 7, solid line), and after (Fig. 6, dotted and dashed lines) efflux of 12.5 mM ANTS and/or 45 mM DPX from EYPC-SUVs caused similar changes. The extent of blockage in first (Fig. 7) and second (Fig. 6) SUV population was nearly identical. The concentration dependence of Mg2+ blockage exhibited saturation at 77% (Fig. 8). Hill plot of the binding isotherm gave an apparent KD = 2.9 mM (Fig. 8A). A Hill coefficient n = 1.16 can be interpreted as cooperative and/or higher-order complex formation (29). The curved Scatchard plot (Fig. 8B) was similarly implicative for the presence of higher-order complexes (29).
Figure 7.

Relative emission intensity
I of ANTS at
λem = 510 nm (λex = 353 nm) as a
function of time in presence (dotted/solid lines) and absence
(○) of Mg(OAc)2 (final c = 20 mM) added to
ANTS/DPX-entrapped EYPC-SUVs in 1950 μl of buffer (10 mM Mes/100
mM NaCl, pH 6.0) either before (t ≈ 40 sec,
dotted line) or after (t ≈ 100 sec, solid line)
p-octiphenyl 1 (t ≈ 60 sec,
final c = 50 nM) at pH 6.0, followed by final lysis
(t ≈ 300 sec).
of ANTS at
λem = 510 nm (λex = 353 nm) as a
function of time in presence (dotted/solid lines) and absence
(○) of Mg(OAc)2 (final c = 20 mM) added to
ANTS/DPX-entrapped EYPC-SUVs in 1950 μl of buffer (10 mM Mes/100
mM NaCl, pH 6.0) either before (t ≈ 40 sec,
dotted line) or after (t ≈ 100 sec, solid line)
p-octiphenyl 1 (t ≈ 60 sec,
final c = 50 nM) at pH 6.0, followed by final lysis
(t ≈ 300 sec).
Figure 8.

Percent blockage IB of ANTS/DPX leakage mediated by 14 as a function of Mg(OAc)2 concentration. (A) Hill plot with linear curve fit (KD = 2.9 mM, n = 1.16, r = 0.996). (B) Scatchard plot with linear curve fit (r = 0.956).
The presence of 20 mM Mg(OAc)2 had no effect on the first CD Cotton effect at 307 nm of membrane-bound pore 14 at pH 6.0 (Fig. 4B, solid line vs. ○). This observation demonstrated that Mg2+ blockage is not caused by Mg2+-induced suprastructural changes of pore 14. (Note that the significance of the third Cotton at 240 nm including the apparent increase after addition of Mg2+ is questionable because of the onset of a too high pore absorption around this wavelength.)
In the presence of membrane-bound Mg2+-loaded pore 14, an induced CD (ICD) Cotton effect for the low-energy transition of ANTS around 370 nm was clearly detectable at c ≥ 10 mM (Fig. 9, solid line and ●). The Cotton effects seen for the high-energy ANTS transitions below 300 nm are near cut-off and thus of questionable significance. The CD Cotton effects of p-octiphenyl-(GLDLDL-NH2)8 1 are below detection limit for c < 10 μM when measured in the CD cells with l = 1.0 mm (rather than l = 1.0 cm as in Fig. 4). The millimolar ANTS concentrations needed to observe an ICD corresponded qualitatively to the internal ANTS concentration needed to observe blockage of pore 14. It was, however, not possible to reach saturation under experimentally accessible conditions.
Figure 9.

CD spectra of 0 (dashed line), 5 (dotted line), 10 (solid line), and 20 mM ANTS (●/○) in presence of p-octiphenyl 1, EYPC-SUVs, and 0 (○) and 20 mM Mg(OAc)2 (dashed, dotted, and solid lines/●). Cell, l = 0.1 cm.
The induced CD Cotton effect for ANTS was observed with transmembrane pore 14 in the presence (●) but not in the absence (○) of 20 mM Mg(OAc)2 (Fig. 9). This ability of Mg2+ to direct ANTS into the chiral environment provided by pore 14 (Fig. 9, ● vs. ○), together with its inability to change the suprastructure of pore 14 (Fig. 4B, solid vs. ○), indicated that binding of extravesicular Mg2+ and intravesicular ANTS to internal D-quartets accounted for Mg2+ blockage of intact pores 14.
Discussion
Although overinterpretation of experimental data must, in general, be avoided, we believe that discussion of possible pore formation and blockage mechanisms may be helpful for readers with regard to (i) future design strategies of supramolecular synthetic pores with variable internal active sites and (ii) the potential for molecular recognition and catalysis within pore 14.
The usefulness of internal charge repulsion (ICR) to create large, stable, and functionalized space within transmembrane rigid-rod β-barrels first has been proposed and confirmed as design strategy for giant ion channels with cationic interior (32). The present study on rigid-rod β-barrels with anionic interior reveals remarkable scope and relevance of the ICR model. According to the ICR model, the precise pH-gating of suprastructure (Fig. 3B, ●) and function (Fig. 3A, ●) of open pore Po formed by p-octiphenyl β-barrel 14 are direct consequences of insufficient and excessive ICR between fully protonated and fully deprotonated internal aspartates at pH ≤ 5.0 and pH ≥ 7.0, respectively (Fig. 10). Consistent with previous findings on self-assembly as well as programmed assembly (24, 31), this interpretation identifies a negative first Cotton effect at 300–330 nm as dominant CD characteristic for intact rigid-rod β-barrels.
Figure 10.

Putative mechanism for regulation of pore 14 by pH and concentration (Po, open pore with properties consistent with the transmembrane barrel-stave suprastructure 14 in Fig. 1; Pc, closed pore of unknown structure with contracted trimeric and “imploded” tetrameric barrels as possibilities).
According to the ICR model, inactivation of Po at pH ≥ 7.0 occurs by “barrel explosion” after internal overcharging into polyanionic, presumably aqueous monomers p-octiphenyl-(GLDLDL-NH2)8 1 with a less distinct CD spectrum. Inactivation at pH ≤ 5.0 would then originate from complementary “barrel implosion” into ultimately neutral closed pores Pc (Fig. 10). “Imploded tetramers” and “contracted trimers” can be imagined for the unknown suprastructure of Pc, characterized by a dominant second positive CD Cotton effect at 274 nm. The clearly different CD spectra at pH ≤ 5.0 in water and membranes would then suggest that barrel implosion with insufficient ICR occurs only after pressure from a surrounding liquid crystalline phase.
These findings forced us to accept that rigid-rod β-barrels 14 with hydrophobic sides may self-assemble in water as well as at pH ≤ ≈6.0. This counterintuitive conclusion may become more convincing considering the poor solubility [c (1) ≈ 3 μM] compared with “hydrophilic” barrels (25, 31) and even 2-methylpropane (c = 850 μM) in water (33), and the observation of aqueous “hydrophobic” barrels designed to have cationic interior by atomic force microscopy (34). Although the more appealing, alternative “inside-out” β-barrels with external aspartates can be excluded for steric reasons (21), we failed so far to secure more direct structural evidence for putative “hydrophobic” pores 14 in water because of their poor nanomolar solubility.
According to the biphasic dependence of active pore Po on the concentration of monomer p-octiphenyl-(GLDLDL-NH2)8 1, the CAC of rigid-rod β-barrels 14 in water is around 30 nM. The concentration dependence of ion transport by supramolecular pores 1n is given by Eq. 5,
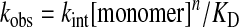 |
5 |
where the observed rate constant kobs depends on an intrinsic rate constant kint as well as KD and assembly number n of supramolecule 1n (2, 32). According to Eq. 5, n ≈ 4 at c < CAC indicates thermodynamically unfavorable pore formation and supports weakly that four monomers 1 are needed to form an active pore 14. Linear concentration dependence above CAC corroborated the thermodynamically favored pore formation from preformed tetramers 14 of comparable suprastructure in water that was directly observed by CD spectroscopy (Figs. 3 and 4).
The proposed pore formation mechanism summarizing the above discussion is given in Fig. 10. A putative pore blockage mechanism is given in Fig. 11. The proposed formation of an inclusion complex 14 ⊃ (ANTSn ⋅ Mg2+n′)n′′ with apparent KD = 2.9 mM is based on up to 77% blockage of pore 14 in presence of intravesicular ANTS and extravesicular Mg2+ (Figs. 6–9).
Figure 11.

Putative general mechanism for regulation of pore 14 by inorganic cations (exemplified by Mg2+) and organic anions (exemplified by ANTS). The stoichiometry of the proposed inclusion complex, e.g., 14 ⊃ (ANTSn ⋅ Mg2+n)n′′, is chosen arbitrarily to illustrate the concept.
An alternative explanation for Mg2+ blockage of 14 based on changes in ion selectivity of pore 14 rather than inclusion complex formation was considered. Because the activity of the polyanionic pore 14 in the APTS/DPX assay is likely to originate from efflux of the cationic quencher DPX, increasing anion selectivity of pore 14 ⊃ Mg2+n after Mg2+ binding within the transmembrane channel is likely to hinder DPX efflux. This alternative mechanism for Mg2+ blockage is, however, inconsistent with unfavorable ANTS efflux through a cationic “metallopore” 14 ⊃ Mg2+n. Preliminary results confirmed that Mg2+n ⊃ 14 can mediate efflux of the bulkier organic anion 5(6)-carboxyfluorescein. Although additional contributions from reduced DPX efflux possibly contribute to the experimental outcome, we thus concluded that formation of an inclusion complex 14 ⊃ (ANTSn ⋅ Mg2+n′)n′′ accounts chiefly for blockage of pore 14. Experimental support for inclusion complex 14 ⊃ (ANTSn ⋅ Mg2+n′)n′′ formation was secured from structural studies, indicating that (i) binding of Mg2+ does not affect the suprastructure of barrel 14 (Fig. 4B), (ii) ANTS does not bind to barrel 14 without Mg2+ (Fig. 9, ○), and, most importantly, (iii) ANTS does bind to barrel 14 in the presence of Mg2+ (Fig. 9, ●).
Conclusion
In summary, we report that novel p-octiphenyl β-barrels designed to contain a central channel decorated with precisely placed aspartate residues, i.e., D-quartets, mediate efflux of organic ions from spherical bilayer membranes at low nanomolar concentrations in a controllable manner. Biphasic concentration dependence indicates a CAC ≈ 30 nM in water, thermodynamically favored pore formation from these preformed aqueous β-barrels of presumably similar but not experimentally confirmed suprastructure above the CAC, and likely formation of tetrameric supramolecules.
Significant regulatory mechanisms without precedence for synthetic ion channels and pores formed by barrel-stave supramolecules are consistent with functional internal D-quartets: Exceptionally narrow, doubly pH-gated activity (5 < pH < 7) and blockage by Mg2+–ANTS complexes (KD = 2.9 mM). This doubly pH-gated Mg2+-sensitive activity at 5 < pH < 7 of barrel-stave supramolecule 14 with internal aspartates is in noteworthy contrast to Szoka and colleagues' (35) de novo “α-barrels” with internal glutamates and hydrophobic exterior active at pH < 5. Doubly pH-gated pore formation of 14 is explained with the internal charge repulsion model, i.e., pore “implosion” and “explosion” after insufficient and excessive internal charge, respectively. The latter characteristic suggests usefulness of synthetic pores with internal carboxylate clusters to bind (and eventually convert) organic anions by means of inorganic cations (Fig. 11). Preliminary results on pore blockage by matching organic (poly)anions with minimal outer diameter < ≈1.2 nm are in support of these expectations as well as the inner diameter ≈1.2 nm anticipated for tetrameric suprastructure 14. The central role of D-clusters in biological processes as diverse as biomineralization (36) and terpenoid biosynthesis (37), together with perspectives to possibly perform such chemical processes within confined and anisotropic transmembrane space, indicates intriguing and diverse future applicability of the p-octiphenyl β-barrels with internal D-quartets described here.
Supplementary Material
Acknowledgments
We thank D. Gerard for assistance in synthesis; N. Sakai for experimental assistance and advice; V. Cardona and R. E. Offord (Centre Médical Universitaire, Geneva) for matrix-assisted laser desorption ionization–time-of-flight measurements; A. Pinto, J.-P. Saulnier, the group of F. Gülaçar, and H. Eder for NMR, MS, and elemental analyses, respectively; and the Swiss National Science Foundation (21-57059.99 and National Research Program “Supramolecular Functional Materials” 4047-057496) for financial support.
Abbreviations
- ANTS
8-aminonaphtalene-1,3,6-trisulfonate
- CD
circular dichroism
- DPX
p-xylenebis(pyridinium)bromide
- EYPC
egg yolk phosphatidylcholine
- ICR
internal charge repulsion
- SUV
small unilamellar vesicle
- CAC
critical assembly concentration
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Bayley H, Cremer P S. Nature (London) 2001;413:226–230. doi: 10.1038/35093038. [DOI] [PubMed] [Google Scholar]
- 2.Bandyopadhyay P, Janout V, Zhang L-H, Regen S L. J Am Chem Soc. 2001;123:7691–7696. doi: 10.1021/ja010926m. [DOI] [PubMed] [Google Scholar]
- 3.Wright A J, Matthews S E, Fischer W B, Beer P D. Chem Eur J. 2001;7:3474–3481. doi: 10.1002/1521-3765(20010817)7:16<3474::aid-chem3474>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Yoshino N, Satake A, Kobuke Y. Angew Chem Int Ed Engl. 2001;40:457–459. [PubMed] [Google Scholar]
- 5.Waser P, Rueping M, Seebach D, Duchardt E, Schwalbe H. Helv Chim Acta. 2001;84:1821–1845. [Google Scholar]
- 6.Bong D T, Clark T D, Granja J R, Ghadiri M R. Angew Chem Int Ed Engl. 2001;40:988–1001. [PubMed] [Google Scholar]
- 7.Gokel G W. Chem Eur J. 2001;7:33–39. doi: 10.1002/1521-3765(20010105)7:1<33::aid-chem33>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 8.Fyles T M, Hu C, Knoy R. Org Lett. 2001;3:1335–1337. doi: 10.1021/ol015713g. [DOI] [PubMed] [Google Scholar]
- 9.Biron E, Voyer N, Meillon J C, Cormier M-E, Auger M. Biopolymers. 2001;55:364–372. doi: 10.1002/1097-0282(2000)55:5<364::AID-BIP1010>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 10.Arndt H D, Knoll A, Koert U. Angew Chem Int Ed Engl. 2001;40:2076–2078. doi: 10.1002/1521-3773(20010601)40:11<2076::AID-ANIE2076>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 11.Kirkovits G J, Hall C D. Adv Supramol Chem. 2000;7:1–47. [Google Scholar]
- 12.Pérez C, Espínola C G, Foces-Foces C, Núñez-Coello P, Carrasco H, Martín J D. Org Lett. 2000;2:1185–1188. doi: 10.1021/ol005534j. [DOI] [PubMed] [Google Scholar]
- 13.Choma C, Gratkowski H, Lear J D, DeGrado W F. Nat Struct Biol. 2000;7:161–166. doi: 10.1038/72440. [DOI] [PubMed] [Google Scholar]
- 14.Sakai N, Matile S. Chem Eur J. 2000;6:1731–1737. doi: 10.1002/(sici)1521-3765(20000515)6:10<1731::aid-chem1731>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 15.Wang D, Guo L, Zhang J, Jones L R, Chen Z, Pritchard C, Roeske R W. J Peptide Res. 2001;57:301–306. doi: 10.1046/j.1397-002x.2000.00823.x. [DOI] [PubMed] [Google Scholar]
- 16.Whitesides G M, Simanek E E, Mathias J P, Seto C T, Chin D N, Mammen M, Gordon D M. Acc Chem Res. 1995;28:37–44. [Google Scholar]
- 17.Lawrence D S, Jiang T, Levett M. Chem Rev. 1995;95:2229–2260. [Google Scholar]
- 18.Prins L J, Reinhoudt D N, Timmerman P. Angew Chem Int Ed Engl. 2001;40:2382–2426. doi: 10.1002/1521-3773(20010702)40:13<2382::aid-anie2382>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 19.Rebek J., Jr Acc Chem Res. 1999;32:278–286. [Google Scholar]
- 20.Sanders J K M. Chem Eur J. 1998;4:1378–1383. [Google Scholar]
- 21.Matile S. Chem Rec. 2001;1:162–172. doi: 10.1002/tcr.6. [DOI] [PubMed] [Google Scholar]
- 22.Matile S. Chem Soc Rev. 2001;30:158–167. [Google Scholar]
- 23.Sakai N, Gerard D, Matile S. J Am Chem Soc. 2001;123:2517–2524. doi: 10.1021/ja003141+. [DOI] [PubMed] [Google Scholar]
- 24.Sakai N, Brennan K C, Weiss L A, Matile S. J Am Chem Soc. 1997;119:8726–8727. [Google Scholar]
- 25.Baumeister B, Matile S. Chem Eur J. 2000;6:1739–1749. doi: 10.1002/(sici)1521-3765(20000515)6:10<1739::aid-chem1739>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 26.Weiss L A, Sakai N, Ghebremariam B, Ni C, Matile S. J Am Chem Soc. 1997;119:12142–12149. [Google Scholar]
- 27.Wyman T B, Nicol F, Zelphati O, Scaria P V, Planck C, Szoka F C., Jr Biochemistry. 1997;36:3008–3017. doi: 10.1021/bi9618474. [DOI] [PubMed] [Google Scholar]
- 28.Haugland R P. Handbook of Fluorescent Probes and Research Chemicals. 6th Ed. Eugene OR: Mol. Probes; 1996. [Google Scholar]
- 29.Connors K A. Binding Constants. New York: Wiley; 1987. [Google Scholar]
- 30.Hecht M H. Proc Natl Acad Sci USA. 1994;91:8729–8730. doi: 10.1073/pnas.91.19.8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das G, Matile S. Chirality. 2001;13:170–176. doi: 10.1002/1520-636X(2001)13:3<170::AID-CHIR1016>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 32.Baumeister B, Sakai N, Matile S. Angew Chem Int Ed Engl. 2000;39:1955–1959. doi: 10.1002/1521-3773(20000602)39:11<1955::aid-anie1955>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 33.Hine J, Mookerjee P K. J Org Chem. 1975;40:292–298. [Google Scholar]
- 34.Das G, Ouali L, Adrian M, Baumeister B, Wilkinson K J, Matile S. Angew Chem Int Ed Engl. 2001;40:4657–4661. doi: 10.1002/1521-3773(20011217)40:24<4657::aid-anie4657>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 35.Parente R A, Nir S, Szoka F C., Jr Biochemistry. 1990;29:8720–8728. doi: 10.1021/bi00489a031. [DOI] [PubMed] [Google Scholar]
- 36.Baeuerlein E, editor. Biomineralization. New York: Wiley–VCH; 2000. [Google Scholar]
- 37.Wang K, Ohuma S. Trends Biochem Sci. 1999;24:445–451. doi: 10.1016/s0968-0004(99)01464-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.